
Abstract
Background
The progression of normal cells through the cell cycle is meticulously regulated by checkpoints guaranteeing the exact replication of the genome during S-phase and its equal division at mitosis. A prerequisite for this achievement is synchronized DNA-replication and centrosome duplication. In this context the expression of cyclins A and E has been shown to play a principal role.
Results
Our results demonstrated a correlation between centrosome amplification, cell cycle fidelity and the level of mRNA and protein expression of cyclins A and E during the part of the cell cycle defined as G1-phase by means of DNA content based histogram analysis. It is shown that the normal diploid breast cell line HTB-125, the genomically relatively stable aneuploid breast cancer cell line MCF-7, and the genomically unstable aneuploid breast cancer cell line MDA-231 differ remarkably concerning both mRNA and protein expression of the two cyclins during G1-phase. In MDA-231 cells the expression of e.g. cyclin A mRNA was found to be ten times higher than in MCF-7 cells and about 500 times higher than in HTB-125 cells. Topoisomerase II α showed high mRNA expression in MDA compared to MCF-7 cells, but the difference in protein expression was small. Furthermore, we measured centrosome aberrations in 8.4% of the MDA-231 cells, and in only 1.3% of the more stable aneuploid cell line MCF-7. MDA cells showed 27% more incorporation of BrdU than reflected by S-phase determination with flow cytometric DNA content analysis, whereas these values were found to be of the same size in both HTB-125 and MCF-7 cells.
Conclusions
Our data indicate that the breast cancer cell lines MCF-7 and MDA-231, although both DNA-aneuploid, differ significantly regarding the degree of cell cycle disturbance and centrosome aberrations, which partly could explain the different genomic stability of the two cell lines. The results also question the reliability of cytometric DNA content based S-phase determination in genomically unstable tumor cell populations.
Similar content being viewed by others

Background
The centrosome is the major microtubule-organising centre in mammalian cells and is a principal part of the cytoskeleton. It consists of two barrel-shaped centrioles surrounded by pericentriolar material [1] and reproduces during interphase of the cell cycle. During mitosis the centrosome functions as spindle pole, assembling spindle microtubules to guarantee correct chromosome separation [2, 3]. The centrosome duplication cycle proceeds in exact coordination with other cell cycle events, including DNA replication [4]. Recent studies report that centrosome duplication requires phosphorylation of pRb, E2F transcription factors and cdk2 activity [3]. In late G1, prior to the initiation of DNA synthesis, cdk2 associates with cyclin E, which is a vital prerequisite for entrance into S-phase and also centrosome duplication [5, 6]. Evidence has been obtained that overexpression of cyclin E accelerates the G1/S passage [7, 8]. At this point cyclin A can be detected, which also binds to and activates cdk2. Cyclin A increases during S and G2-phase and is degenerated in mitosis [9–11]. Deviating expression of cyclin A and E results in abolished coordination between initiation of centrosome duplication and DNA replication [12]. It has been previously demonstrated that cyclin E protein and mRNA overexpression is common in human breast cancers [13, 14].
The highly conserved topoisomerase II α [15, 16] is a nuclear enzyme that controls the topological state of DNA and is indispensable for DNA replication, recombination and possibly transcription [17, 18]. It also directs condensation and segregation of mitotic chromosomes. Topoisomerase II α expression is low in G1-phase, increases in S-phase and reaches its maximum in G2/M-phase [16, 19]. Failure in coordination of the centrosome duplication and DNA replication cycles inevitably leads to centrosome aberrations, followed by impaired distribution of chromosomes at cell division, and thus to genomic instability [20, 21]. Numerical and qualitative changes of centrosomes are common in human cancers, and are suggested to be one of the principal causes of genomic instability [22, 23].
Image cytometric measurements in serum starved diploid HTB-125 and aneuploid MCF-7 and MDA-231 breast cell lines demonstrated a clear difference in the percentage of cells with nonmodal DNA content values (Fig. 1) as manifestation of genomic instability. Remarkably this difference was also evident comparing the two aneuploid cell lines in demonstrating MCF-7 cells as being genomically more stable than the MDA-231 cell line.

Image cytometry Nuclear DNA content of the cell lines HTB-125, MCF-7 and MDA-231, exposed to serum starvation, and measured by image cytometry of Feulgen stained cells (2c denotes diploid DNA content). We measured 204 cells of the MDA-231 cell line, and 150 cells in MCF-7 and HTB-125 each. G1 indicates the percentage of cells in G1-phase of the cell cycle. S and G2 indicate the percentage of cells in S and G2-phase, respectively. G2 Exc is the percentage of cells with DNA content values exceeding twice the modal value plus 1c.
The aim of our investigation is focused on the expression of cyclins A, E and topoisomerase II α in relation to centrosomal abnormalities and genomic instability, and the significance of these cell cycle abnormalities on the reliability of cytometric S-phase determination.
Results
Image cytometry
Figure 1 illustrates the image DNA histograms of HTB-125, MCF-7 and MDA-231 cultured for 72 hours in medium containing 0.1% FBS. The diploid HTB-125 and the aneuploid MCF-7 showed distinct stemlines without any S-phase fraction and other nonmodal DNA content values. The aneuploid MDA-231 cell line however was characterized by both an unchanged high S-phase and DNA content values clearly deviating from the modal value of the stemline (notice in figure 1 values to the left of the stemline and values exceeding 6c).
mRNA levels of cyclins A, E and topoisomerase II α
Real time quantitative PCR is a very precise method to determine mRNA and DNA amounts. We used this technique to measure the relative mRNA expression levels of cyclins A, E and topoisomerase II α in MDA-231, MCF-7 and HTB-125 cells, which were assorted by centrifugal elutriation to be in G1-phase. The proportion of the G1 cells according to the cellular DNA measurement was 99%, 97%, and 93% for the HTB-125, MCF-7 and MDA-231 cells respectively. The intra-assay CVs (Coefficient of Variation) of the real time measurements were cyclin A 1.8%, cyclin E 1.9%, topoisomerase II α 2.3%. The inter-assay CVs were cyclin A 4.1%, cyclin E 4.3% and topoisomerase II α 4.4%.
Figure 2 demonstrates a high cyclin A mRNA level in MDA-231 cells. The amount of transcripts was 10 times higher than in MCF-7 cells and about 500 times higher than in the HTB-125 cell line. The mRNA levels of cyclin E and topoisomerase II α were also high in the MDA-231 cell line, as compared with the corresponding transcript levels in MCF-7 and HTB-125 cells. Thus, cyclin A and topoisomerase II α, decisive for DNA synthesis and cyclin E, regulating the onset of DNA replication, showed a strong upregulation of their mRNA levels in G1-phase cells of the aneuploid breast cancer cell line MDA-231. In MCF-7 cells this upregulation was rather moderate. That could give a hint at MCF-7 cells still being more under cell cycle control than MDA-231 cells.

Real time qPCR and immunoassays Relative amount of cyclin A, E and topoisomerase II α mRNA determined by running real time quantitative PCR on reverse transcribed total RNA, extracted from HTB-125, MCF-7 and MDA-231 cells in G1-phase, which were assorted by centrifugal elutriation. The algorithm, chosen for calculating the relative amount of target mRNA gives values without any units of measurement. The detection range of the instrument is in our case five orders of magnitude. For the protein determination we calculated the percentage of positively stained G1-phase cells that were assorted by flow cytometry.
An important by-product of our investigation was the confirmation of 18S rRNA being an endogenous control for real time PCR with stable expression levels throughout the cell cycle. There was no detectable difference in 18S rRNA amount between MDA-231 cells in G1-phase and asynchronous MDA-231 cells.
Cyclins A, E and topoisomerase II α protein expression
HTB-125, MCF-7 and MDA-231 cells were stained indirectly with anticyclin A, E and anti topoisomerase II α monoclonal antibodies, and propidium iodide. We analysed and compared specific staining only in the region corresponding to G1-phase.
Figure 2 illustrates 58% (SE (standard error of the mean) = 2.1%) of positive cells for cyclin A in MDA-231, 16 times more than we measured in MCF-7 cells (SE = 1.6%). Cyclin E protein expression was about two times higher in MDA-231 (SE = 3.9%) than in MCF-7 cells (SE = 4.1%), which was still in quite good accordance with the corresponding mRNA measurements. In the case of topoisomerase II α however, the upregulation between MCF-7 and MDA-231 cells on the mRNA level (16 times) differed considerably from those of the protein level (1.2 times). It should be noticed that mRNA and protein expression are not always directly related to each other.
Determination of S-phase fraction by single parameter DNA cytometry as compared with two-parameter DNA-BrdU cytometry
The upper part of figure 3 shows DNA histograms (a, c and e) of the three cell lines that are compared with results from the same cell material double stained for DNA-BrdU (b, d, f) in the lower part of the figure. In case of the diploid cell line the S-phase fraction (SE = 1.2%) calculated from the DNA histogram is in good agreement with the fraction of BrdU positive cells (SE = 1.5%). Also in the aneuploid MCF-7 cell line there is only a minor difference between the S-phase fractions determined by both methods (SE = 3.1%, 1.8% respectively). In contrast, the genomically unstable aneuploid cell line MDA-231 showed a substantial difference between the fraction of BrdU incorporating cells (SE = 1.5%) and the calculated S-phase population (SE = 1.9%). Thus, according to BrdU incorporation there were 27% more cells synthesizing DNA (referred to BrdU incorporation as total amount of DNA synthesis) than detected, using DNA histogram based S-phase fraction determination.

Flow cytometry and BrdU incorporation DNA histograms of HTB-125 (a), MCF-7 (c), MDA-231 (e) cell lines and two parameter histograms (BrdU incorporation vs. DNA content) of HTB-125 (b), MCF-7 (d) and MDA-231 (f) cell lines. As one can see the S-phases in HTB-125 and MCF-7 are very similar with both methods, but in the unstable aneuploid cell line MDA-231, there is a very substantial difference between the measurements with the two methods.
Figure 3f shows DNA synthesizing cells in a range of more than two orders of magnitude, suggesting a genomically unstable cell population. In addition DNA synthesizing cells, which were equally distributed between the early, intermediate and late part of S-phase in the case of the more stable cell line MCF-7, displayed a clear shift towards the early part of the S-phase in MDA-231 cells (picture not shown).
Centrosome staining and analysis
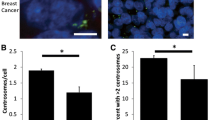
Frequencies of spontaneous centrosome aberrations in the diploid cell line HTB-125 and in the two human breast cancer cell lines MCF-7 and MDA-231 were analysed as described in Materials and Methods (Fig. 4). In HTB-125, only 0.3% (5 of 1601 cells examined) of the cells showed aberrant centrosomes, three of which had three centrosomes per cell and two were irregular in shape. In contrast, in cancer cells, significantly higher percentages of aberrant centrosomes were detected. MCF-7 and MDA-231 showed 1.3% (20 of 1501), and 8.4% (138 of 1643) cells with hyperamplified centrosomes respectively. In the MDA-231 cell line up to 9 centrosomes per cell were observed. These abnormally amplified centrosomes were irregular in shape, or had a bigger size compared with normal ones.

Centosome measurements Percentage of cells that show more than two centrosomes per cell in the cell lines HTB-125, MCF-7 and MDA-231, examined with a Zeiss Axioskop fluorescence microscope.
Discussion
In this study we measured with real time quantitative PCR the mRNA amount of cyclins A, E and topoisomerase II α in centrifugal elutriated G1-phase cells in the normal breast tissue derived diploid cell line HTB-125, the genomically relatively stable aneuploid breast cancer cell line MCF-7 and the genomically unstable aneuploid breast cancer cell line MDA-MB-231. The respective protein amount was determined by indirect immunofluorescence. Furthermore, we stained centrosomes in these three cell lines for hyperamplifications, and we did a comparison between the assessment of S-phase according to FCM and BrdU incorporation.
The three cell lines were chosen as a model for gradually reduced genomic stability as judged from DNA content analysis (Fig. 1). In particular the model should demonstrate different stages of stability among the aneuploid cell lines, manifested by aberrant expression of proliferation markers. Whereas the MCF-7 cells showed only moderate disturbance in regulation, compared to HTB-125 cells, the MDA-231 cell line revealed strongly upregulated cyclin A, E and topoisomerase II α mRNA and cyclin A, E protein levels. Topoisomerase II α showed high mRNA expression in MDA compared to MCF-7 cells, but the difference in protein expression was small. Taking into account that cyclin E and A are crucial for G1/S transition, their upregulation in MDA-231 cells might imply severe disturbance of regulating this checkpoint. An additional support of this assumption is the upregulated topoisomerase II α mRNA level, during the G1-phase of the cell cycle, as determined by cytometric DNA content measurements. There are other reports about cyclin A upregulation on the protein level in breast cancer cell lines and in aneuploid breast carcinomas [13, 24]. Furthermore, there are data reporting aberrant cyclin E mRNA and protein expression in breast cancer [14, 25].
The data obtained in the present study, indicate the existence of two different types of aneuploid species represented by the breast cancer cell line MCF-7, which is genomically relatively stable, as reflected in DNA content measurements, and the other breast cancer cell line MDA-231 that is genomically unstable. One reason for aneuploidy can be unequal distribution of chromosomes among the daughter cells based on abnormally amplified centrosomes. P53 seems to be involved in regulation of centrosome duplication [26, 27], which might be one cause for mutant p53 MDA-231 cells [28] showing, in our investigation, centrosome hyperamplifications in 8.4% of the cells. In contrast, MCF-7 cells with wild type p53 showed only in 1.3% of their cells centrosomal aberrations. Another reason for cells to become aneuploid is to undergo endoreduplication. Treating cells with a compromised G1/S checkpoint or cells that are Rb or p21 negative with microtubule inhibitors, will make them replicate the genome without going through mitosis and thus achieving the status of polyploidy. These cells prove to be unstable, losing chromosomes randomly to become aneuploid [29, 30]. Furthermore, cyclin A and E seem to be involved in the process of centrosome duplication [3, 5, 31, 32]. If cyclin A and E are overexpressed during G1, centrosome duplication and DNA replication cycles might occur asynchronously.
Our results show that upregulation of cyclins A, E and topoisomerase II α as well as centrosome hyperamplification might be signs of disturbed G1/S checkpoint regulation preferentially in the unstable aneuploid cell line MDA-231. In addition, our data indicate the existence of different graduations of erroneous checkpoint regulation. MCF-7 cells, although aneuploid, seem to exhibit minor checkpoint control aberrations in showing also only few centrosome hyperamplifications.
A further aspect of this work is the comparison between the assessment of S-phase according to the FCM method and to the number of BrdU incorporating cells. The basis of the BrdU method is that cells are pulse-labeled with the thymidine analogue BrdU, resulting in labeling of DNA synthesizing cells. Incorporated BrdU is detected by the use of primary BrdU antibodies, secondary fluorescein isothiocyanate (FITC)-conjugated antibodies and is then quantified by FCM analysis. We found that the two separate measurements did not give any significant difference in the normal breast tissue derived cell line and in MCF-7 cells. However, in MDA cells there was 27% more incorporation of BrdU than reflected by S-phase determination with the FCM method. According to the results presented here, this might at least in part be due to a substantial number of cells synthesizing DNA, but being located in the G1 compartment of the DNA content histogram. Obviously, the broadened trapezoidal model used for fitting of the S-phase underestimates this fraction of BrdU positive cells in G1. In a recent study, we got positive correlation between 9 years survival and S-phase fraction in diploid breast tumors, but not in aneuploid ones (to be published). A possible explanation for this discrepancy may be that S-phase measurement by FCM does not prove to be precise and reliable in genomically unstable cells. The clinical implications are critical since estimation of S-phase fraction as a measure of tumor growth activity is frequently decisive for choice of therapy.
Conclusions
Taken together our results indicate that the aneuploid breast cancer cell lines MCF-7 and MDA-MB-231, although both aneuploid, differ significantly regarding the fraction of cells expressing cyclins A and E and topoisomerase II α (on the mRNA level) during the part of the cell cycle defined as G1-phase by means of DNA content based histogram analysis. The degree of this disturbance seems to be correlated with centrosome hyperamplification and may cover aspects of genomic instability. The results also question the reliability of cytometric DNA content based S-phase determination in genomically unstable tumor cell populations.
Methods
Cell lines and culture conditions
The DNA-diploid Hs 578Bst cell line (ATCC-HTB-125) was derived from normal human breast tissue peripheral to an infiltrating ductal carcinoma. The cells were grown in modified Dulbecco's medium with 30 ng/ml EGF (90%), 50 μg/ml penicillin, 50 μg/ml streptomycin, supplemented with 10% FBS.
The DNA-hypotetraploid MCF-7 cell line (ATCC-HTB-22) originally came from a human breast adenocarcinoma. The cells were grown in minimum essential medium (Gibco BRL) with 2 mM L-glutamine, 0.1 mM non-essential amino acids, 1.0 mM sodium pyruvate, 50 μg/ml streptomycin and 10% FBS.
The DNA-hypotriploid MDA-MB-231 cell line (ATCC-HTB-26) was derived from a human breast adenocarcinoma. These cells were grown in Dulbecco's MOD eagle medium (Gibco BRL) with 2 mM L-glutamine, 1.0 mM sodium pyruvate, 50 μg/ml penicillin, 50 μg/ml streptomycin, and 10% FBS.
The cells were incubated at 37°C in a humid atmosphere of 5% CO2 in 325 cm2 flasks and were subcultured when having reached confluence, using 0.25% trypsin-0.03% EDTA solution (Gibco BRL). For experimental purposes, cells were seeded at a concentration 10–20,000 cells/cm2 and harvested before having reached confluence. The medium was removed and the cells were then washed once in 0.25% trypsin (Gibco). After removing trypsin the cells were kept at room temperature for 10 minutes. Then they were shaken vigorously, 10 ml of PBS was added and the mixture was shaken again. The cell suspension was transferred to a 15 ml tube. After 5 minutes of centrifugation at 3,000 RPM, the supernatant was removed and the cells kept at 4°C until further treatment. Serum starvation was performed by culturing the cells in medium containing 0.1% FBS during 72 hours.
Centrifugal elutriation
Separation of cells on the basis of different cell size was made in a Beckman JE-G centrifugal elutriator rotor. Exponentially growing cells were harvested by trypsination, washed, and resuspended in respective medium. Approximately 2 × 108 cells were elutriated in each run at 4°C. The rotor speed was 2,780 rpm, and the initial flow rate was 12 ml/min. Five fractions were obtained by stepwise increase of the flow rate up to 45 ml/min. The first fraction containing partly non-viable cells was discarded. The cells of the second fraction containing entirely G1 cells were collected by centrifugation and resuspended in PBS. Since about 4–6 × 106 cells were needed for the mRNA measurement, fractions from several elutriation runs were stored in PBS at -20°C. Aliquots were fixed in buffered formalin for flow-cytometric analysis of the cellular DNA content as described earlier [33].
Determination of mRNA
G1 cells assorted by centrifugal elutriation were used for RNA extraction, cDNA synthesis and real time quantitative PCR with an amplicon for cyclins A, E and topoisomerase II α.
RNA extraction and DNase treatment
Total RNA was extracted using the Trizol reagent, followed by treatment with DNAse I (both reagents from Life Technologies, Paisley, UK). After 15 minutes of incubation at room temperature, the enzyme was inactivated with 25 mM EDTA, followed by heating for 10 minutes at 70°C to denature DNase.
cDNA synthesis
We reverse transcribed 2 μg of total RNA in a final reaction volume of 20 μl, containing 1xFirst Strand Buffer (50 mM Tris-HCl, 75 mM KCl, 3 mM MgCl2), 100 ng random hexamers, 0.1 M DTT, 10 mM dNTP mix, Applied Biosystems (ABI), 200 units Superscript II RNase H- Reverse Transcriptase and DEPC treated water (all other reagents from Life Technologies). First, the mixture of total RNA, random hexamers and DEPC treated water was kept at 70°C for 10 minutes and afterwards put on ice. Then the mix containing buffer, DTT and dNTPs was added, followed by incubation at 25°C for 10 minutes. After two minutes at 42°C, the Superscript II was spiked in for the remaining 50 minutes of reverse transcription. We stopped the reaction by heating to 70°C for 15 minutes.
Real time quantitative PCR
With real time PCR the measurement is taking place at a quite early stage during the polymerase chain reaction. At the exponential phase of the amplification process, all reagents are still abundant and there is no danger of limiting conditions, which might interfere with a precise quantitation. This is an advantage over endpoint analysis, where small variations in starting reaction conditions may escalate and lead to totally different final product yield for the same starting copy number of template [34]. Concerning real time quantitation the crucial value is the threshold cycle (CT), which is the fractional cycle number during PCR at which the amount of amplified target in each sample reaches a fixed threshold above the background noise [35, 36]. The product accumulation is directly proportional to the increase in fluorescent emission that occurs if the Taq polymerase is performing a 5'-3' nucleolytic digestion of the dual-labeled fluorogenic probe, which has to be hybridized to the template. On the other hand, the CT-value is inversely proportional to the logarithm of the initial template copy number [37–39]. As long as the 5'-reporter dye (distinct fluoresceins: FAM, VIC, TET) is in a certain spatial proximity to the 3'-quencher dye (TAMRA, 6-carboxy-tetramethylrhodamine), no fluorescence is detectable [36].
Relative quantitation of cyclin A, E and topoisomerase II α mRNA levels
Cyclin A, E and topoisomerase II α mRNA levels were determined by performing real time PCR. We did a relative quantitation according to the comparative CT method, 18S rRNA served as the endogenous external control and our inter-run standard was cDNA prepared from asynchronous MDA-MB-231 cells. All PCR-reactions were performed on a 7700 Sequence Detector (ABI). We ran each amplicon (cyclins A, E, topoisomerase II α and 18S rRNA) in triplicates for all cell lines. From the mean CT-values of the respective triplicates the mean CT-value of the 18S rRNA amplicon was subtracted. The purpose of the endogenous control is to correct for differences in pipetting the cDNA from the cell lines and to compensate for putative reverse transcription and PCR inhibition. This subtraction gave us ΔCT-values. The CT-value of the inter-run standard was subtracted from all the other ΔCT-values, and that resulted in ΔΔCT-values. The last step in getting the relative amount of target mRNA is to calculate 2-ΔΔCT [40].
Primers, probes and the PCR reaction
Primers and probes for the targets cyclin A, E and topoisomerase II α were found with the aid of Primer Express 1.0 (ABI, Foster City, CA), a computer program specialized in designing short amplicons for real time PCR. To exclude an amplification from genomic DNA, we placed one primer of each amplicon across an exon/exon junction in getting not more than six bases of the 3'-end on one exon and the remaining nucleotides of this primer on the adjoining one. The probes we purchased from ABI, Warrington, Cheshire, UK; primers were ordered from Life Technologies. The complete 18S rRNA amplicon was already designed by ABI.
The reaction volume for the PCR was 25 μl. Each master mix comprised 1xTaqMan buffer A, 6 mM MgCl2, 200 μM dATP, dCTP, dGTP, dTTP and 0.625 units of AmpliTaq Gold DNA polymerase (all reagents from ABI). We used the probe for cyclins A, E and topoisomerase II α at a concentration of 100 nM and the probe for 18S rRNA at a concentration of 75 nM. The primers for cyclins A, E and topoisomerase II α had the concentration of 200 nM, in the case of the 18S rRNA the concentration of the forward primer was 50 nM, those of the reverse was 40 nM. Per 25 μl reaction volume 25 ng reverse transcribed total RNA was added. The thermal cycling conditions were as follows: 10 minutes at 95°C (activation of the Taq polymerase), and 40 cycles at 95°C for 15 seconds and 62°C for 1 minute. All target genes and the endogenous control were run in triplicates for all cell lines. Additionally we ran non-template controls in triplicates for all targets and the endogenous control.
Image cytometry
Image cytometry was performed on Feulgen stained cells. The staining and standardization were based on methods previously described [41].
A cell line was defined as genomically relatively stable provided that it showed a) confluence and/or serum starvation induced growth arrest in G0/G1, b) low percentage of cells with nonmodal DNA content values (Fig. 1). Consequently, a genomically unstable cell line was defined by a) unchanged high proliferative activity at confluence and/or upon serum starvation, b) high percentage of cells with nonmodal DNA content values (Fig. 1).
Flow cytometry
Single parameter DNA flow cytometry
For DNA histograms, cells were harvested and analysed as described previously [33]. Briefly, cells were fixed in 4% buffered formaldehyde for 18 hours at room temperature. Formaldehyde was removed by 95% ethanol for 1 hour followed by rehydration in distilled water for 1 hour. After treatment with subtilisin Carlsberg solution [0.1% Sigma protease XXIV, 0.1 M Tris and 0.07 M NaCl (pH 7,5)] and staining with DAPI-Sulforhodamine solution [8 μM DAPI, 50 μM Sulforhodamine 101, 0.1 M Tris and 0.07 M NaCl (pH 7.5)], samples were analysed using a PAS II flowcytometer (Partec, Münster, Germany) equipped with a 100 W mercury arc lamp HBO 100. DAPI fluorescence was measured above 435 nm. The multicycle program for cell cycle analysis (Phoenix flow systems, San Diego, CA) was used for histogram analysis; the number of nuclei/histogram was 40,000. The S-phase was fitted to a broadened trapezoidal model. For background correction the sliced-nuclei model was applied.
Two parameter DNA-BrdU analysis
For BrdU (5'-Bromo-2'-deoxyuridine) labeling, cells were precultured for 24 hours, pulse labeled for 10 minutes with 20 μM BrdU, washed twice with culturing medium and then recultured with new medium for 20 minutes, followed by harvesting as before and fixation in 4% buffered formaldehyde. Immunostaining was carried out using a modified technique described by Sangfelt et al. [42] with 2 mg/ml monoclonal anti-BrdU antibodies (Boehringer Mannheim Scandinavia AB, Bromma, Sweden) for 1 hour at room temperature and FITC-conjugated anti mouse IgG antibodies (F-0257, Sigma), diluted 1/100 in PBT (PBS, 2% BSA, 0,5% Tween 20) for 1 hour in the dark at room temperature. DNA was stained with 10 μM propidium iodide (PI) for 20 minutes. A Nikon fluorescence microscope equipped with a mercury arc lamp HBO 100 was used for control of the quality of immunostaining. The samples were analysed in a FACScalibur flowcytometer (Becton Dickinson, Sweden) for two-parameter dot plot histogram analysis (BrdU incorporation vs. DNA content).
Cell staining and indirect immunofluorescence
Indirect immunofluorescence was performed by the method of Gong et al. [43]. As negative control, we used FITC-conjugated mouse IgE monoclonal antibody (13834X, Pharmingen). All four antibodies: cyclin E (HE 121, Santa Cruz Biotechnology), cyclin A (6E6, Novocastra Laboratories UK), topoisomerase II α (Novocastra Laboratories UK) and IgE were incubated for 1 hour at room temperature, followed by incubation with the secondary antibody (FITC-conjugated IgG anti mouse antibody) for 1 hour in the dark at room temperature diluted 1/100 in PBT. DNA was stained with propidium iodide (10 μg/ml in PBS+RNAse 1 mg/ml). The samples were kept in the dark for 20 minutes before running them in the FACS. The FACS was equipped with an argon laser; the FITC fluorescence was analysed at 515 nm and the PI at 488 nm. 20,000 events were stored for every sample. The Cell Quest (Becton Dickinson) program was used for the two-parameter dot plot histogram analysis.
Centrosome staining
Cells were seeded onto sterile glass slides in culture dishes and cultivated for 3 days. After rinsing with PBS, cells were fixed in methanol at -20°C for 10 min and in acetone at -20°C for 6 min. After each incubation cells were extensively rinsed with PBS. All procedures were done at room temperature. Blocking solution (1% normal goat serum, 0.1% Tween 20/PBS) was applied to the cells for 30 min at 37°C. Cells were incubated with anti γ-tubulin antibody (Sigma T6557), diluted 1:1000 in PBS, containing 2% normal goat serum, for 1 hour. Antibody-antigen complexes were detected by FITC-conjugated anti mouse IgG. Stained slides were mounted with Vectashield mounting medium. With a Zeiss Axioskop fluorescence microscope, the proportion of cells with more than two centrosomes was counted in more than 500 cells of each cell line.
References
Paintrand M, Moudjou M, Delacroix H, Bornens M: Centrosome organization and centriole architecture: their sensitivity to divalent cations. J Struct Biol. 1992, 108 (2): 107-128.
Kellogg DR, Moritz M, Alberts BM: The centrosome and cellular organization. Annu Rev Biochem. 1994, 63: 639-674. 10.1146/annurev.bi.63.070194.003231.
Meraldi P, Lukas J, Fry AM, Bartek J, Nigg EA: Centrosome duplication in mammalian somatic cells requires E2F and Cdk2-cyclin A. Nat Cell Biol. 1999, 1 (2): 88-93. 10.1038/10054.
Sluder G, Hinchcliffe EH: The apparent linkage between centriole replication and the S phase of the cell cycle. Cell Biol Int. 1998, 22 (1): 3-5. 10.1006/cbir.1997.0249.
Hinchcliffe EH, Li C, Thompson EA, Maller JL, Sluder G: Requirement of Cdk2-cyclin E activity for repeated centrosome reproduction in Xenopus egg extracts. Science. 1999, 283 (5403): 851-854. 10.1126/science.283.5403.851.
Wimmel A, Lucibello FC, Sewing A, Adolph S, Muller R: Inducible acceleration of G1 progression through tetracycline-regulated expression of human cyclin E. Oncogene. 1994, 9 (3): 995-997.
Morgan DO: Cyclin-dependent kinases: engines, clocks, and microprocessors. Annu Rev Cell Dev Biol. 1997, 13: 261-291. 10.1146/annurev.cellbio.13.1.261.
Reed SI: Control of the G1/S transition. Cancer Surv. 1997, 29: 7-23.
Geley S, Kramer E, Gieffers C, Gannon J, Peters JM, Hunt T: Anaphase-promoting complex/cyclosome-dependent proteolysis of human cyclin A starts at the beginning of mitosis and is not subject to the spindle assembly checkpoint. J Cell Biol. 2001, 153 (1): 137-148. 10.1083/jcb.153.1.137.
Girard F, Strausfeld U, Fernandez A, Lamb NJ: Cyclin A is required for the onset of DNA replication in mammalian fibroblasts. Cell. 1991, 67 (6): 1169-1179.
Pagano M, Pepperkok R, Verde F, Ansorge W, Draetta G: Cyclin A is required at two points in the human cell cycle. Embo J. 1992, 11 (3): 961-971.
Lacey KR, Jackson PK, Stearns T: Cyclin-dependent kinase control of centrosome duplication. Proc Natl Acad Sci U S A. 1999, 96 (6): 2817-2822. 10.1073/pnas.96.6.2817.
Keyomarsi K, Pardee AB: Redundant cyclin overexpression and gene amplification in breast cancer cells. Proc Natl Acad Sci U S A. 1993, 90 (3): 1112-1116.
Nielsen NH, Arnerlov C, Emdin SO, Landberg G: Cyclin E overexpression, a negative prognostic factor in breast cancer with strong correlation to oestrogen receptor status. Br J Cancer. 1996, 74 (6): 874-880.
Lynch BJ, Guinee DG, Holden JA: Human DNA topoisomerase II-alpha: a new marker of cell proliferation in invasive breast cancer. Hum Pathol. 1997, 28 (10): 1180-1188.
Woessner RD, Mattern MR, Mirabelli CK, Johnson RK, Drake FH: Proliferation- and cell cycle-dependent differences in expression of the 170 kilodalton and 180 kilodalton forms of topoisomerase II in NIH-3T3 cells. Cell Growth Differ. 1991, 2 (4): 209-214.
Osheroff N, Zechiedrich EL, Gale KC: Catalytic function of DNA topoisomerase II. Bioessays. 1991, 13 (6): 269-273.
Wang JC: DNA topoisomerases. Annu Rev Biochem. 1985, 54: 665-697. 10.1146/annurev.bi.54.070185.003313.
Goswami PC, Roti Roti JL, Hunt CR: The cell cycle-coupled expression of topoisomerase IIalpha during S phase is regulated by mRNA stability and is disrupted by heat shock or ionizing radiation. Mol Cell Biol. 1996, 16 (4): 1500-1508.
Brinkley BR, Goepfert TM: Supernumerary centrosomes and cancer: Boveri's hypothesis resurrected. Cell Motil Cytoskeleton. 1998, 41 (4): 281-288. 10.1002/(SICI)1097-0169(1998)41:4<281::AID-CM1>3.0.CO;2-C.
Orr-Weaver TL, Weinberg RA: A checkpoint on the road to cancer. Nature. 1998, 392 (6673): 223-224. 10.1038/32520.
Levine DS, Sanchez CA, Rabinovitch PS, Reid BJ: Formation of the tetraploid intermediate is associated with the development of cells with more than four centrioles in the elastase-simian virus 40 tumor antigen transgenic mouse model of pancreatic cancer. Proc Natl Acad Sci U S A. 1991, 88 (15): 6427-6431.
Lingle WL, Lutz WH, Ingle JN, Maihle NJ, Salisbury JL: Centrosome hypertrophy in human breast tumors: implications for genomic stability and cell polarity. Proc Natl Acad Sci U S A. 1998, 95 (6): 2950-2955. 10.1073/pnas.95.6.2950.
Collecchi P, Santoni T, Gnesi E, Giuseppe Naccarato A, Passoni A, Rocchetta M, Danesi R, Bevilacqua G: Cyclins of phases G1, S and G2/M are overexpressed in aneuploid mammary carcinomas. Cytometry. 2000, 42 (4): 254-260. 10.1002/1097-0320(20000815)42:4<254::AID-CYTO6>3.3.CO;2-J.
Stanta G, Bonin S, Losi L, Eusebi V: Molecular characterization of intraductal breast carcinomas. Virchows Arch. 1998, 432 (2): 107-111. 10.1007/s004280050142.
Fukasawa K, Choi T, Kuriyama R, Rulong S, Vande Woude GF: Abnormal centrosome amplification in the absence of p53. Science. 1996, 271 (5256): 1744-1747.
Tarapore P, Horn HF, Tokuyama Y, Fukasawa K: Direct regulation of the centrosome duplication cycle by the p53-p21Waf1/Cip1 pathway. Oncogene. 2001, 20 (25): 3173-3184. 10.1038/sj.onc.1204424.
Xu J, Loo G: Different effects of genistein on molecular markers related to apoptosis in two phenotypically dissimilar breast cancer cell lines. J Cell Biochem. 2001, 82 (1): 78-88. 10.1002/jcb.1147.
Niculescu AB, Chen X, Smeets M, Hengst L, Prives C, Reed SI: Effects of p21(Cip1/Waf1) at both the G1/S and the G2/M cell cycle transitions: pRb is a critical determinant in blocking DNA replication and in preventing endoreduplication. Mol Cell Biol. 1998, 18 (1): 629-643.
Stewart ZA, Leach SD, Pietenpol JA: p21(Waf1/Cip1) inhibition of cyclin E/Cdk2 activity prevents endoreduplication after mitotic spindle disruption. Mol Cell Biol. 1999, 19 (1): 205-215.
Hinchcliffe EH, Sluder G: "It takes two to tango": understanding how centrosome duplication is regulated throughout the cell cycle. Genes Dev. 2001, 15 (10): 1167-1181. 10.1101/gad.894001.
Meraldi P, Nigg EA: Centrosome cohesion is regulated by a balance of kinase and phosphatase activities. J Cell Sci. 2001, 114 (Pt 20): 3749-3757.
Castro J, Heiden T, Wang N, Tribukait B: Preparation of cell nuclei from fresh tissues for high-quality DNA flow cytometry. Cytometry. 1993, 14 (7): 793-804.
Orlando C, Pinzani P, Pazzagli M: Developments in quantitative PCR. Clin Chem Lab Med. 1998, 36 (5): 255-269.
Gibson UE, Heid CA, Williams PM: A novel method for real time quantitative RT-PCR. Genome Res. 1996, 6 (10): 995-1001.
Heid CA, Stevens J, Livak KJ, Williams PM: Real time quantitative PCR. Genome Res. 1996, 6 (10): 986-994.
Holland PM, Abramson RD, Watson R, Gelfand DH: Detection of specific polymerase chain reaction product by utilizing the 5'----3' exonuclease activity of Thermus aquaticus DNA polymerase. Proc Natl Acad Sci U S A. 1991, 88 (16): 7276-7280.
Lee LG, Connell CR, Bloch W: Allelic discrimination by nick-translation PCR with fluorogenic probes. Nucleic Acids Res. 1993, 21 (16): 3761-3766.
Higuchi R, Fockler C, Dollinger G, Watson R: Kinetic PCR analysis: real-time monitoring of DNA amplification reactions. Biotechnology (N Y). 1993, 11 (9): 1026-1030.
User Bulletin #2. ABI PRISM Sequence Detection System. 1997, [http://www.ukl.uni-freiburg.de/core-facility/taqman/user_bulletin_2.pdf]
Auer GU, Caspersson TO, Wallgren AS: DNA content and survival in mammary carcinoma. Anal Quant Cytol. 1980, 2 (3): 161-165.
Sangfelt O, Erickson S, Castro J, Heiden T, Gustafsson A, Einhorn S, Grander D: Molecular mechanisms underlying interferon-alpha-induced G0/G1 arrest: CKI-mediated regulation of G1 Cdk-complexes and activation of pocket proteins. Oncogene. 1999, 18 (18): 2798-2810. 10.1038/sj.onc.1202609.
Gong J, Traganos F, Darzynkiewicz Z: Growth imbalance and altered expression of cyclins B1, A, E, and D3 in MOLT-4 cells synchronized in the cell cycle by inhibitors of DNA replication. Cell Growth Differ. 1995, 6 (11): 1485-1493.
Acknowledgements
This work was supported by grants from the Swedish Cancer Society (Cancerfonden) and the Cancer Society in Stockholm (Cancerföreningen). UJR receives a research grant from the Medical University of Luebeck (N14-2001).
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
UK carried out the real time qPCR experiments and the image cytometry, and did the main work on the manuscript. JC carried out the centrifugal elutriation, the immunofluorescent determination (cyclins, topoisomerase II α, BrdU) and the FCM. UJR assisted in carrying out the experiments and helped with the manuscript. KF did the centrosome staining. CÖ, FF and NL-L participated in doing the immunoassays and the FCM. BT helped with the interpretation of the FCM data analysis. GA planned and coordinated the study.
Ulrike Kronenwett, Juan Castro contributed equally to this work.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Kronenwett, U., Castro, J., Roblick, U.J. et al. Expression of Cyclins A, E and Topoisomerase II α correlates with centrosome amplification and genomic instability and influences the reliability of cytometric S-phase determination. BMC Cell Biol 4, 8 (2003). https://doi.org/10.1186/1471-2121-4-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2121-4-8