
Abstract
Background
For many RNA molecules, secondary structure rather than primary sequence is the evolutionarily conserved feature. No programs have yet been published that allow searching a sequence database for homologs of a single RNA molecule on the basis of secondary structure.
Results
We have developed a program, RSEARCH, that takes a single RNA sequence with its secondary structure and utilizes a local alignment algorithm to search a database for homologous RNAs. For this purpose, we have developed a series of base pair and single nucleotide substitution matrices for RNA sequences called RIBOSUM matrices. RSEARCH reports the statistical confidence for each hit as well as the structural alignment of the hit. We show several examples in which RSEARCH outperforms the primary sequence search programs BLAST and SSEARCH. The primary drawback of the program is that it is slow. The C code for RSEARCH is freely available from our lab's website.
Conclusion
RSEARCH outperforms primary sequence programs in finding homologs of structured RNA sequences.
Similar content being viewed by others


Introduction
Ribonucleic acid (RNA) can fold back onto itself to form a base-paired secondary structure. This phenomenon confers functional specificity to a wide range of RNA molecules. For some protein-coding genes, secondary structure signals present in the messenger RNA help regulate the gene. Examples of such control elements include the iron-responsive element in genes involved in iron metabolism, the selenocysteine insertion sequence that signals selenocysteine should be incorporated into the amino acid, and riboswitches that directly alter gene expression in response to the concentration of small molecules such as thiamin [1–7]. Other genes do not code for protein; the transcripts of these noncoding RNA (ncRNA) genes are the biochemically functional end product in the cell [8, 9].
We are interested in the problem of finding homologs of such RNA sequences. For both protein and RNA, homology is most readily inferred at the tertiary structure level. For most proteins and RNAs, however, we only have primary sequence data and do not know the tertiary structure. For RNA, secondary structure confers much functional specificity, and potential folds are readily discernible from the primary sequence. Therefore, we can obtain increased power in homology searching by considering the secondary structure of RNA sequences [10].
It is useful to distinguish three classes of alignment algorithms that can be used to find homologs of RNA sequences. The first class only uses primary sequence information to align the query sequence to the target database. Such searches are exemplified by the Smith-Waterman algorithm and its heuristic approximations found in programs like BLAST and FASTA. These sequence alignment programs are O(N2) in time and memory [11–13], where N is the length of the sequences being analysed. The second class consists of a search with a known RNA structure against a sequence database. Such searches have been implemented with profile stochastic context free grammars (SCFGs) and require O(N3) memory and O(N4) time [14–18]. Alternatively, such searches can be performed by defining an RNA structural pattern, though this approach works best on highly conserved secondary structures, and patterns have to be developed by hand [19–22]. A third type of approach consists of a search with a query sequence with an unknown secondary structure, where the algorithm searches over all possible foldings of the query aligned to the target. Sankoff described such an algorithm, which is O(N4) in memory and O(N6) in time [23].
While it is convenient to distnguish among these three classes of algorithms, the boundaries between them are not absolute. Various constrained versions of the Sankoff algorithm have been published that allow it to run in a reasonable amount of time [24–26]. One such algorithm constrains the possible alignments [25], while the other two constrain the foldings allowed [24, 26]. Holmes and Rubin introduced the idea of a "fold envoelope," which allows the algorithm to be constrained to a subset of folds. It can be argued that the profile SCFG approach to searching a database with an RNA of known structure is the limiting case where the fold envelope only includes one structure.
Three types of scoring functions can be used with these search algorithms. When only a single query sequence is given, log-odds position independent substitution matrices are used to give the alignment scores. These are analogous to the BLOSUM matrices used in protein searches [27]. In the pattern search approach, a binary match/doesn't match scoring function is generally used where all allowed letters at each position are enumerated. This is analogous to PROSITE patterns used to analyze amino acid sequences [28]. Finally, a profile-based scoring scheme can be used where position dependent log-odds scores are derived from the observed frequencies in a multiple sequence alignment. This is analogous to the profile approach used in many protein database search programs, including profile hidden Markov models [29–31]. For RNA sequences, only the pattern approach [19–22] and the profile approach [14, 15, 17, 18] to finding homologs of an RNA sequence in a nucleotide sequence database have been described to date.
Here we are specifically interested in the problem of finding structural homologs of a single RNA sequence. Since the alignment algorithm is essentially independent of the scoring system, developing such a tool is just a matter of developing an appropriate pairwise substitution matrix and combining it with one of the aforementioned alignment algorithms. We could, for example, derive a single nucleotide matrix and use it in BLASTN searches. Such a primary sequence search would lose much information, much like doing a BLASTN search for homologs of a protein-coding sequence would. When RNAs have conserved secondary structure, we want to consider the intramolecular base pairs that provide this structure to find homologs optimally [10]. While using the Sankoff algorithm would be ideal, as we often do not know the correct secondary structure of a single query RNA sequence, its cost in time and memory is so prohibitive as to make it impractical at this time for sequence database searching. Therefore, we have chosen to focus on the case where we know the secondary structure of the query sequence.
Here we describe RSEARCH, a program that, given a query sequence with a known secondary structure, searches a nucleotide sequence database for similar RNAs on the basis of both primary sequence and secondary structure. Its core alignment algorithm is identical to profile SCGG alignment [14, 16, 18]. Since alignments are pairwise, alignments are scored using appropriate pairwise substitution matrices. Furthermore, analogous to BLAST, the program calculates statistical confidence values for all hits [32]. It is still quite slow; for the time being, we deal with this problem through brute force by parallelizing the search program for clustered computing using the Message Passing Interface (MPI) library [33].
Implementation
RIBOSUM substitution matrices
In order to perform database searches with a single, folded RNA sequence query, a 16 × 16 substitution matrix for scoring aligned base pairs and a 4 × 4 matrix for single aligned nucleotides are needed. Such matrices should give the log-odds ratio for observing a given substitution relative to background nucleotide frequencies [34]. Specifically, for the 4 × 4 single nucleotide matrix, the individual scores are given by

where i and j are the two aligned nucleotides, f ij is the empirically observed frequency of i aligned to j in homologous RNAs, and g i and g j are the background frequencies of the individual nucleotides. Similarly, for the 16 × 16 base pairing matrix, the individual scores are given by

where i is basepaired to j, k is basepaired to l, i is aligned with k, and j is aligned with l. In this case,  is the observed frequency of the two base pairs i - j and k - l aligned to each other in homologous RNAs. g again is the background frequency of the individual nucleotides. Note that g is used for individual nucleotides and not base pairs; the null model in this case is an identical and independently distributed (i.i.d.) model consisting of unaligned random sequences that do not base pair.
is the observed frequency of the two base pairs i - j and k - l aligned to each other in homologous RNAs. g again is the background frequency of the individual nucleotides. Note that g is used for individual nucleotides and not base pairs; the null model in this case is an identical and independently distributed (i.i.d.) model consisting of unaligned random sequences that do not base pair.
The key question, then, is how to find the values for f, f', and g needed to calculate these matrices. The values in f and f' are conditional on evolutionary divergence time; a shorter divergence time implies higher scores for identities and lower scores for mismatches. Two methods exist to account for evolutionary divergence time. The first method, used by Dayhoff to construct the PAM matrices, infers a rate matrix from closely related sequences. This rate matrix is then used to calculate an exponential family of matrices at different evolutionary distances [35]. The second method, used to construct the BLOSUM family of matrices, filters and weights sequences in a multiple sequence alignment to approximate a range around some time point [27]. Matrices produced using the latter method have been found to perform better [36], though it is in dispute whether this is an effect of the algorithm or the underlying data used to generate the matrices [37]. Several evolutionary models and a rate matrices have been published for RNA evolution [38–40]. Because BLOSUM-style matrices are argued to be better for finding distant homology relationships [36], we have chosen to forgo the pre-existing RNA rate matrices and construct BLOSUM-style matrices instead.
The algorithm starts with a structurally annotated alignment of multiple RNA sequences to be used as training data. The consensus secondary structure is mapped onto individual sequences by removing any base pairs from the secondary structure for an individual sequence that align with a gap in that sequence. Sequences are then weighted by grouping all sequences more than a certain percentage identical using single-linkage clustering; all sequences in a group are given equal weights that sum to 1. This is identical to the clustering used in constructing the BLOSUM matrices [27]. The percent identity used in this clustering is the first number in the matrix name. In order to allow for a shorter evolutionary distance than would be allowed by following the BLOSUM algorithm exactly, we added a second percentage identity cutoff not found in the original BLOSUM algorithm. Only pairs of sequences whose percent identity meet or exceed this cutoff are counted at all. This threshold is the second number in the matrix name. It should be noted that this second threshold does not necessarily have to be less than the first, clustering percent identity. If that is the case, then one would be counting weighted pairs within clusters; no intercluster pairs would be counted.
Let each of i, j, k, l represent a nucleotide (1 ≤ i, j, k, l ≤ 4). Then, two triangular count matrices are initialized using c
ij
= 0 (1 ≤ i ≤ j ≤ 4),  = 0 (1 ≤ 4i + j ≤ 4k + l ≤ 16), where c is the count matrix for single-stranded regions and c' is the count matrix for basepaired nucleotides (an ij basepair aligned to a kl basepair). Triangular matrices are used because nucleotide (base pair) X in sequence 1 aligned to nucleotide (base pair) Y in sequence 2 should count the ssame as nucleotide (base pair) Y in sequence 1 aligned to nucleotide (base pair) X in sequence 2. However, we assume that an X-Y base pair may not be equivalent to a Y-X base pair in the context of the entire RNA molecule and therefore count these pairs separately. A count vector d
i
= 0(1 ≤ i ≤ 4) is also initialized for background nucleotide frequencies. Each pair of sequences is then examined. If the pair does not meet the minimal percent identity criterion, it is skipped and the next pair is examined. Otherwise, the weight of this pairing, w is set to be the average of the weights given to the two individual sequences. (Arguably, this weight should be set to be the product rather than the average of the individual weights. Though we did not fully explore this possibility, preliminary evidence suggests the method of calculating this weight does not appreciably influence performance.) For each aligned base pair (ij, kl) in the alignment, w is added to , d
i
, d
j
, d
k
, and d
l
; for all other aligned nucleotides (i, j), w is added to c
ij
, d
i
, and d
j
. The counts are then converted to empirical frequencies using:
= 0 (1 ≤ 4i + j ≤ 4k + l ≤ 16), where c is the count matrix for single-stranded regions and c' is the count matrix for basepaired nucleotides (an ij basepair aligned to a kl basepair). Triangular matrices are used because nucleotide (base pair) X in sequence 1 aligned to nucleotide (base pair) Y in sequence 2 should count the ssame as nucleotide (base pair) Y in sequence 1 aligned to nucleotide (base pair) X in sequence 2. However, we assume that an X-Y base pair may not be equivalent to a Y-X base pair in the context of the entire RNA molecule and therefore count these pairs separately. A count vector d
i
= 0(1 ≤ i ≤ 4) is also initialized for background nucleotide frequencies. Each pair of sequences is then examined. If the pair does not meet the minimal percent identity criterion, it is skipped and the next pair is examined. Otherwise, the weight of this pairing, w is set to be the average of the weights given to the two individual sequences. (Arguably, this weight should be set to be the product rather than the average of the individual weights. Though we did not fully explore this possibility, preliminary evidence suggests the method of calculating this weight does not appreciably influence performance.) For each aligned base pair (ij, kl) in the alignment, w is added to , d
i
, d
j
, d
k
, and d
l
; for all other aligned nucleotides (i, j), w is added to c
ij
, d
i
, and d
j
. The counts are then converted to empirical frequencies using:



The score matrices s and s' are then calculated using equations 1 and 2.
In order to collect these counts, we need high-quality structure-annotated alignments. We decided to use the small subunit ribosomal RNA alignment from the European Ribosomal RNA Database [41]. Specifically, we pruned the 1995 version of the database by removing sequences in which either more than 5% of the nucleotides are ambiguous or less than 50% of the base-paired positions are present. The resultant alignment consists of 2492 sequences ranging from 610 to 2305 nucleotides in length. When all pairs of sequences are counted, approximately 2.30 × 109 aligned single nucleotides and 1.06 × 109 aligned base pairs are counted and used to calculate the matrix. We created 170 unique matrices by varying the percent identity level at which clustering occurs and the minimal percent identity for a pair of sequences to be counted. We have chosen to call this series of matrices the RIBOSUM matrices (RIBOsomal rna Substitution Matrix).
Construction of a covariance model from a single RNA query
For these matrices to be useful, we need a good algorithm to perform alignment between an RNA query and a nucleotide database. Like primary sequence alignment, we need to consider both homologous regions of sequence that align and insertion and deletion events that put gaps into the alignment. Unlike primary sequence alignment, we also have to consider the nucleotide correlations within each sequence that make up the secondary structure. This structure can be modeled as a bifurcating tree, with each branch terminating in the loop of a stem-loop. Whatever algorithm we use must unambiguously pair each nucleotide in the query with either a nucleotide in the target or a gap, and vice versa. Our algorithm is based on profile stochastic context-free grammars (SCFGs) [14, 16, 18]. While this formulation was initially described in the framework of probabilisitc modeling of profiles, it can deal with arbitrary, non-probabilistic scores just as well. We therefore use the term "covariance model" to describe both the profile SCFG form of the model [14, 16, 18], and the single-sequence, non-probabilistic form presented here.
A covariance model produces ("emits") a nucleotide sequence. The model consists of a set of interconnected states. The states form a tree-like structure, with the root customarily being drawn at the top. As one moves down the tree, nucleotides are filled in from both the left and the right until they meet in the middle. Each state can emit either no nucleotides, a nucleotide on the left side, a nucleotide on the right side, or a base pair consisting of two nucleotides, one on each side. Bifurcations result in a split in the sequence, with each half being filled in from both sides along one of the two bifurcated branches. The model is traversed by following a series of transitions from one state to the next after each emission. Each transition is governed by a score, and only a limited set of transitions are allowed at all. Given a parameterized covariance model, algorithms exist for searching a database for homologous sequences and aligning the model to hits found in the database [14, 16, 18].
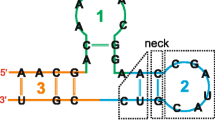
For our purposes, there are two separable steps in the creation of a covariance model. First, the model architecture needs to be determined from the given secondary structure. It is easiest to think of the model as being composed of modular nodes, where each node contains a characteristic arrangement of states. The node architecture of the model follows from the secondary structure of the query. A sample RNA secondary structure and its corresponding "guide tree" of nodes is given in Figure 1. There are eight types of nodes. A ROOT node marks the start of the model. A BIF node marks a bifurcation in the tree, and is always followed by a BEGL node on the left branch and a BEGR node on the right branch. All branches end with an END node. The remaining nodes are match nodes; these nodes represent either a base-pair (MATP) or a single nucleotide (MATL and MATR) in the secondary structure. (For a profile SCFG built from a multiple sequence alignment, only those positions thought to be conserved by some measure correspond to match nodes; for our single-sequence covariance models, all positions correspond to match nodes.) For consistency, MATL is always preferred over MATR when possible. Each secondary structure yields one and only one model architecture, and a model architecture implies a unique secondary structure. As each node is associated with a static arrangement of states, this architecture also gives the final arrangement of states in the model. A more detailed exposition of this algorithm for profile SCFGs is given in reference [18] and the C code used to construct a covariance model from a given secondary structure can be found in the modelmaker.c file of the Infernal package [42].

An example SCFG architecture. The sequence at the top folds into the specified secondary structure. At the bottom, the nodal architecture of the model that would produce this sequence is shown. Shaded triangles represent base pair emitting nodes, and point to the base pair they emit. Open triangles represent single nucleotide emitting nodes, and point to the nucleotide they emit.
The second step in the algorithm is parameterization of the model. This is best thought of in terms of the possible "node-states," i.e. the various state types present in each node type. A list of all possible node-states is given in Table 1, along with what they signify in the pairwise alignments we are creating here. In a profile SCFG, emission scores are log-odds scores as shown in Table 1. When we only have a single sequence, we cannot infer emission probabilities from the data given. We instead set these scores from the log-odds RIBOSUM matrix (Table 1). In the MP (match pair), ML (match left), and MR (match right) states, emission scores are log-odds scores for both profiles and single sequence models. In the IL (insert left) and IR (insert right) states, emission scores for a profile SCFG are based on observed nucleotide frequencies in insertions. For our single-sequence model, emission scores are 0 because we assume that the nucleotide distribution in insertions follows the null model.
Transition scores are set for transitions from one node-state to another node-state. In a profile SCFG, the log transition probabilities are derived from the observed frequencies of the various transitions. In the single-sequence case, we derive a transition score using the standard affine gap penalty formulation. We parameterize the overall penalty for a gap as α + βn where α is the gap open penalty, β is a gap residue penalty, and n is the size of the gap. We take half the α penalty on opening a gap and the other half on closing it. The β penalty is taken for each residue in a gap. Gaps emitted on both sides simultaneously (i.e. through a MATP_D node-state) are taken as two independent gaps. We also want to use a separate set of gap penalties for gaps within a base-paired region. If both node-states in a transition are in MATP nodes, the α parameter is replaced by a different parameter, α'. Similarly, for transitions from a MATP_D state to another MATP node, β is replaced by β'. α and β are used for transitions between base-paired and single-stranded regions.
To map this formulation onto the covariance model, we classify all node-states into one of six classes (Table 1): M_cl, for an aligned match or mismatch between the query and target; IL_cl, for a gap in the query sequence on the left; IR_cl, for a gap in the query sequence on the right; DL_cl, for a gap in the target sequence on the left; DR_cl, for a gap in the target sequence on the right; and DB_cl, for a gap in the target sequence on both sides. All classes other than M_cl represent some sort of gap that requires a gap penalty. The exact parameterization of transitions between classes is given in Table 2. For some transitions, the gap penalty presented in Table 2 represents the sum of penalties for several different gaps. All gap penalties are multiplied by -1 to get the transition score used in the model. These four gap parameters are empirically determined as described later in the paper. They are not normalized to have their exponentials sum to 1; therefore the resultant scores using these models cannot be directly interpreted probabilistically.
Local alignment searches
The model as described above can only perform global alignment with respect to the query sequence. The model is modified slightly to allow for local alignment as well. Two different types of locality are allowed. The first we call "begin locality," and resembles local alignment as implemented in the Smith-Waterman algorithm [11]. In this case, a penalty – beginsc – is taken if the alignment begins inside the model, i.e. the states representing the outermost parts of the RNA secondary structure are not included. This is analogous to the convention in the Smith-Waterman algorithm that there is no penalty (score of 0) for a local rather than a global alignment [11]. Following that convention, the beginsc penalty is set by default to 0. The second type of locality is "end locality." In this case, a penalty – endsc – is taken to allow the subtree of a model below the current state to be ignored, and replaced by an insertion of arbitrary size in the target sequence. There are many known examples of stems whose length changes dramatically or even completely disappear in homologous RNA sequences. One such example is the P8 stem of Archaeal RNase P, which is not present in the RNase P RNA of Methanocaldococcus jannaschii and Archaeoglobus fulgidus, but is present in other Archaeal RNase P sequences [43]. Examples of both kinds of locality are shown in Figure 2.

The two classes of local alignment. Each example shows how the nodal guide tree best aligns to the target sequence. At the bottom is the RSEARCH output for the alignment. On the left is an example of begin locality, while on the right is an example of end locality. The numbers next to the query sequence represent positions relative to the entire query; the numbers next to the target sequence represent positions relative to the subsequence defined in the "Target =" line.
These modifications are easily accommodated in the standard scanning algorithm for covariance models, which is described in detail elsewhere [14, 16]. The beginsc parameter is modeled as a transition from the root state to the consensus states (MATP_MP, MATL_ML, MATR_MR, BIF_B). The endsc parameter is modeled as a transition from each of the consensus states (MATP_MP, MATL_ML, MATR_MR, BEGL_S, BEGR_S) to a special "EL" (end-local) state, which generates residues at the background residue frequency and thus has a zero score for any subsequence after the transition cost, endsc, has been paid. (In actuality, version 1.0 of RSEARCH allowed transitioning to any state from the root with a beginsc penalty, and allows transitioning from any state to EL with an endsc penalty. More recent versions implement the algorithm as described. This slight difference does not appear to significantly alter performance [data not shown].)
The scanning algorithm takes a covariance model with M states (including B bifurcations), parameterized as described above, as well as a target database sequence of length L. Theoretically, the best alignment could have the nucleotide at position 1 in the database base pair with any nucleotide at position 2 through L. If the database includes large genomic contigs, L could be on the order of tens of megabases, which is much larger than we would expect any RNA to ever be. There is no need to check for base-pairings further apart than the longest RNA we would expect to find. To reduce time and memory requirements, we limit the total length of sequence in the target database for a single hit to a parameter D. Then, only positions 2 through D will need to be checked for a base pair to position 1. D needs to be set small enough for efficient performance but large enough so as not to miss any real homologs. By default, D is set to be two times the query length. The algorithm has a time complexity of O((M - B)LD + BLD2) and a memory complexity of O((M - B)D + BD2) [16]. A greedy algorithm is used to resolve these scores into a maximally scoring set of K non-overlapping hits (i1, j1), (i2, j2), ... (i K , j K ) on the target sequence, where i x and j x are the starting and ending coordinates of the hit on the target sequence, respectively. Alignments are then determined using the previously reported divide-and-conquer algorithm [18]. For each hit greater than a specified threshold, the score, alignment positions in the query and the target, the alignment, and E-values and P-values (calculated as described below) are reported.
Statistics
In order to determine statistical significance, we need to know what distribution RSEARCH scores follow. Much work has been done on the statistics of primary sequence alignment [32, 44–49]. All these approaches rest on the proposition, proven for the ungapped case and empirically true for the gapped case, that local alignment scores follow the Gumbel distribution [32, 50]. For a specific query sequence, the expected number of hits (E) with score greater than or equal to a given score (x) is given by the formula E = KNe-λx, where N is the size of the database and K and λ are characteristic parameters dependent on the query sequence and the base composition of the database. (It should be noted that this equation is often seen written as E = KMNe-λx, where M is the size of the query sequence. As we have chosen to recalculate λ and K for each individual query sequence, we have incorporated the M parameter into our K.) This formula can also be written as E = e-λ(x - μ), where  . The probability (P) that a score greater than or equal to a given score (x) is observed by chance is then given by P = 1 - e-E= 1 - exp(KNe-λx). Thus, calculating the E-value and P-value for a given score is simple provided a reasonable procedure for determining λ and K is found.
. The probability (P) that a score greater than or equal to a given score (x) is observed by chance is then given by P = 1 - e-E= 1 - exp(KNe-λx). Thus, calculating the E-value and P-value for a given score is simple provided a reasonable procedure for determining λ and K is found.
In the absence of a theory for the distribution of gapped structural alignment scores, we have chosen to determine K and λ empirically through maximum likelihood fitting of a Gumbel distribution to the score histogram obtained from alignment to random, simulated sequences. A large number (usually 1000) of i.i.d. sequences of length 2 × D (where D is the maximum possible length of a hit) are generated. The G+C content of these sequences are set as described below. The query is searched against each random sequence, and the best score is recorded in a histogram. A maximum-likelihood method is then used to determine λ and μ for a database of length 2 × D from these data [44, 49, 51, 52]. We can then calculate K using the formula  .
.
We initially created the random sequences using an i.i.d. model assuming a single, fixed G+C content for all sequences. As will be described below, this proved to be inadequate, as many databases have heterogeneous G+C contents. We then randomly choose a G+C content for each random sequence based on the distribution of G+C contents in the genome. We determine the G+C content in the target database measured in adjacent, non-overlapping windows of 100nt each, and use the distribution of these contents to select randomly a G+C content for each random sequence. For some databases where the range of frequently observed G+C contents is large, one pair of values for (λ, K) is not enough to accurately calculate E values. To allow for multiple values of (λ, K) partition points in the G+C content distribution can be set. For N partition points, the distribution is divided into N+l bins, and λ and K are calculated for each bin. For instance, if a partition point of 50 is set, λ and K are first calculated for random sequences with G+C contents sampled from the portion of the G+C content distribution with G+C content < 50%, and λ and K are then calculated again with G+C content sampled from the part of the distribution where the G+C content is ≥ 50%. Then, for a given database hit, the G+C content of the sequence of the hit is calculated and used to select the appropriate λ and K for calculating statistics. Thus, if partitions are used, the rank order of hits based on score and rank order of hits based on statistics may be different.
Implementation and parallelization
RSEARCH was implemented in C. Source code is available from our web site [53] and is available free of charge under the terms of the GNU General Public License (GPL). Version 1.0 was used for all experiments reported here. Timings and benchmarks reported were performed on a 1 GHz Pentium III Linux workstation with the Mandrake distribution, using the Intel C compiler version 6.0 with options "-O3 -static -mpl -xK" to compile the program. Because the RSEARCH algorithm is time-consuming, we also implemented a data-parallel version of RSEARCH using the Message-Passing Interface (MPI) [33].
Data sets and parameters
Several different data sets were used for testing and analysis, as described below. Sequence and structures for ribonuclease P RNA were taken from the RNase P database [54]. Signal Recognition Particle (SRP) sequences and structures were taken from the SRP database [55]. Three different human SRP sequences appear in the database. We chose to use sequence A, which corresponds to the originally sequenced RNA molecule. (This sequence was taken from GenBank accession X01037, but has two nucleotides that are different from the current GenBank version X01037.1). We used an Asn tRNA from Archaeoglobus fulgidus (GenBank AE001087.1, positions 4936–5008) [56] with the structure proposed by tRNAscan-SE [57]. For a representative yeast (S. cerevisiae) tRNA, we took the genomic sequence of the Ala tRNA originally sequenced by Holley [58, 59] (GenBank accession Z28265.1, positions 1117–1189). The precursor to the C. elegans miRNA mir-40 was also used [60] (GenBank accession AL110499.1, positions 17411–17507). Unless otherwise noted, the full length of each gene was used as the query sequence.
Three different databases were used for searches. The yeast genome was downloaded from http://www.yeastgenome.org and dated August 29, 2001 [59]. The database of 11 Archaeal genomes was previously described [61]. The Arabidopsis thaliana genome was downloaded from ftp://ftp.tigr.org/pub/data/a_thaliana/ath1/SEQUENCES[62].
For testing BLAST, WU-BLAST 2.0MP, dated October 20, 2002, was used with the -W3 and -kap options [12, 63]. For SSEARCH (an implementation of the full Smith-Waterman algorithm), version 3.4t05 was used with default parameters [11, 64].
Results
Optimal parameter set
We first asked what set of parameters – matrix, gap penalties, beginsc, and endsc – would be optimal to use as the defaults in RSEARCH. To assess this, we decided to choose the set of parameters that gives the lowest minimum error rate for a set of two test searches. The minimum error rate is defined as the minimal possible sum of false positives and false negatives for a search taken over all possible cutoff scores. The first search we used was the genomic copy of the alanine tRNA from S. cerevisiae folded using tRNAscan-SE searched against the yeast genome to identify the 295 tRNAs present there. The second search we used was M. jannaschii RNase P searched against a database of 11 Archaeal genomes to identify the 11 RNase P homologs found there. As doing the real searches for all the parameters we wanted to test would have been computationally infeasible, we estimated the false negative rate in many cases by searching a smaller database and extrapolating to the size of the full database. To abbreviate the yeast tRNA search, we took chromosome VII as a proxy for the whole genome. For the RNase P search, we created a smaller database of similar G+C content. After several rounds of iterative trial and error optimizing different parameters, we decided to use RIBOSUM85-60 as the default matrix with α = 10.00, β = 5.00, α' = 0.00, β' = 15.00, beginsc = 0.00, and endsc = -15.00. We might have been able to derive a more robust parameter set had we used a more comprehensive set of tests, but the long running time required by RSEARCH makes such an approach infeasible.
RIBOSUM85-60 has several characteristics typical of these matrices (Figure 3). It consists of two matrices – one 16 × 16 for base pair substitutions and the other 4 × 4 for single nucleotide substitutions. In the singlue nucleotide substitution matrix, the A-A identity has a score (2.22) much larger than the other single nucleotide identities. This suggests that conserved As are especially common in single stranded regions of 16S ribosomal RNA. Unlike typical nucleotide or amino acid substitution matrices, not all values on the identity diagonal of the 16 × 16 matrix are positive. This reflects the specificity of base pairing. Canonical Watson-Crick and G-U pairs are observed much more often than non-canonical pairs. Since non-canonical pairs occur less often than expected on the basis of individual nucleotide probabilities, the log-odds score for these pairs aligned to themselves is negative. Second, substitution of one canonical pair for another usually gives a positive score (e.g. A-U to C-G has a score of 1.47). Therefore, the RIBOSUM matrices resemble what we intuitively assume a good base pairing substitution matrix would look like.

The RIBOSUM85-60 matrix. The 16 × 16 matrix is used to get scores for aligning base pairs. The 4 × 4 matrix is used to get scores for aligning single-stranded regions. Positive scores are shaded.
We compared the minimum error rates at these parameter choices to the performance of BLAST and SSEARCH on the same search problems. For the problem of finding yeast tRNAs using the alanine yeast tRNA as a query, the minimum error rate for the BLAST search was 194, while SSEARCH gave a minimum error rate of 223. The minimum error rate observed using RSEARCH was 50. Instead of the default matrices in BLAST and SSEARCH, we also tried other matrices and gap penalties, both made in a similar fashion to RIBOSUM85-60 and as suggested by others [65]. None of these changes resulted in a significant improvement in performance for either BLAST or SSEARCH. For the M. jannaschii RNase P search, both BLAST and SSEARCH give a minimum error rate of 4, while RSEARCH gives a minimum error rate of 2. These tests indicate that RSEARCH, using secondary structure, is capable of outperforming primary sequence search programs.
To help insure that the above results were not the result of overtraining on those two specific searches, we performed similar tests with another tRNA and RNase P query sequence. We first asked how well an Asn tRNA from A. fulgidus could find the 494 tRNAs present in the database of Archaea genomes. The minimum error rates for BLAST and SSEARCH were 305 and 373, respectively. The RSEARCH search had a minimum error rate of 66. We also used the P. furiosus RNase P sequence to search the database of Archaeal genomes for homologs. The minimum error rate for BLAST was 6 and for SSEARCH was 5. The minimum error rate using RSEARCH was a perfect 0. These data reinforce our conclusion that RSEARCH can outperform primary sequence search programs.
Statistics
Calculation of minimum error rates requires prior knowledge of all homologs of the query sequence in the database. As we wish to use RSEARCH to search a database when such information is still unknown, we need a method for evaluating the statistical significance of a hit. We assumed that RSEARCH scores would follow the Gumbel distribution, just as scores from primary sequence search programs like BLAST do [32, 45, 46]. We therefore asked whether the scores produced by a search of random sequence do in fact follow the Gumbel distribution. The distribution of scores from one such search of random sequences is shown in Figure 4a. It is clear from these plots that the score distribution more closely fits the Gumbel distribution than the normal Gaussian distribution.

RSEARCH statistics, a Distribution of scores for a search against random sequences. We searched a database of 10,000 random sequences of 10,000 nucleotides each with a GC composition of 50% using the precursor to the C. elegans miRNA mir-40 as the query [60]. We took the best score found for each of the 10,000 sequences in the database and plotted their distribution. We then calculated the mean and standard deviation and plotted the Gaussian distribution for those values. We also calculated K and λ for the Gumbel distribution and plotted that distribution. b Average observed number of hits with E-value less than a cutoff versus reported E-value for searches of various RNase P queries against database of Archaeal genomes. E-values were computed using partition points of 40% and 60% G+C content.
We next assessed whether, on average, the E-values reported are an accurate reflection of the false positive rate. We examined six searches of the Archaeal genome database with M. jannaschii, P. furiosus, E. coli, B. subtilis, S. cerevisiae, and H. sapiens RNase P sequences as the queries. For the six searches, we then computed the average observed E-value (observed number of false positives) at various calculated E-value cutoffs. If the statistical model is correct, we expect the calculated E-value cutoff to be equal to the average number of observed false positives scoring better than the cutoff. We first calculated E-values using random sequences with a fixed G+C content of 45.8%, which is the overall G+C content of the Archaeal database. Under this model, there were 246 ± 257 false positives at an E-value cutoff of 1. Therefore, this statistical model was inadequate.
Looking more closely at the data led us to hypothesize that the statistical method was failing because the target database consists of a heterogeneous population of sequences with widely varying G+C contents. We first tried correcting for this by randomly picking a G+C content for each random sequence used in the simulation to calculate λ and K. This G+C content was picked from the distribution of G+C contents observed in the database. With this model, there were 8 ± 8 false positives at an E-value cutoff of 1. While the average number of false positives is closer to that predicted by the E-value, and the standard deviation is much smaller, we wished to improve the statistics even further. Since the G+C content distribution of the database has a large variance, we decided to partition the G+C distribution into 3 bins: one for G+C contents less than 40%, one for contents between 40% and 60%, and one for G+C contents greater than 60%. We calculated separate values of λ and K for each of these bins. With this statistical model, there are 2 ± 3 false positives at an E-value of 1. Observed E-values between 1 and 100 never deviate significantly from the computed E-value (Figure 4b), especially for observed E-values less than 10. Therefore, this statistical model was used for further searches of the Archaeal database. Since partitions are only necessary for databases with a large variance, and since the optimal partitions vary from database to database, the default statistical model in RSEARCH is to calculate a single λ and K without using any partitions.
Examples of Performance
We then wished to assess how well RSEARCH would perform in additional realistic scenarios. To study this, we chose an RNA molecule which was not part of the training set at all – the Signal Recognition Particle (SRP) RNA. We tested a variety of SRP query sequences against several database genomes. Each test was designed to look across phylogenetic domains or kingdoms. In each case, we compared its performance to BLAST and SSEARCH. In some cases, RSEARCH performed as well as these primary sequence search programs. In one rare case, using Pyrococcus horikoshii SRP as the query, SSEARCH and BLAST outperformed RSEARCH. Some examples where RSEARCH does outperform primary sequence searches are given below.
In one example, we searched for the 11 SRP genes in the Archaeal genomes using SRP from the Eubacteria B. subtilis as the query. No hits with an E-value less than 10 were observed with BLAST. SSEARCH found 13 hits at an E-value cutoff of 10, three of which were true homologs and 10 of which were false positives. No hits were observed with an E-value less than 0.05 using SSEARCH. In contrast, 16 hits with an E-value less than 10 were observed with RSEARCH, six of which are true homologs. Two of these true positives, but none of the false positives, had an E-value less than 0.05 (E = 0.0064 for M. jannaschii and E = 0.0067 for A. fulgidus).
If we instead use H. sapiens (a eukaryote) SRP as the query to find homologs in the Archaeal genomes, BLAST found seven hits with an E-value less than 10, none of which are true homologs. SSEARCH found nine hits with an E-value less than 10, only one of which was a true homolog. SSEARCH did not find any hits with an E-value less than 0.05. RSEARCH, on the other hand, found four hits, two of which are true homologs, with an E-value less than 10. The two true homologs, but not the two false positives, had E-values less than 0.05 (E= 0.0067 for Methanobacterium thermoautotrophicum and E = 0.0081 for A. fulgidus).
As a final test, we searched the genome of the plant A. thaliana with H. sapiens (an animal) SRP. There are at least eight copies of SRP in the genome; we take a significant hit to any of these eight copies as indicative of an ability to find SRP [66]. Neither BLAST nor SSEARCH can find any of these copies with an E-value less than 10. In contrast, several copies of SRP can be found using RSEARCH, with the most significant hit having an E-value of 9.6 × 10-6. Taken together, these data suggest that if we knew about either H. sapiens or B. subtilis SRP, we would be able to find SRP genes in distantly related genomes in other phylogenetic domains or kingdoms with confidence using RSEARCH, but not with either SSEARCH or BLAST.
Timings
As mentioned previously, the time complexity of the scanning algorithm in RSEARCH is O((M - B)LD + BLD2). We know that D is set to be 2M by default, and assume that in the unrealistic worst case, every position in the query structure represents a bifurcation. Then, the worst-case running time of the scanning algorithm is O (NM3), for a query of length M and database of length N, though actual running time will be less based on the number of bifurcations. Calculation of the statistics, which is O(M4), takes an additional amount of time. Therefore, for a large database where M <<N, the algorithm scales linearly with the size of the database but as the cube of the length of the query sequence. It takes 2.9 CPU days to search E. coli SRP (113 nt) against the 2.1 × 107 nucleotide Archaeal database. Approximately 2% of that time is spent calculating values for K and λ. In contrast, the P. furiosus RNase P sequence (330nt) requires 38 CPU days to search the same database. For this search, approximately 7% of the time is spent calculating values for K and λ. These searches would take 26 CPU years and 340 CPU years respectively to search the non-redundant nucleotide database of GenBank (6.9 × 109 nucleotides). Actual running times can be reduced by using a large-scale clustered computing facility. Actual running times for the above searches on a parallel cluster are 33 minutes for finding homologs of E. coli SRP in the Archaea (128 CPUs), and 7.4 hours for finding homologs of P. furiosus RNase P in the Archaea (124 CPUs). Therefore, use of RSEARCH is currently practical only when a large multiprocessor computing facility is available.
Discussion
Here we have presented RSEARCH, a program for finding homologs of a single RNA sequence given its secondary structure. RSEARCH extends previous profile SCFG implementations in three ways, each of which contributes to its superior performance over BLAST and SSEARCH [14, 16]. First, RSEARCH allows the use of a single sequence as a query, by incorporating a substitution matrix and gap penalties to set the parameters of the covariance model. Second, RSEARCH includes local alignment. Third, RSEARCH includes empirically derived values for statistical significance. Combined, these improvements make RSEARCH a useful tool for finding homologs of biologically important RNAs.
There are three areas in which future development efforts should be focused to improve RSEARCH's performance. First, the quality of the substitution matrix influences the performance of the program. Here we built the matrix using only a single class of RNA molecules and chose the best matrix based on only two sample tests. Using additional classes of RNA molecules for both building the matrix and choosing the best default may improve RSEARCH's performance. Alternative algorithms for clustering and weighting sequences should also be explored. Finally, an exponential family of matrices (like the PAM matrices) rather than an empirical family (like the BLOSUM matrices) may be worth considering as well. The rate matrix of Knudsen and Hein would be useful in this approach [40].
Second, RSEARCH is quite slow. Many searches are infeasible on a single CPU. We worked around this problem by performing searches in parallel using a clustered computing environment. This solution is not ideal due to the resources required for such an environment. Advances in computing technology will gradually make more and more searches practical on a single workstation; a new workstation purchased today is two to three times as fast as the machines used in this paper. More importantly, heuristic improvements to RSEARCH may speed it up significantly, just as BLAST and FASTA are significant speed improvements to the Smith-Waterman algorithm.
Finally, the requirement that the secondary structure of the query sequence is known must be addressed. Even a one base pair misprediction can significantly alter the results of the search (data not shown). This is not a problem if one is searching for homologs of an RNA sequence whose structure is well established (e.g. tRNA, RNase P, SRP). As RNA secondary structures are established through the sequencing of many homologs and comparative analysis [67], there is less need for a program that can handle a single sequence query rather than a large sequence family in these cases. The power of RSEARCH comes from being able to do searches when we only know of a single member of an RNA sequence family (e.g., novel noncoding RNA genes recently discovered in E. coli and various Archaea [61, 68–73]). In these cases, ideally we would like to be able to accurately predict secondary structure starting only with a single sequence. Recent work shows promise in simultaneously aligning and folding a pair of RNA sequences [23, 25, 26, 74]. These algorithms predict structure more accurately than single-sequence RNA folding algorithms. Many RNA genefinding approaches take advantage of comparative data. Close homologs of novel RNAs can often be found by primary sequence search programs. These homologs can then be used in a pairwise RNA folder to get a structure for the query sequence. Improvements in such algorithms and an understanding of how best to predict the folding of a query sequence for RSEARCH should allow us to use RSEARCH to find homologs of these novel RNAs.
Availability and requirements
Source code of RSEARCH is available from our web site [53] and is available free of charge under the terms of the GNU General Public License (GPL). In should compile under any Unix system with a C compiler.
References
Hentze MW, Caughman SW, Casey JL, Koeller DM, Rouault TA, Harford JB, Klausner RD: A model for the structure and functions of iron-responsive elements. Gene 1988, 72: 201–8. 10.1016/0378-1119(88)90145-X
Schlegl J, Gegout V, Schlager B, Hentze MW, Westhof E, Ehresmann C, Ehresmann B, Romby P: Probing the structure of the regulatory region of human transferrin receptor messenger RNA and its interaction with iron regulatory protein-1. RNA 1997, 3: 1159–72.
Lambert A, Lescure A, Gautheret D: A survey of metazoan selenocysteine insertion sequences. Biochimie 2002, 84: 953–9. 10.1016/S0300-9084(02)01441-4
Wilting R, Schorling S, Persson BC, Bock A: Selenoprotein synthesis in Archaea: identification of an mRNA element of Methanococcus jannaschii probably directing selenocysteine insertion. J Mol Biol 1997, 266: 637–41. 10.1006/jmbi.1996.0812
Miranda-Rios J, Navarro M, Soberon M: A conserved RNA structure (thi box) is involved in regulation of thiamin biosynthetic gene expression in bacteria. Proc Natl Acad Sci USA 2001, 98: 9736–41. 10.1073/pnas.161168098
Stormo GD, Ji Y: Do mRNAs act as direct sensors of small molecules to control their expression? Proc Natl Acad Sci USA 2001, 98: 9465–7. 10.1073/pnas.181334498
Winkler W, Nahvi A, Breaker RR: Thiamine derivatives bind messenger RNAs directly to regulate bacterial gene expression. Nature 2002, 419: 952–6. 10.1038/nature01145
Erdmann VA, Barciszewska MZ, Hochberg A, de Groot N, Barciszewski J: Regulatory RNAs. Cell Mol Life Sci 2001, 58: 960–77.
Eddy SR: Non-coding RNA genes and the modern RNA world. Nat Rev Genet 2001, 2: 919–29. 10.1038/35103511
Eddy SR: Computational genomics of noncoding RNA genes. Cell 2002, 109: 137–40. 10.1016/S0092-8674(02)00727-4
Smith TF, Waterman MS: Comparison of biosequences. Adv Appl Math 1981, 2: 482–9.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ: Basic local alignment search tool. J Mol Biol 1990, 215: 403–10. 10.1006/jmbi.1990.9999
Pearson WR, Lipman DJ: Improved tools for biological sequence comparison. Proc Natl Acad Sci USA 1988, 85: 2444–8.
Eddy SR, Durbin R: RNA sequence analysis using covariance models. Nucleic Acids Res 1994, 22: 2079–88.
Sakakibara Y, Brown M, Hughey R, Mian IS, Sjolander K, Underwood RC, Haussler D: Stochastic context-free grammars for tRNA modeling. Nucleic Acids Res 1994, 22: 5112–20.
Durbin R, Eddy S, Krogh A, Mitchison G: Biological Sequence Analysis Cambridge University Press, Cambridge 1998.
Gautheret D, Lambert A: Direct RNA motif definition and identification from multiple sequence alignments using secondary structure profiles. J Mol Biol 2001, 313: 1003–11. 10.1006/jmbi.2001.5102
Eddy SR: A memory-efficient dynamic programming algorithm for optimal alignment of a sequence to an RNA secondary structure. BMC Bioinformatics 2002, 3: 18. 10.1186/1471-2105-3-18
Gautheret D, Major F, Cedergren R: Pattern searching/alignment with RNA primary and secondary structures: an effective descriptor for tRNA. Comput Appl Biosci 1990, 6: 325–31.
Billoud B, Kontic M, Viari A: Palingol: a declarative programming language to describe nucleic acids' secondary structures and to scan sequence database. Nucleic Acids Res 1996, 24: 1395–403. 10.1093/nar/24.8.1395
Pesole G, Liuni S, D'Souza M: PatSearch: a pattern matcher software that finds functional elements in nucleotide and protein sequences and assesses their statistical significance. Bioinformatics 2000, 16: 439–50. 10.1093/bioinformatics/16.5.439
Macke TJ, Ecker DJ, Gutell RR, Gautheret D, Case DA, Sampath R: RNAMotif, an RNA secondary structure definition and search algorithm. Nucleic Acids Res 2001, 29: 4724–35. 10.1093/nar/29.22.4724
Sankoff D: Simultaneous solution of the RNA folding, alignment, and protosequence problems. SIAM J Appl Math 1985, 45(5):810–825.
Gorodkin J, Heyer LJ, Stormo GD: Finding the most significant common sequence and structure motifs in a set of RNA sequences. Nucleic Acids Res 1997, 25: 3724–32. 10.1093/nar/25.18.3724
Mathews DH, Turner DH: Dynalign: an algorithm for finding the secondary structure common to two RNA sequences. J Mol Biol 2002, 317: 191–203. 10.1006/jmbi.2001.5351
Holmes I, Rubin GM: Pairwise RNA structure comparison with stochastic context-free grammars. In Pac Symp Biocomput 2002, 163–74.
Henikoff S, Henikoff JG: Amino acid substitution matrices from protein blocks. Proc Natl Acad Sci USA 1992, 89: 10915–9.
Gattiker A, Gasteiger E, Bairoch A: ScanProsite: a reference implementation of a PROSITE scanning tool. Applied Bioinformatics 2002, 1: 107–108.
Gribskov M, McLachlan AD, Eisenberg D: Profile analysis: detection of distantly related proteins. Proc Natl Acad Sci USA 1987, 84: 4355–8.
Krogh A, Brown M, Mian IS, Sjolander K, Haussler D: Hidden Markov models in computational biology. Applications to protein modeling. J Mol Biol 1994, 235: 1501–31. 10.1006/jmbi.1994.1104
Eddy SR: Profile hidden markov models. Bioinformatics 1998, 14: 755–63. 10.1093/bioinformatics/14.9.755
Karlin S, Altschul SF: Methods for assessing the statistical significance of molecular sequence features by using general scoring schemes. Proc Natl Acad Sci USA 1990, 87: 2264–8.
Pacheco PS: Parallel Programming with MPI Morgan Kaufmann, San Francisco 1997.
Altschul SF: Amino acid substitution matrices from an information theoretic perspective. J Mol Biol 1991, 219: 555–65.
Dayhoff MO, Schwartz RM, Orcutt BC: A model of evolutionary change in proteins. In Atlas of Protein Sequence and Structure (Edited by: Dayhoff MO). National Biomedical Research Foundation, Washington DC 1978, 345–352.
Henikoff S, Henikoff JG: Performance evaluation of amino acid substitution matrices. Proteins 1993, 17: 49–61.
Pearson WR: Comparison of methods for searching protein sequence databases. Protein Sci 1995, 4: 1145–60.
Muse SV: Evolutionary analyses of DNA sequences subject to constraints of secondary structure. Genetics 1995, 139: 1429–39.
Tillier ER, Collins RA: High apparent rate of simultaneous compensatory base-pair substitutions in ribosomal RNA. Genetics 1998, 148: 1993–2002.
Knudsen B, Hein J: RNA secondary structure prediction using stochastic context-free grammars and evolutionary history. Bioinformatics 1999, 15: 446–54. 10.1093/bioinformatics/15.6.446
Yves Van de Peer, Ilse Van den Broeck, Peter De Rijk, Rupert De Wachter: Database on the structure of small ribosomal subunit RNA. Nucleic Acids Res 1994, 22: 3488–3494.
Infernal – inference of RNA secondary structure alignments[http://infernal.wustl.edu/]
Harris JK, Haas ES, Williams D, Frank DN, Brown JW: New insight into RNase P RNA structure from comparative analysis of the archaeal RNA. RNA 2001, 7: 220–32. 10.1017/S1355838201001777
Mott R: Maximum-likelihood estimation of the statistical distribution of Smith-Waterman local sequence similarity scores. Bull Math Biol 1992, 54(1):59–75.
Altschul SF, Gish W: Local alignment statistics. Methods Enzymol 1996, 26: 460–80.
Pearson WR: Empirical statistical estimates for sequence similarity searches. J Mol Biol 1998, 276: 71–84. 10.1006/jmbi.1997.1525
Olsen R, Bundschuh R, Hwa T: Rapid assessment of extremal statistics for gapped local alignment. In Proceedings of Seventh International Conference on Intelligent Systems for Molecular Biology (Edited by: Lengauer T, Schneider R, Bork P, Brutlag D, Glasgow J, Mewes H-W, Zimmer R). Menlo Park, AAAI Press 1999, 211–222.
Altschul SF, Bundschuh R, Olsen R, Hwa T: The estimation of statistical parameters for local alignment score distributions. Nucleic Acids Res 2001, 29: 351–61. 10.1093/nar/29.2.351
Bailey TL, Gribskov M: Estimating and evaluating the statistics of gapped local-alignment scores. J Comput Biol 2002, 9: 575–93. 10.1089/106652702760138637
Gumbel EJ: Statistics of Extremes Columbia University Press, New York 1958.
Lawless JF: Chapter 4. Statistical Models and Methods for Lifetime Data John Wiley & Sons 1982, 141–202.
Maximum likelihood fitting of extreme value distributions[ftp://ftp.genetics.wustl.edu/pub/eddy/papers/evd.pdf]
Sean Eddy lab homepage[http://www.genetics.wustl.edu/eddy/software]
Brown JW: The ribonuclease P database. Nucleic Acids Res 1999, 27: 314. 10.1093/nar/27.1.314
Gorodkin J, Knudsen B, Zwieb C, Samuelsson T: SRPDB (signal recognition particle database). Nucleic Acids Res 2001, 29: 169–70. 10.1093/nar/29.1.169
Klenk HP, Clayton RA, Tomb JF, White O, Nelson KE, Ketchum KA, Dodson RJ, Hickey EK, Peterson JD, Richardson DL, Kerlavage AR, Graham DE, Kyrpides NC, Fleischmann RD, Quackenbush J, Lee NH, Sutton GG, Gill S, Kirkness EF, McKenney K, Adams MD, Loftus B, Peterson S, Reich CI, McDonald L, Utterback T, Cotton MD, Spriggs T, Artiach P, Kaine BP, Sykes SM, Fraser CM, Smith HO, Woese CR, Venter JC: The complete genome sequence of the hyperthermophilic, sulphate-reducing archaeon Archaeoglobus fulgidus . Nature 1997, 390: 364–70. 10.1038/37052
Lowe TM, Eddy SR: tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 1997, 25: 955–64. 10.1093/nar/25.5.955
Holley RW, Apgar J, Everett GA, Madison JT, Marquisse M, Merrill SH, Penswick JR, Zamir A: Structure of a ribonucleic acid. Science 1965, 147: 1462–1465.
Goffeau A, Barrell BG, Bussey H, Davis RW, Dujon B, Feldmann H, Galibert F, Hoheisel JD, Jacq C, Johnston M, Louis EJ, Mewes HW, Murakami Y, Philippsen P, Tettelin H, Oliver SG: Life with 6000 genes. Science 1997, 275: 1051–2.
Lau NC, Lim LP, Weinstein EG, Bartel DP: An abundant class of tiny RNAs with probable regulatory roles Caenorhabditis elegans . Science 2001, 294: 858–62. 10.1126/science.1065062
Klein RJ, Misulovin Z, Eddy SR: Noncoding RNA genes identified in AT-rich hyperthermophiles. Proc Natl Acad Sci USA 2002, 99: 7542–7. 10.1073/pnas.112063799
Arabidopsis Genome Initiative: Analysis of the genome sequence of the flowering plant Arabidopsis thaliana . Nature 2000, 408: 796–815. 10.1038/35048692
WU-BLAST[http://blast.wustl.edu/]
Pearson WR: Searching protein sequence libraries: comparison of the sensitivity and selectivity of the Smith-Waterman and FASTA algorithms. Genomics 1991, 11: 635–50.
States DJ, Gish W, Altschul SF: Improved sensitivity of nucleic acid database searches using application-specific scoring matrices. METHODS: A Companion to Methods in Enzymology 1991, 3: 66–70.
Regalia M, Rosenblad MA, Samuelsson T: Prediction of signal recognition particle RNA genes. Nucleic Acids Res 2002, 30: 3368–77. 10.1093/nar/gkf468
James BD, Olsen GJ, Pace NR: Phylogenetic comparative analysis of RNA secondary structure. Methods Enzymol 1989, 18: 227–39.
Argaman L, Hershberg R, Vogel J, Bejerano G, Wagner EG, Margalit H, Altuvia S: Novel small RNA-encoding genes in the intergenic regions of Escherichia coli . Curr Biol 2001, 11: 941–50. 10.1016/S0960-9822(01)00270-6
Wassarman KM, Repoila F, Rosenow C, Storz G, Gottesman S: Identification of novel small RNAs using comparative genomics and microarrays. Genes Dev 2001, 15: 1637–51. 10.1101/gad.901001
Rivas E, Klein RJ, Jones TA, Eddy SR: Computational identification of non-coding RNAs in E. coli by comparative genomics. Curr Biol 2001, 11: 1369–73. 10.1016/S0960-9822(01)00401-8
Carter RJ, Dubchak I, Holbrook SR: A computational approach to identify genes for functional RNAs in genomic sequences. Nucleic Acids Res 2001, 29: 3928–38.
Tang TH, Bachellerie JP, Rozhdestvensky T, Bortolin ML, Huber H, Drun-gowski M, Elge T, Brosius J, Huttenhofer A: Identification of 86 candidates for small non-messenger RNAs from the archaeon Archaeoglobus fulgidus . Proc Natl Acad Sci USA 2002, 99: 7536–41. 10.1073/pnas.112047299
Schattner P: Searching for RNA genes using base-composition statistics. Nucleic Acids Res 2002, 30: 2076–82. 10.1093/nar/30.9.2076
Perriquet O, Touzet H, Dauchet M: Finding the common structure shared by two homologous RNAs. Bioinformatics 2003, 19: 108–16. 10.1093/bioinformatics/19.1.108
Acknowledgements
We wish to thank Robin Dowell, Elena Rivas, Shawn Stricklin, and Warren Gish for helpful discussions and Goran Ceric for administering our computer systems. RJK is a predoctoral fellow of the Howard Hughes Medical Institute. This work was supported by the Howard Hughes Medical Institute and NIH R01-HG01363.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
RJK developed and tested this software as a graduate student under the supervision of SRE.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Klein, R.J., Eddy, S.R. RSEARCH: Finding homologs of single structured RNA sequences. BMC Bioinformatics 4, 44 (2003). https://doi.org/10.1186/1471-2105-4-44
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2105-4-44