
Abstract
Background
Gene expression profiling and other genome-scale measurement technologies provide comprehensive information about molecular changes resulting from a chemical or genetic perturbation, or disease state. A critical challenge is the development of methods to interpret these large-scale data sets to identify specific biological mechanisms that can provide experimentally verifiable hypotheses and lead to the understanding of disease and drug action.
Results
We present a detailed description of Reverse Causal Reasoning (RCR), a reverse engineering methodology to infer mechanistic hypotheses from molecular profiling data. This methodology requires prior knowledge in the form of small networks that causally link a key upstream controller node representing a biological mechanism to downstream measurable quantities. These small directed networks are generated from a knowledge base of literature-curated qualitative biological cause-and-effect relationships expressed as a network. The small mechanism networks are evaluated as hypotheses to explain observed differential measurements. We provide a simple implementation of this methodology, Whistle, specifically geared towards the analysis of gene expression data and using prior knowledge expressed in Biological Expression Language (BEL). We present the Whistle analyses for three transcriptomic data sets using a publically available knowledge base. The mechanisms inferred by Whistle are consistent with the expected biology for each data set.
Conclusions
Reverse Causal Reasoning yields mechanistic insights to the interpretation of gene expression profiling data that are distinct from and complementary to the results of analyses using ontology or pathway gene sets. This reverse engineering algorithm provides an evidence-driven approach to the development of models of disease, drug action, and drug toxicity.
Similar content being viewed by others


Background
Molecular profiling technologies have enabled the collection of large, exploratory data sets consisting of measurements for tens of thousands of molecular entities. These rich data sets hold promise for understanding the molecular bases of disease, drug action, and drug toxicity, but do not often lead to a reasonable short list of potential molecular mechanisms that can be investigated further by targeted experiments. For example, gene expression profiling experiments frequently result in a list of hundreds or thousands of gene expression differences that characterize a comparison of biological states like diseased versus normal tissue or treatment versus control. The development of methods to interpret these lists of differential measurements and extract testable hypotheses is necessary to realize the full potential of these large data sets.
The use of prior knowledge in the form of functional groupings of genes into gene sets is central to many methods for interpreting molecular profiling data. Generally, a collection of gene sets is assessed to identify those for which differentially expressed genes from the data set are over-represented (reviewed in [1-3]). Genes can be grouped into sets based on a variety of criteria including: (1) functional annotation, (2) pathway maps, (3) regulatory or structural motifs, and (4) common response to an experimental perturbation.
Analyses using gene sets based on pathway maps (e.g., KEGG, [4]) or functional annotation (e.g., Gene Ontology, [5]) rely on the assumption that differential RNA expression is equivalent to differential protein activity. This assumption can be problematic because RNA abundances are not generally a good measure for the abundance or activity of the corresponding proteins. The correlation of RNA expression to protein expression is variable, with changes in RNA levels accounting for only a modest portion of changes in levels of the corresponding proteins [6-8]. As a group, signal transduction proteins are more poorly correlated with their corresponding RNAs than are structural proteins, thus extrapolation of RNA microarray data to infer changes in signal transduction requires caution [6, 7]. Protein abundance is regulated via translation and proteolysis in addition to regulation via transcription and stability of mRNA. Moreover, protein expression levels are frequently not the key determinant of protein activity; many pathways are regulated by post-translational events such as protein modification or binding, and only secondarily by protein abundance.
Experimental result-based gene sets, for example the L2L, GeneSigDB, and MsigDB chemical and genetic perturbations (CGP) gene sets [9-11], do not evaluate differentially expressed RNAs based on the function of the proteins they encode, but rather by a common mechanism controlling their expression (e.g., the experimental perturbation). This type of gene set has the additional advantage that the function of the corresponding gene products need not be well defined to be considered. One limitation of experimental result-based gene sets is that the sets of genes up-regulated and down-regulated within an experiment are often either handled as distinct “up” and “down” sets or combined without regards to direction. Additionally, gene sets from experiments with similar chemical or genetic perturbations are not generally integrated into a single cohesive gene set.
In this paper, we present a detailed description of Reverse Causal Reasoning (RCR), a reverse engineering algorithm to identify biological mechanisms that are statistically significant explanations for differential measurements in a molecular profiling data set. This approach uses prior knowledge in the form of a large network of biological cause and effect relationships, from which smaller networks representative of discrete biological mechanisms are derived. These smaller networks are essentially structured gene sets, where the upstream node represents an experimental perturbation like a chemical, protein, or protein activity, the downstream nodes represent entities such as RNAs that have been measured in the data set, and the edges specify an “increased”, “decreased”, or “ambiguous” relationship. These mechanism networks are evaluated for both overrepresentation and consistency with respect to differential measurements from a data set.
This general approach has been in use by Selventa since 2005 to identify and evaluate molecular mechanisms involved in diverse biological processes, but not formally described or tested using a publically available knowledge base [12-19]. A related approach has been recently reported [20]. In addition, an approach using the same network structure to represent mechanisms has been developed to quantify and allow comparison of the level of activity of a mechanism between related data sets [21].
We provide an implementation of RCR, Whistle (available at https://github.com/Selventa/whistle), specifically designed for analysing gene expression profiling data, and apply Whistle to three example gene expression profiling data sets. Whistle uses prior knowledge in the form of Biological Expression Language (BEL) statements compiled into a causal graph. BEL is a language for representing causal and correlative biological relationships from the scientific literature in a computable form. The BEL Framework, which provides tools for compiling and managing BEL statements, has recently been made available to the general public as open source along with a starter corpus of statements, used in this paper to analyse the example data sets (https://github.com/OpenBEL/openbel-framework).
Methods
Overview of RCR and whistle
RCR interprets molecular profiling data sets by inferring a set of molecular mechanisms with associated direction (increased or decreased) that can serve as potential causes for the observed profiling differences between two sample groups (e.g., RNAs differentially expressed in diseased vs. normal tissue). This algorithm requires a large knowledge base consisting of prior biological knowledge in the form of cause-and-effect relationships, where the entities measured by the molecular profiling experiment are included as targets of the cause-and-effect relationships.
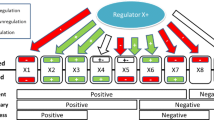
Whistle is an implementation of RCR that uses a BEL knowledge assembly model (KAM) as the knowledge base to analyse a gene expression profiling data set comparing two sample groups. Whistle uses the following general strategy (Figure 1):

Overview of whistle. Whistle evaluates molecular mechanisms as potential explanations for gene expression data by mapping measurements and differentially expressed genes to a directed network of prior scientific knowledge.
-
1.
A KAM is assembled from biological findings
-
2.
Small networks representative of a mechanism (HYPs) are derived from the KAM; each KAM node immediately upstream of a minimum number of measured RNA abundance nodes represents a HYP.
-
3.
Differential mRNA abundance measurements are mapped to mechanism networks and a direction (increased, decreased, or none) is assigned for each network.
-
4.
Evaluation statistics (richness and concordance) are calculated for each mechanism network.
Term definitions related to Whistle input and output are provided in Table 1.
Whistle evaluates prior knowledge as HYPs
The prior knowledge structures used by Whistle are termed ‘HYPs’ (derived from hypotheses). A HYP is a small, directed acyclic network containing an upstream node and downstream nodes that map to measured entities (Figure 2). A HYP can be compared to a qualitative Bayesian network or qualitative probabilistic network [22, 23]. Each edge in a HYP represents a qualitative influence of increases (+), decreases (−), or ambiguous (?). Unlike qualitative probabilistic networks, zero influences (0) are not included within HYPs. Like a naïve Bayes classifier, all downstream nodes in a HYP are assumed independent from each other, given a fixed value of the upstream node; the downstream nodes are assumed to be uncorrelated with each other.

Mapping of differential measurements to a HYP network. A HYP consists of an upstream node, U, and downstream nodes, designated r(A) - r(F), that represent abundances of RNAs measured in the experiment. This example network has six measured downstream nodes (possible), five of which map to significantly increased or decreased genes (observed). Node E is not significantly changed in expression. Three nodes, r(A), r(D), and r(F), support increased U (correct). One node, r(B), supports decreased U (contra). One node, r(C), is connected to U by both causal increase and causal decrease edges (ambiguous). On the basis of the mapped measurements, the direction “increased” is assigned to U.
In the case of Whistle, HYPs are essentially gene sets structured as directed networks, such that both the enrichment of differentially expressed genes and consistency of the directions (increased, decreased) can be evaluated. Each Whistle HYP is derived from a KAM. An increases edge connecting two nodes within a HYP means that the nodes are connected by an increases or directlyIncreases BEL relationship in the KAM. Similarly, a decreases HYP edge is derived from a decreases or directlyDecreases KAM relationship. An ambiguous HYP edge means that the KAM contains multiple, conflicting edges connecting an upstream and downstream node, e.g., both an increases and decreases edge. Other classes of HYPs, e.g., those using other edge types, paths consisting of multiple edges, and/or other types of measured downstream nodes, can be derived from KAMs and evaluated by RCR, but they are not described here or evaluated by Whistle.
Mapping probe sets to the KAM
First, Whistle maps the set of measured RNAs for a data set to the KAM. This mapping is used to generate HYPs for evaluation, determine the downstream nodes for each HYP (possible), and determine the set of downstream nodes for all HYPs evaluated (the population).
Probe set names or other identifiers (e.g., gene symbol or Entrez gene ID) are mapped to RNA abundance nodes in the KAM using the equivalence files provided by the BEL Framework (see http://wiki.openbel.org/ for documentation of BEL Framework namespaces and equivalence files). Next, each KAM node with at least four causally downstream measured RNA abundance nodes is selected as the upstream node for a HYP, resulting in a set of HYPs from the KAM for evaluation against the data set.
Mapping differential measurements to HYP networks
Next, the data set of differential measurements is mapped to the HYP networks and basic metrics including direction, correct, and contra are calculated (see Table 1). Differential gene expression data is processed such that each measurement is expressed as one of three discrete states: significant increase, significant decrease, or no significant change, based on user-defined significance criteria. These criteria generally include thresholds for fold change, adjusted p-value, and average abundance (see Data analysis for examples).
A direction of increased, decreased, or none is assigned to each HYP, representing the inferred state of the upstream node of the network based on the states of the downstream nodes mapped from the data set. The direction is the state consistent with the majority of the significantly increased and decreased downstream nodes of the HYP network. For each significantly increased or decreased HYP downstream node, the causal linkage(s) to the upstream node determines if the observed state is consistent with an increased, decreased, or neither (ambiguous) state of the upstream node. For example, if upstream node A is connected to RNA abundance node B by a causal decrease, a significant increase in B would be consistent with a decrease in A. If upstream node A is connected to B by an ambiguous edge, an increase in B would be considered an ambiguity.
Evaluation statistics (concordance and richness)
Two evaluation statistics, concordance and richness, are calculated for each HYP. These statistics allow the HYPs to be prioritized based on the strength and consistency of their support in the data set. Lower concordance and richness p-values reduce the number of false positive, but increase the number of false negative HYP inferences. As is common for other prior knowledge-driven approaches [3], both the concordance and richness statistics are biased in favour of HYP networks with a larger number of downstream nodes. For most statistic-based metrics, values associated with a larger number of measurements can achieve higher levels of significance than those associated with a small number of measurements (see Results - Bias in evaluation statistics by HYP size). Thus, we use these metrics primarily as a filter to identify potential explanations for the gene expression data, and not as a strict ranking for the most interesting explanations. A threshold of p < 0.1 for both concordance and richness generally limits both false negatives and false positives to an acceptable level, and provides a manageable set of HYPs for manual evaluation (see Results).
Concordance
The concordance statistic is calculated as a p-value that characterizes the consistency of the observed states of the downstream nodes with the direction assigned to the HYP upstream node. Concordance is calculated as a binomial distribution where the directions expected for the downstream RNA abundance nodes, based on the assigned direction of the HYP upstream node and the causal relationships to the downstream nodes are tested. Downstream nodes with the assigned state no significant change and nodes connected to the upstream node by an ambiguous relationship are excluded. For the set of significantly increased or decreased nodes comprising the correct and contra for a HYP, the concordance statistic is the probability of getting at least the number of state changes consistent with the direction (correct).
The binomial distribution can be written as f(k;n,p) where:
k is the number of successful predictions (correct),
n is the number of trials (observed), and
p is the probability of achieving a result, 0 ≤ p ≤ 1.
The point probability of getting exactly k successful predictions is calculated as:
For the purposes of evaluating a HYP, the probability of getting the direction of prediction correct, p, is 0.5; the network can either predict the observed direction for a state change correctly or incorrectly. For some HYP x i , the number of trials, n i , corresponds to the number of downstream nodes for the mechanism that are mapped to state changes (observed). The number of successful predictions k i is the number of downstream nodes of x i , mapped to state changes that are consistent with the HYP direction (correct). Let l i be the number of downstream nodes for which the predicted direction cannot be determined (ambiguous).
The point probability for some HYP, x i , for some k i is then calculated as:
The concordance p-value is a cumulative probability based on the area under the curve of a probability distribution function. Thus, concordance for some HYP, x i , is the sum of prob ij for all j = k i , k i + 1, …, min(n i − l i , m). The concordance for x i is then given as:
Richness
The richness statistic for a HYP indicates the enrichment of nodes that have an observed significantly increased or decreased state in the downstream nodes of the network (possible) compared to the number of significantly increased or decreased RNAs (state changes) in the total population of measured RNAs mapped to HYPs. Richness is calculated as a p-value using a hypergeometric probability distribution, a method commonly used to characterize the degree to which a subset of a whole is particularly notable.
The hypergeometric distribution can be written as f(k,N,m,n) where:
k is the number of notable items in the subset (observed),
N is the number of items in the full set (population),
m is the number of notable items in the full set (state changes), and
n is the size of the subset (possible).
The probability that the subset is not due to chance can be calculated as:
For the richness calculation, the size of the full set, N, corresponds to the number of observations in the experiment that can be identified and mapped to at least one downstream node in the set of all possible HYPs (for a given HYP generation algorithm); m is the cardinality of the set of state changes. These values are constant for all mechanisms x i , i = 1,2,…,n. For any mechanism, x i , n i is the number of downstream nodes that are mapped to measurements in the experiment (possible), and the number of notable items, k i , corresponds to the number of downstream nodes also found in the set of m state changes (observed).
The point probability for some HYP, x i , for some k i is then calculated as:
The richness p-value is a cumulative probability based on the area under the curve of a probability distribution function. Thus, richness for a HYP, x i , is the sum of prob ij for all j ≥ k i . Additionally, j is bounded by the size of the subset n i and number of notable events m. The complete richness calculation is then given as:
which determines the cumulative probability over the range j = k i , k i + 1, …, min(n i , m).
Data analysis for examples
Three gene expression data sets were used as examples to evaluate Whistle output: (1) a mouse high fat diet insulin resistance model [24]; (2) human microvascular endothelial cells treated with TNF [25]; and (3) human breast epithelial cells treated with a PI3 kinase inhibitor [26]. Microarray data sets used as input for the TNF (GSE2638) and PIK3CA (GSE17785) examples were downloaded from Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/gds). The high fat diet (E-MEXP-1755) data set was downloaded from ArrayExpress (http://www.ebi.ac.uk/arrayexpress/). GSE2638 used the Affymetrix Human Genome U133A, GSE17985 uses Affymetrix Human Genome U133 Plus 2.0, and E-MEXP-1755 used the Affymetrix Mouse genome 430 2.0 Array. Raw RNA expression data for each data set was analysed using the “affy” (2.10.0) and “limma” (2.10.1) packages of the Bioconductor suite of microarray analysis tools available for the R statistical environment [27-29]. Robust Microarray Analysis (RMA) background correction and quantile normalization were used to generate microarray expression values. An overall linear model was fit to the data for all sample groups, and specific contrasts of interest were evaluated to generate raw p-values for each probe set on the expression array [30].
Unless otherwise specified, state changes for probe sets were generated using the following significance criteria: (1) adjusted p-values of less than or equal to 0.05, corrected for multiple testing effects using Benjamini-Hochberg FDR, (2) absolute fold changes of at least 1.3, and (3) an average normalized Affymetrix abundance of at least 32. Other thresholds were used to determine differential probe sets as specified. Genes represented by multiple probe sets were considered to have changed if at least one probe set met criteria for differential expression. Gene expression changes that met these criteria are considered to be significantly differentially expressed and have the directional qualities of increased or decreased, i.e., they were upregulated or downregulated, respectively, in response to the experimental perturbation.
Knowledge base
The “BEL Large Corpus”, version 1.4, (available at http://resource.belframework.org/belframework/1.0/knowledge/large_corpus.bel), was used as the prior knowledge source for the examples. The Large Corpus contains approximately 80,000 causal statements manually curated from the published biomedical literature, each statement representing an observation from experiments performed in human, mouse, or rat. Each statement is associated with a citation and key experimental context information (species, cell line, tissue, etc.). The Large Corpus was compiled into a Knowledge Assembly Model (KAM) using the BEL Framework, version 3.0.0 (available at http://download.openbel.org/OpenBEL_Framework-3.0.0-preview.zip) with the default compiler settings. For the examples, KAM nodes representing orthologous nodes were collapsed to either mouse or human as appropriate, using the BEL Framework API.
Because a BEL KAM is used as the prior knowledge source, each HYP is referred to by the BEL term label of its upstream node. BEL terms follow the general format f(ns:v), where f is a function like ‘proteinAbundance’ (short form ‘p’) or ‘biologicalProcess’ (short form ‘bp’); ns is a reference to a namespace like ‘MGI’ (Mouse Genome Informatics symbol) or ‘EGID’ (Entrez Gene Id); and v is a value from the indicated namespace, like ‘Akt1’. A list of BEL functions and namespace abbreviations used in the examples is provided in Additional file 1. Full BEL language documentation can be found in the OpenBEL Portal (http://www.openbel.org/content/bel-lang-language).
Results
Example data sets
To highlight the utility of RCR for generating testable mechanistic hypotheses from gene expression profiling data, we provide examples of the application of Whistle v1.0 to three published gene expression data sets using a network compiled from the BEL Large Corpus as the prior knowledge source. Each data set was analysed to identify significantly differentially expressed probe sets (see Methods) and the resulting data analysed using Whistle and the Large Corpus KAM (Table 2). These examples demonstrate the use of Whistle to identify potential molecular upstream controllers of observed differential gene expression from experimental data sets.
Example 1 - high fat diet diabetes model
We applied Whistle to data set E-MEXP-1755, the liver gene expression profile from mice fed a high fat diet, a model for impaired glucose tolerance and type 2 diabetes [31]. For this data set, C57BL/6 J male mice were fed either a high fat diet or standard chow for 15 weeks, and RNA was extracted from the liver for profiling [24].
Mapping of measurements from the Affymetrix MG-430 2.0 microarray supported the generation of 606 HYPs for evaluation from the mouse Large Corpus KAM (see Additional file 2), with a population of 7594 unique downstream RNA abundance nodes. The 372 probe sets meeting criteria for significant differential expression between high fat diet and normal diet mouse livers map to 193 unique RNA abundance nodes in the population (Table 2).
Of the 606 HYPs evaluated from the mouse Large Corpus KAM, 13 met the standard richness and concordance p-value significance thresholds of 0.1 (Table 3) (see Randomized data sets for threshold selection). For example, the HYP bp(GO:“response to endoplasmic reticulum stress”) is inferred to be significantly increased for the high fat data set (Figure 3). This mechanism, representing the biological process defined by the GO term ‘response to endoplasmic reticulum stress’, is causally upstream from 27 measured RNA abundances in the mouse-orthologized Large Corpus KAM (possible). Of the 193 significantly increased or decreased RNA abundance nodes resulting from high fat diet, seven map to the bp(GO:“response to endoplasmic reticulum stress”) HYP, representing a significant enrichment in endoplasmic reticulum stress-regulated RNA abundance nodes (richness p = 3.5E-6). Of these seven, six are in a direction consistent with increased response to endoplasmic reticulum stress, and one is consistent with decreased response. Thus, the direction increased is assigned to the bp(GO:“response to endoplasmic reticulum stress”) HYP. The concordance p-value, which evaluates the directions of the observed states of the downstream nodes against the predictions made by the HYP, is 6.3E-2, supporting the inference of increased bp(GO:“response to endoplasmic reticulum stress”).

Scored HYP example. The HYP with the upstream node bp(GO:“response to endoplasmic reticulum stress”), scored for the E-MEXP-1755 high fat diet data set. This network contains 27 measured RNA abundance nodes (possible), represented as ovals coloured by differential expression (red - significantly increased, green - significantly decreased, grey - no significant change). A total of seven differentially expressed RNAs mapped to the network (observed), including six supporting increased mechanism activity (correct) and one supporting decreased activity (contra, marked with an ‘X’ on the edge).
Several of the significant mechanisms inferred by Whistle are consistent with the dietary changes and insulin resistance associated with this mouse model. Increased food intake (path(SDIS:“food intake”)) and decreased response to starvation (bp(GO:“response to starvation”)) are consistent with the dietary differences characterizing the high-fat diet group and the control group. Circulating insulin levels are reported to be increased in the high fat diet fed mouse model [31], and several HYPs representing increased insulin (p(MGI:Ins2), a(CHEBI:insulin), and a(SCHEM:“serum insulin”)) are inferred by Whistle. Increased response to endoplasmic reticulum stress (bp(GO:“response to endoplasmic reticulum stress”) is consistent with reports of ER stress induction in mouse obesity models [32]. The decreased transcriptional activity of Foxa2 protein (tscript(p(MGI:Foxa2))) is consistent with the reported inactivation of Foxa2 in insulin-resistant mouse models [33].
Several of the highly relevant HYPs inferred by Whistle for the E-MEXP-1755 data set represent the abundance or activity of proteins for which the corresponding RNAs are not differentially expressed. These mechanisms would not be identified by standard overrepresentation analyses based on the functions of the proteins encoded by the differentially expressed genes. For example, insulin (represented by the HYPs p(MGI:Ins2), a(CHEBI:insulin), and a(SCHEM:“serum insulin”)) is inferred increased, but probe sets for the mouse insulin genes Ins1 and Ins2, and the insulin receptor gene Insr do not meet criteria for differential expression. Insulin is produced by the pancreas, and is not expected to be regulated by gene expression in the liver. Similarly, the HYP representing the transcriptional activity of Foxa2, tscript(p(MGI:Foxa2)), is inferred decreased, but the Foxa2 RNA (probe set 1422833_at) is not differentially expressed. Foxa2 is an insulin-regulated liver transcription factor whose activation leads to oxidation of fatty acids and secretion of triacyglycerols, and whose impairment has been implicated in diabetes [34]. Foxa2 regulation by insulin occurs via phosphorylation [33], consistent with the lack of observed regulation of Foxa2 RNA.
Example 2 - TNF treatment of human microvascular endothelial cells
We applied Whistle to a gene expression profile of the response of human microvascular endothelial cells to TNF. This data set (GSE2638) was generated by stimulating HMEC-1 cells with 2 ng/ml TNF for 5 hours [25]). In endothelial cells, TNF, a potent mediator of inflammatory signalling, acts through its cognate receptors to initiate downstream signalling, including activation of the transcription factor NF-κB, resulting in increased transcription of cell surface receptors involved in leukocyte adhesion [35-37].
Mapping of measurements from the Affymetrix U133A microarray supported the generation of 593 HYPs from the human Large Corpus KAM (see Additional file 2), with a population of to 6644 unique downstream RNA abundance nodes. The 557 probe sets meeting criteria for significant differential expression between TNF stimulated and untreated cells map to 330 unique RNA abundance nodes in the population.
Of the 593 HYPs evaluated from the human Large Corpus KAM, 58 met the standard richness and concordance p-value significance thresholds of 0.1. The top 20 significant mechanisms as ranked by the concordance statistic are shown in Table 4. These significant mechanisms include several molecules and processes canonically associated with TNF signalling. Transcriptional activity of NF-κB complex (tscript(complex(PFH:“Nfkb Complex”))) is inferred increased, consistent with the complex’s role as a key mediator of TNF-induced transcription [36]. TNF reduces endothelial cell lifespan [38], consistent with the inferred increase of replicative cell aging (bp(GO:“replicative cell aging”)). TNF activates p38 MAPK in endothelial cells [37], and increased kinase activity of p38 MAPK (kin(p(PFH:“p38 MAPK Family”))) is inferred by Whistle. Interestingly, while the inferred increases of lipopolysaccharide (a(CHEBI:lipopolysaccharide)) bacterial infection (path(SDIS:“bacterial infection”)) seem biologically implausible to occur during the 5 hour TNF treatment used to generate the data set, these mechanisms are aligned with reports that LPS and TNF activate very similar transcriptional responses in endothelial cells [39]. A HYP representing TNF itself was not evaluated, as TNF is not connected to sufficient downstream RNA abundance nodes in the Large Corpus KAM.
Example 3 - PI3K inhibitor treatment of human breast epithelial cells
We applied Whistle to data set GSE17785, the gene expression profile of human breast epithelial cells treated with a PI3K inhibitor. For this data set, a clone of cell line MCF10A with a knock-in of the activating PIK3CA mutation H1047R was treated with either the PI3K inhibitor GDC-0941 or the vehicle DMSO for 4 hours [26]. The phosphatidylinositol 3-kinase (PI3K) signalling pathway is a key mediator of cell survival and proliferation [40].
Mapping of measurements from the Affymetrix U133 Plus 2.0 microarray supported the generation of 603 HYPs from the human Large Corpus KAM (see Additional file 1), with a population of 7500 unique downstream RNA abundance nodes. The 3410 probe sets meeting criteria for significant differential expression between PI3K inhibitor-treated and DMSO-treated cells map to 1126 unique RNA abundance nodes in the population.
Of the 603 mechanisms evaluated from the human Large Corpus KAM, 45 met the standard richness and concordance p-value significance thresholds of 0.1, including decreased PI3K complex activity (kin(complex(NCH:“p85/p100 PI3Kinase Complex”))) and several mechanisms canonically associated with inhibition of PI3K signalling, e.g., increased activity of FOXO transcription factors FOXO1, FOXO3, and FOXO4 (tscript(p(HGNC:FOXO1)), tscript(p(HGNC:FOXO3)), tscript(p(HGNC:FOXO4))); decreased AKT kinase activity (kin(p(PFH:“AKT Family”))), and decreased insulin signalling (a(SCHEM:“serum insulin”)) [40]. The top 20 significant mechanisms as ranked by the concordance statistic are shown in Table 5.
Randomized data sets
Random data are not expected to produce a biological signal. To further investigate the significance of the mechanisms inferred by Whistle, we generated a set of 1,000 matched random data sets for each example data set and compared the Whistle results to those for the matched real data. These random data sets were generated such that the population was the same as the matched example data set and that the total number of significantly increased and decreased RNAs in the randomized data matched that of the real data set.
We used the random data Whistle results to guide the selection of a generally applicable threshold for richness and concordance p-values to use to identify significant HYPs, minimizing false positive and false negative significant HYPs. Whistle results for the randomized data sets supported the selection of 0.1 as a standard threshold for richness and concordance p-values. Applying thresholds of p < = 0.1 for both richness and concordance to all three example data sets and their matched random data yields a median number of significant HYPs inferred for the 1,000 random data sets equal to 5-10% of the number of HYPs inferred for the corresponding real data at the same threshold (Table 6, Figure 4). Thus, we expect this threshold to yield less than 10% false positive inferred HYPs for most data sets. While selection of a more stringent p-value threshold such as 0.05 reduced the number of HYPs meeting significance criteria in the random data sets, it resulted in false negatives, i.e., the loss of several biologically relevant mechanisms. Specifically, lowering the p-value threshold from 0.1 to 0.05 for the high fat diet data set resulted in the loss of eight of the thirteen HYPs including increased bp(GO:“response to endoplasmic reticulum stress”), decreased bp(GO:“response to starvation”), and decreased tscript(p(MGI:Foxa2)). As the mechanisms represented by these HYPs are highly biologically relevant for the high fat data set (see Example 1), we chose to apply the relatively permissive p < = 0.1 richness and concordance evaluation statistic threshold.

Numbers of significant HYPs at different richness and concordance thresholds in randomized data sets. Boxplot showing the number of HYPs (mechanisms) meeting the specified richness and concordance threshold across 1,000 randomized data sets. The number of significant HYPs at each threshold for the matching real data is plotted for comparison in green. (A) High Fat Diet, (B) TNF treatment, (C) PI3K inhibitor.
Bias in evaluation statistics by HYP size
We further evaluated the richness and concordance statistics by examining bias of both metrics to the number of downstream RNAs (possible) in the HYP within the randomized data sets. Because these data sets do not contain true biological signal, the correlation of either metric with the HYP size can be used to examine the expected bias.
Richness
Minimal correlation between richness and HYP size (possible) was observed in the randomized data (Pearson correlation coefficient −0.125; see Additional file 3). This weak degree of correlation suggests that the HYP size has minimal influence on richness.
Concordance
A higher level of correlation between concordance and HYP size was observed in the randomized data (Pearson correlation coefficient −0.457; see Additional file 3: Figure S5).
To correct this bias, we fit a LOESS curve to the log concordances from a subset of the randomized data matching the example data sets (n = 180,300 HYP scores) and computed an adjusted concordance measure by subtracting the fit from the log raw concordance. This correction reduced the overall correlation of concordance to the HYP size (possible) in each of the three example data sets (see Additional file 3: Figure S7). The Spearman correlation of the top 50 HYPs by concordance versus adjusted concordance was 0.80, 0.97, and 0.92 for the High Fat Diet (E-MEXP-1755), TNF (GSE2638), and PI3K Inhibitor (GSE17785) data sets, respectively. The corrected concordance values had minimal effect on the ranking of the top HYPs by concordance in the three example data sets.
Moreover, limiting the HYPs to those which are associated with a minimum number of differentially expressed RNAs removes much of the concordance bias in the randomized data. Concordance evaluates the consistency of the direction of the significant RNA expression changes with the HYP sub-network and thus is most relevant when applied to HYPs which have several significantly changing RNAs. The richness statistic is used to evaluate if the number of differentially expressed RNAs for a given mechanism is significant. In practice, concordance is critically evaluated only for those mechanisms for which at least four downstream RNAs are differentially expressed (observed), as this number is the minimum required for concordance to meet the 0.1 threshold. In the randomized data sets, only about 20% of total scored HYPs have at least four observed. Limiting to these scores reduces the correlation coefficient between concordance and possible in the randomized data from −0.457 to −0.181 (see Additional file 3: Figure S6). While the concordance bias is significant, it has limited impact on the use of concordance to filter the HYPs to a list of interesting mechanisms for a data set or to use concordance to rank the most interesting HYPs. Given the limited influence of the concordance bias within the range of interest, we have not implemented correction of concordance in Whistle.
Discussion
RCR is a reverse engineering algorithm that reasons from observed effects to potential causes, transforming lists of differentially expressed genes into mechanistic hypotheses. The use of a knowledge base structured as a directed, causal network provides RCR with some key advantages over other analysis techniques relying on prior knowledge: (1) RCR does not rely on the assumption that changes in RNA expression are equivalent to changes in the activity of the corresponding protein, (2) the structuring of gene sets as networks (‘HYPs’) allows evaluation of genes up- and down-regulated by the same mechanism as a cohesive, causally consistent mechanism, and (3) flexibility to generate HYP networks for evaluation from the knowledge base network, potentially combining related upstream nodes to a single HYP or dividing HYPs based on knowledge context.
The integration of qualitative causal relationships in Reverse Causal Reasoning fundamentally distinguishes it from other techniques in which gene expression profiling data is interpreted via over-representation analysis of functionally related sets of genes. The HYP networks assessed by RCR group measurable quantities based on a shared upstream controller, and specify the direction of control for each measurable quantity. Gene sets for over-representation analysis derived from pathway maps such as KEGG [4] or gene ontology (GO) annotation [5], differ sharply from the HYPs used by RCR in that the genes within a gene set are not related by a specified common regulator. Gene sets derived from experimental data (e.g., L2L, [9]) are more similar to mechanism networks because they are selected by a common cause (the perturbation in the experiment) but they generally do not express the causal information in a single structure. The incorporation of this causal information allows RCR to assess genes both up- and down-regulated by a controller as a common mechanism, and evaluate the causal consistency of the network against the observed data using the concordance metric.
RCR provides a qualitative assessment of significance of a mechanism, ideal for exploratory analyses to provide qualitative hypotheses about molecular mechanisms connecting the perturbation (e.g., disease or drug treatment) to the observed gene expression. In contrast, Network Perturbation Amplitude (NPA) methods, which use the same network structure as RCR to evaluate mechanisms, provide quantitative comparison of the activity of interesting mechanisms between data sets [21]. The RCR scores and evaluation metrics do not indicate relative strength of activation or inhibition of a mechanism, only the direction and significance. HYPs with lower richness and concordance p-values are more likely to be biologically relevant. We found that filtering to p-values below 0.1 typically results in a reasonable set of mechanisms for further manual evaluation, with an acceptably low level of false positives. Of note, HYPs with a relatively small number of downstream RNAs (possible) are intrinsically biased towards less significant p-values than larger HYPs due to the smaller sample size.
RCR shares a limitation with other analysis approaches that require prior knowledge that only those mechanisms or pathways that are represented in the body of prior knowledge can be inferred to be active. For example, the Whistle analysis of the TNF data set (Example 2) did not infer TNF itself to be significant. TNF is not connected to sufficient downstream RNA abundance nodes in the Large Corpus KAM, thus no HYP representing TNF was evaluated. The knowledge base used for the example data sets, the Large Corpus KAM, is significantly smaller than the knowledge base that Selventa routinely uses with this algorithm. The species-orthologized Large Corpus KAM generates approximately 600 mechanism networks. In contrast, the full Selventa knowledge base permits evaluation of more than 2000 mechanisms, and contains more downstream elements for each mechanism. We selected the Large Corpus KAM to use here because it is publically available and sufficiently large for demonstration purposes.
The derivation of HYP networks from a larger causal network provides two immediate logical extensions to RCR. First, individual HYP networks can be causally connected within the larger network (KAM). Each HYP represents a node in the KAM, thus HYPs that meet significance criteria can be joined through causal relationships in the KAM to generate more complex explanatory networks. For example, for the PI3K inhibitor treatment data set, decreased PI3K activity (kin(complex(NCH:“p85/p110 PI3Kinase Complex”))), decreased AKT kinase activity (kin(p(PFH:“AKT Family”))), and increased activity of FOXO transcription factors FOXO1, FOXO3, and FOXO4 (tscript(p(HGNC:FOXO1)), tscript(p(HGNC:FOXO3)), tscript(p(HGNC:FOXO4))) can be causally joined. Second, while Whistle is designed to create and score very simple HYP networks from a larger network, HYPs representing more complex networks can be derived from the KAM and evaluated by RCR. For example, algorithms could be created to generate HYPS representing transcriptional motifs like sets of genes co-regulated by multiple transcription factors, or signalling pathway motifs like a ligand and receptor and their combined set of downstream RNA targets.
Conclusions
RCR, a reverse engineering algorithm, provides mechanistic explanations for molecular profiling data sets, reducing data complexity and providing hypotheses to guide follow-up experimentation. This approach of identifying upstream causes that can explain the data offers some advantages over methods that map differential entities to gene ontology categories or pathway maps, because it does not rely on the assumption that changes in RNA expression leads to comparable changes in active protein.
To identify potential explanations for a set of differential measurements obtained by comparison of two sample groups, this algorithm evaluates small networks representing biological mechanisms. These small networks are comparable to gene sets, but differ in their network structure that incorporates the direction of causal influence (increase or decrease) of the mechanism on the measured quantity. This network structure enables both evaluations incorporating the direction of influence and the possibility to link multiple significant networks into a larger explanatory network.
Whistle, an implementation of RCR focused on the interpretation of gene expression data that uses prior knowledge in BEL format, is available at (https://github.com/Selventa/whistle).
References
Huang DW, Sherman BT, Lempicki RA: Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37: 1-13. 10.1093/nar/gkn923.
Wu MC, Lin X: Prior biological knowledge-based approaches for the analysis of genome-wide expression profiles using gene sets and pathways. Stat Methods Med Res. 2009, 18: 577-593. 10.1177/0962280209351925.
Khatri P, Sirota M, Butte AJ: Ten years of pathway analysis: current approaches and outstanding challenges. PLoS Comput Biol. 2012, 8: e1002375-10.1371/journal.pcbi.1002375.
Kanehisa M, Goto S, Furumichi M, Tanabe M, Hirakawa M: KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2010, 38: D355-D360. 10.1093/nar/gkp896.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G: Gene ontology: tool for the unification of biology. The gene ontology consortium. Nat Genet. 2000, 25: 25-29. 10.1038/75556.
Shankavaram UT, Reinhold WC, Nishizuka S, Major S, Morita D, Chary KK, Reimers MA, Scherf U, Kahn A, Dolginow D, Cossman J, Kaldjian EP, Scudiero DA, Petricoin E, Liotta L, Lee JK, Weinstein JN: Transcript and protein expression profiles of the NCI-60 cancer cell panel: an integromic microarray study. Mol Cancer Ther. 2007, 6: 820-832. 10.1158/1535-7163.MCT-06-0650.
Guo Y, Xiao P, Lei S, Deng F, Xiao GG, Liu Y, Chen X, Li L, Wu S, Chen Y, Jiang H, Tan L, Xie J, Zhu X, Liang S, Deng H: How is mRNA expression predictive for protein expression? A correlation study on human circulating monocytes. Acta Biochim Biophys Sin (Shanghai). 2008, 40: 426-436. 10.1111/j.1745-7270.2008.00418.x.
Schwanhäusser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M: Global quantification of mammalian gene expression control. Nature. 2011, 473: 337-342. 10.1038/nature10098.
Newman JC, Weiner AM: L2L: a simple tool for discovering the hidden significance in microarray expression data. Genome Biol. 2005, 6: R81-10.1186/gb-2005-6-9-r81.
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP: Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005, 102: 15545-15550. 10.1073/pnas.0506580102.
Culhane AC, Schwarzl T, Sultana R, Picard KC, Picard SC, Lu TH, Franklin KR, French SJ, Papenhausen G, Correll M, Quackenbush J: GeneSigDB-a curated database of gene expression signatures. Nucleic Acids Res. 2010, 38: D716-D725. 10.1093/nar/gkp1015.
Pollard J, Butte AJ, Hoberman S, Joshi M, Levy J, Pappo J: A computational model to define the molecular causes of type 2 diabetes mellitus. Diab Technol Therap. 2005, 7: 323-336. 10.1089/dia.2005.7.323.
Smith JJ, Kenney RD, Gagne DJ, Frushour BP, Ladd W, Galonek HL, Israelian K, Song J, Razvadauskaite G, Lynch AV, Carney DP, Johnson RJ, Lavu S, Iffland A, Elliott PJ, Lambert PD, Elliston KO, Jirousek MR, Milne JC, Boss O: Small molecule activators of SIRT1 replicate signaling pathways triggered by calorie restriction in vivo. BMC Syst Biol. 2009, 3: 31-10.1186/1752-0509-3-31.
Kumar R, Blakemore SJ, Ellis CE, Petricoin EF, Pratt D, Macoritto M, Matthews AL, Loureiro JJ, Elliston K: Causal reasoning identifies mechanisms of sensitivity for a novel AKT kinase inhibitor, GSK690693. BMC Genomics. 2010, 11: 419-10.1186/1471-2164-11-419.
Laifenfeld D, Gilchrist A, Drubin D, Jorge M, Eddy SF, Frushour BP, Ladd B, Obert LA, Gosink MM, Cook JC, Criswell K, Somps CJ, Koza-Taylor P, Elliston KO, Lawton MP: The role of hypoxia in 2-butoxyethanol-induced hemangiosarcoma. Toxicol Sci. 2010, 113: 254-266. 10.1093/toxsci/kfp213.
Westra JW, Schlage WK, Frushour BP, Gebel S, Catlett NL, Han W, Eddy SF, Hengstermann A, Matthews AL, Mathis C, Lichtner RB, Poussin C, Talikka M, Veljkovic E, Van Hooser AA, Wong B, Maria MJ, Peitsch MC, Deehan R, Hoeng J: Construction of a computable cell proliferation network focused on non-diseased lung cells. BMC Syst Biol. 2011, 5: 105-10.1186/1752-0509-5-105.
Schlage WK, Westra JW, Gebel S, Catlett NL, Mathis C, Frushour BP, Hengstermann A, Van Hooser A, Poussin C, Wong B, Lietz M, Park J, Drubin D, Veljkovic E, Peitsch MC, Hoeng J, Deehan R: A computable cellular stress network model for non-diseased pulmonary and cardiovascular tissue. BMC Syst Biol. 2011, 5: 168-10.1186/1752-0509-5-168.
Toedter G, Li K, Sague S, Ma K, Marano C, Macoritto M, Park J, Deehan R, Matthews A, Wu GD, Lewis JD, Arijs I, Rutgeerts P, Baribaud F: Genes associated with intestinal permeability in ulcerative colitis: changes in expression following infliximab therapy. Inflamm Bowel Dis. 2012, 18: 1399-1410. 10.1002/ibd.22853.
Deehan R, Maerz-Weiss P, Catlett NL, Steiner G, Wong B, Wright MB, Blander G, Elliston KO, Ladd W, Bobadilla M, Mizrahi J, Haefliger C, Edgar A: Comparative transcriptional network modeling of three PPAR-α/γ co-agonists reveals distinct metabolic gene signatures in primary human hepatocytes. PLoS ONE. 2012, 7: e35012-10.1371/journal.pone.0035012.
Chindelevitch L, Ziemek D, Enayetallah A, Randhawa R, Sidders B, Brockel C, Huang ES: Causal reasoning on biological networks: interpreting transcriptional changes. Bioinformatics. 2012, 28: 1114-1121. 10.1093/bioinformatics/bts090.
Martin F, Thomson TM, Sewer A, Drubin DA, Mathis C, Weisensee D, Pratt D, Hoeng J, Peitsch MC: Assessment of network perturbation amplitude by applying high-throughput data to causal biological networks. BMC Syst Biol. 2012, 6: 54-10.1186/1752-0509-6-54.
Wellman MP: Fundamental concepts of qualitative probabilistic networks. Artif Intell. 1990, 44: 257-303. 10.1016/0004-3702(90)90026-V.
Lucas PJF: Bayesian network modelling through qualitative patterns. Artif Intell. 2005, 163: 233-263. 10.1016/j.artint.2004.10.011.
Wilder SP, Kaisaki PJ, Argoud K, Salhan A, Ragoussis J, Bihoreau M-T, Gauguier D: Comparative analysis of methods for gene transcription profiling data derived from different microarray technologies in rat and mouse models of diabetes. BMC Genomics. 2009, 10: 63-10.1186/1471-2164-10-63.
Viemann D, Goebeler M, Schmid S, Nordhues U, Klimmek K, Sorg C, Roth J: TNF induces distinct gene expression programs in microvascular and macrovascular human endothelial cells. J Leukoc Biol. 2006, 80: 174-185. 10.1189/jlb.0905530.
Wallin JJ, Guan J, Edgar KA, Zhou W, Francis R, Torres AC, Haverty PM, Eastham-Anderson J, Arena S, Bardelli A, Griffin S, Goodall JE, Grimshaw KM, Hoeflich KP, Torrance C, Belvin M, Friedman LS: Active PI3K pathway causes an invasive phenotype which can be reversed or promoted by blocking the pathway at divergent nodes. PLoS ONE. 2012, 7: e36402-10.1371/journal.pone.0036402.
Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP: Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003, 4: 249-264. 10.1093/biostatistics/4.2.249.
Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini AJ, Sawitzki G, Smith C, Smyth G, Tierney L, Yang JYH, Zhang J: Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004, 5: R80-10.1186/gb-2004-5-10-r80.
Gentleman R: Bioinformatics and computational biology solutions using R and bioconductor. 2005, New York: Springer Science + Business Media
Smyth GK: Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004, 3 (1): Article 3. doi: 10.2202/1544-6115.1027
Winzell MS, Ahrén B: The high-fat diet-fed mouse: a model for studying mechanisms and treatment of impaired glucose tolerance and type 2 diabetes. Diabetes. 2004, 53 (Suppl 3): S215-S219.
Ozcan U, Cao Q, Yilmaz E, Lee A-H, Iwakoshi NN, Ozdelen E, Tuncman G, Görgün C, Glimcher LH, Hotamisligil GS: Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004, 306: 457-461. 10.1126/science.1103160.
Howell JJ, Stoffel M: Nuclear export-independent inhibition of Foxa2 by insulin. J Biol Chem. 2009, 284: 24816-24824. 10.1074/jbc.M109.042135.
Wolfrum C, Stoffel M: Coactivation of Foxa2 through Pgc-1beta promotes liver fatty acid oxidation and triglyceride/VLDL secretion. Cell Metab. 2006, 3: 99-110. 10.1016/j.cmet.2006.01.001.
Lakshminarayanan V, Drab-Weiss EA, Roebuck KA: H2O2 and tumor necrosis factor-alpha induce differential binding of the redox-responsive transcription factors AP-1 and NF-kappaB to the interleukin-8 promoter in endothelial and epithelial cells. J Biol Chem. 1998, 273: 32670-32678. 10.1074/jbc.273.49.32670.
Kempe S, Kestler H, Lasar A, Wirth T: NF-kappaB controls the global pro-inflammatory response in endothelial cells: evidence for the regulation of a pro-atherogenic program. Nucleic Acids Res. 2005, 33: 5308-5319. 10.1093/nar/gki836.
Zhou Z, Connell MC, MacEwan DJ: TNFR1-induced NF-kappaB, but not ERK, p38MAPK or JNK activation, mediates TNF-induced ICAM-1 and VCAM-1 expression on endothelial cells. Cell Signal. 2007, 19: 1238-1248. 10.1016/j.cellsig.2006.12.013.
Shimada Y, Ito H, Kaji K, Fukuda M: Tumor necrosis factor reduces lifespan of human endothelial cells in vitro. Mech Ageing Dev. 1990, 55: 245-254. 10.1016/0047-6374(90)90152-6.
Magder S, Neculcea J, Neculcea V, Sladek R: Lipopolysaccharide and TNF-alpha produce very similar changes in gene expression in human endothelial cells. J Vasc Res. 2006, 43: 447-461. 10.1159/000095162.
Courtney KD, Corcoran RB, Engelman JA: The PI3K pathway as drug target in human cancer. J Clin Oncol. 2010, 28: 1075-1083. 10.1200/JCO.2009.25.3641.
Acknowledgements
The authors would like to acknowledge the late Navin Chandra, who, as founding CTO of Genstruct, articulated a vision of cause-and-effect analysis to pragmatically address complex issues in molecular biology. His insights were a critical first step leading to the development of RCR. The authors would like also to thank Justin Sun and Tat Chu for important contributions to RCR, Ty Thomson for helpful discussions, and William Hayes for help generating and running the randomized data sets.
The research described in this article was supported by Selventa.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
All authors are either current or former employees of Selventa (formerly Genstruct). Selventa has applied for a patent related to the method described in this manuscript; TS and DP are listed as inventors on the application.
Authors’ contributions
TS, WL, KOE, and DP developed the RCR methodology and initial implementation. AJB and SU developed Whistle. NLC provided requirements for and testing of Whistle and developed the Whistle/RCR use examples. NLC and DP drafted the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12859_2012_6209_MOESM2_ESM.xlsx
Additional file 2: Evaluated HYPs for the TNF (HG- U133A), PI3K inhibitor (HG- U133 Plus 2.0), and high fat diet (MG-430 2.0 Array) data sets.(XLSX 9 MB)
12859_2012_6209_MOESM3_ESM.pdf
Additional file 3: Figure S5: Evaluation of richness correlation with HYP size. Scatter plot (left) and boxplot (right) of richness versus HYP size (possible) for randomized data matched to the example data sets. Pearson correlation coefficient = -0.125. S6. Evaluation of concordance correlation with HYP size. Scatter plot (top left) and boxplot (top right) of concordance versus HYP size (possible) for randomized data matched to the example data sets. Pearson correlation coefficient = -0.457. Boxplot (bottom right) shows reduced correlation for scores limited to those HYPs with at least four RNA expression changes; correlation coefficient -0.181. S7. Evaluation of LOESS fit-adjusted concordance for example data sets.Boxplots for concordance versus HYP size (possible) for the High Fat Diet, TNF, and PI3K inhibitor example data sets (left) and adjusted concordance (right). (PDF 239 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an open access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Catlett, N.L., Bargnesi, A.J., Ungerer, S. et al. Reverse causal reasoning: applying qualitative causal knowledge to the interpretation of high-throughput data. BMC Bioinformatics 14, 340 (2013). https://doi.org/10.1186/1471-2105-14-340
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2105-14-340