
Abstract
Background
Bayesian Network (BN) is a powerful approach to reconstructing genetic regulatory networks from gene expression data. However, expression data by itself suffers from high noise and lack of power. Incorporating prior biological knowledge can improve the performance. As each type of prior knowledge on its own may be incomplete or limited by quality issues, integrating multiple sources of prior knowledge to utilize their consensus is desirable.
Results
We introduce a new method to incorporate the quantitative information from multiple sources of prior knowledge. It first uses the Naïve Bayesian classifier to assess the likelihood of functional linkage between gene pairs based on prior knowledge. In this study we included cocitation in PubMed and schematic similarity in Gene Ontology annotation. A candidate network edge reservoir is then created in which the copy number of each edge is proportional to the estimated likelihood of linkage between the two corresponding genes. In network simulation the Markov Chain Monte Carlo sampling algorithm is adopted, and samples from this reservoir at each iteration to generate new candidate networks. We evaluated the new algorithm using both simulated and real gene expression data including that from a yeast cell cycle and a mouse pancreas development/growth study. Incorporating prior knowledge led to a ~2 fold increase in the number of known transcription regulations recovered, without significant change in false positive rate. In contrast, without the prior knowledge BN modeling is not always better than a random selection, demonstrating the necessity in network modeling to supplement the gene expression data with additional information.
Conclusion
our new development provides a statistical means to utilize the quantitative information in prior biological knowledge in the BN modeling of gene expression data, which significantly improves the performance.
Similar content being viewed by others


Background
Reverse engineering of genetic networks will greatly facilitate the dissection of cellular functions at the molecular level [1–3]. The time course gene expression study offers an ideal data source for transcription regulatory network modeling. However, in a typical microarray experiment usually up to tens of thousands of genes are measured in only several dozens or less samples, data from such experiments alone is significantly underpowered, leading to high rate of false positive predictions [4]. Network reconstruction from microarray data is further limited by low data quality, noise and measurement errors [5].
Incorporating other types of data and existing knowledge of gene relationships into the network modeling process is a practical approach to overcome some of these problems. It has been proven that data integration and useful bias with relevant knowledge can improve the network prediction accuracy from gene expression data [6, 7]. Among the various approaches of network modeling, Bayesian Networks (BN) have shown great promise and are receiving increasing attention [8]. BN is a graphic probabilistic model that describes multiple interacting quantities by a directed acyclic graph (DAG). The nodes in the network represent random variables (expression levels), and edges represent conditional dependencies between nodes [9]. Learning a BN structure is to find a DAG that best matches the dataset, namely maximizing the posterior probability of DAG given data D: P (DAG|D). The sound probabilistic schematics allow BN to deal with the inherent stochasticity in gene expressions and the noise brought in by the microarray technology. Furthermore, BN is capable of integrating prior knowledge into the system in a natural way [9, 10].
A number of studies demonstrated that adding prior knowledge to BN improved the performance [4, 11–14]. Many sources of data and information are useful to supplement the gene expression data, and they can be incorporated at different steps of BN simulation, from prior structure definition to structure simulation and evaluation.
Known protein-DNA interaction or other clues of the relationships between transcription factors and their target genes are useful to transcription regulatory network inference. Hartemink et al. included data from the chromatin immunoprecipitation (ChIP) assay [15], and Tamada et al incorporated promoter sequence motif information [16], to define the prior probability of network structures. Information of other types of gene pair relationship has also been explored. Steele et al. developed a gene-pair association score from the correlation of their concept profiles derived from literature, and utilized that to define the prior structure probabilities [12]. Larsen et al defined a Likelihood of Interaction (LOI) score, which measures the statistical significance of two genes interacting with each other according to their shared Gene Ontology (GO) information. They then restricted the candidate network edges (interactions) to those with significant p -values of LOI during the BN structure learning iterations [17, 18]. By doing so, the quantitative information of the likelihood is not fully utilized in the network modeling. Djebbari and Quackenbush utilized literature, high-throughput protein-protein interaction (PPI) data, or the combination of both to define the seed (initial) network structure. They observed an improved ability of the BN analysis to learn gene interaction networks from the expression data [19].
Imoto et al formulated an novel approach to incorporate prior biological knowledge within the BN framework by adopting the energy concepts from statistical physics [20, 21], which was later further extended by Husmeier and Werhli [22, 23]. In this approach an energy function was first defined to measure the agreement between a candidate network and the prior biological knowledge, and prior distribution of network structure is hence calculated using the Gibbs distribution in a canonical ensemble. Using this approach, the two groups examined several types of prior knowledge, including PPI, protein-DNA interaction, binding site information, literature, and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways [22–24]. The algorithms were validated using yeast gene expression data [20, 21], and synthetic data [22].
Existing studies often utilize prior knowledge to construct the prior distribution of network, or initial network structure. It has been demonstrated that the sampling method during simulation also affects the performance of BN structure learning [25]. Though prior knowledge has been utilized to bias the sampling step, it is normally done through restricting the search space to sub regions, for instance, only simulate candidate structures whose significance is above a certain threshold according to prior knowledge [17, 18].
In searching for the network structure (DAG) that maximize P (DAG|D), the Markov Chain Monte Carlo (MCMC) approach is regarded better than greedy searching algorithms, especially for the microarray data with small sample size where there is often no single structure that is prominently better than others [9]. In this study we propose a new approach to incorporate prior knowledge in a quantitative way to bias the MCMC simulation of candidate structure. It utilizes information of functional linkage between gene pairs, assuming that functionally linked genes are likely to interact with each other. It is known that interacting proteins or genes often share similar function, and participate in the same biological pathways and processes [26]. Interaction has been utilized to infer functional linkage and annotate gene functions [27]. Increasing evidence suggests that the reverse is also frequently true [28]. In our algorithm a probability score is first calculated that measures how likely two genes are functionally linked based on prior knowledge; A candidate edge reservoir is then constructed where the number of copies of each edge is proportional to this probability score; The reservoir is in turn used for sampling candidate network structure during the MCMC simulation. This way the quantitative information of the potential gene pair link predicted by prior knowledge is retained.
We will consider two type of prior knowledge: co-citation in PubMed literature and similarity in ontological annotation according GO http://www.geneontology.org/. We will demonstrate they both contain information of functional linkage. The performance of the new algorithm is evaluated using a synthetic data set as well as data from two real microarray experiments: the yeast cell cycle study, and the mouse pancreas development/growth study. We will demonstrate that including the prior knowledge significantly improves the performance of BN modeling of gene expression data.
Results
Algorithm
BN is a graphical model to capture complex relationships among a set of random variables {X1, X2,...,X n } encoding the Markov assumption, each node representing a variable. In the context of gene network modeling, each node represents a gene, while gene interactions are represented by directed edges between nodes. Each variable X i in the DAG is conditionally independent of its non-descendants given its set of parents. Mathematically the joint distribution of the DAG can be decomposed into a product form as:
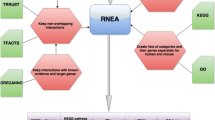
where Π i denotes the parent set of the variable X i . This is referred as the chain rule for BNs [9, 10]. Learning a BN structure is to find a DAG that best matches the dataset, namely maximizing the posterior probability of DAG given data P (DAG|D). Here we adopt the sampling-based approach to Bayesian inference, and sample network structures from a candidate edge reservoir with the MCMC network learning method. In the reservoir the edge representation is proportional to the likelihood of the two genes being functionally linked according to prior knowledge. This way, the edges between the strongly-related gene pairs have higher chance to be proposed as part of the candidate network. The overall design is given in Figure 1. The major steps included:

The framework of our BN modeling to incorporate the quantitative information in prior knowledge.
-
1.
Determine the probability of functional link p link between each gene pairs
1.1 Calculate GO schematic similarity
1.2 Calculate p value of PubMed co-citation.
1.3 Integrate GO and PubMed information using the Naïve BN to determine plink.
-
2.
Construct candidate network edge reservoir in which copy number of each edge is proportional to the p link of the corresponding gene pair.
-
3.
Learn network structure using the MCMC algorithm through sampling the candidate network edge reservoir.
At each step of the iteration, the proposed network is retained with an acceptance probability that is determined by the relative posterior of the proposed versus current network, penalized by the network complexity [29, 30]. In calculating the posterior we use the BDe (Bayesian Dirichlet equivalence) scoring metric [10, 31]. The prior distribution is assumed to be uniform.
To evaluate the performance of our BN algorithm, and the benefit of adding prior knowledge, we compare it to two alternative approaches: (1) Plain BN. In each iteration, a new network is proposed by randomly changing one edge in the current network. (2) The method developed by Husmeier and Werhli [22, 23].
GO schematic similarity and significance of PubMed co-citation
GO annotation and gene citation database (PubMed) were downloaded from ftp://ftp.ncbi.nlm.nih.gov/gene/DATA. Schematic similarity in GO taxonomy was first calculated for each gene pair using the approach proposed by Cao et al[32], which calculates the shared information content of the GO terms. The value of this measure ranges between [0 1], with 0 being no similarity, and 1 being maximum similarity. The GO similarity between each gene pair is defined to be the maximum schematic similarity of all the GO terms they share.
For a given pair of genes, the total number of PubMed abstracts in which each gene appears (n and m, respectively), and in which both appear (k) were determined. The probability of co-citation frequency observed by random chance is calculated by
where , and N is the total number of abstracts in PubMed [1, 2].
Construction of the candidate network edge reservoir
We used the Naïve Bayesian network to integrate the GO and co-citation information, and a simple Bayesian naïve classifier to predict the functional linkage probability p link for all gene pairs. Note that the prior knowledge of functional linkage is undirected, i.e. p link (i, j) = p link (j, i). An edge sampling reservoir was constructed, in which the number of replicates for the edge between gene i and j N (Edge i,j) is in proportion to their plink :
where Ceil(x) is the smallest integer no less than x. In this definition, any gene pair will be represented at least once and at most 10 times. The edges of gene pairs with higher plink will appear more frequently in the edge reservoir, and hence enjoy a higher chance to be selected during the network structure learning.
Implementation
Our BN simulation algorithm is implemented in Matlab utilizing Kevin Murphy's BNT package [33]bnt.googlecode.com, and is summarized in Table 1. Note that steps 1 and 3.1 contain unique features that separate our approach from others. The source code is available upon request. The networks were visualized with Cytoscape [34].
Validation
Utility of GO similarity and PubMed co-citation in discovering functional linkage between gene pairs
Lee et al developed an approach to evaluate if gene-pair functional relationships can be predicted by a certain type of high-throughput genomic data (gene expression, PPI, ChIP-chip, etc) [35, 36]. Assuming that p(L|D) and (~ L|D) denote the probabilities of gene pairs to share or not share functional annotation given that they are linked by data D (for instance, co-expressed, sharing PPI, protein of one gene binds to the promoter of the other, etc), and p(L) and p(~ L) represent the prior probabilities of sharing and not sharing functional annotation, they proposed a log likelihood score [35, 36]:
to describe the utility of data D in functional linkage inference. An LLS close to 0 suggest that the data is not more informative than random pairing, whilst higher positive values of LLS indicates that data D contains more information of functional linkage.
We adopted equation (4) to evaluate whether GO schematic similarity and PubMed co-citation were useful in identifying functional linkage. The KEGG http://www.genome.ad.jp/KEGG and Munich Information Center for Protein Sequences (MIPS, mips.gsf.de/) database [1, 2] were used to construct the benchmarks of functional linkage. These databases were chosen for their high quality [37]. In this study we utilized yeast and mouse gene expression data to validate our algorithm. For each species, the positive control set consists of randomly sampled 5% (43,761 for yeast, and 35,424 for mouse) of all gene pairs that are in the same KEGG pathways [38]. The choice of 5% rather than all is to lower the computational complexity. The negative control set was constructed with gene pairs that encode proteins localized in different cellular compartments, with the underlying assumption that they are functionally unrelated and do not interact with each other. Four categories in the MIPS annotation [39] were utilized: 70.03 cytoplasm, 70.10 nucleus, 70.16 mitochondrion, and 70.27 extracellular/secretion proteins.
Again we only kept 5% of all possible gene pairs, totaling 112,693 for yeast and 531,089 for mouse, respectively. The same benchmark sets were also utilized to train the Naïve Bayesian classifier when calculating plink.
The LLS of co-citation in discovering functional linkage is then determined by:
The LLS of GO schematic similarity was performed in the similar fashion. The LLS value for gene pair sets in different ranges of GO similarity and co-citation p-value were given in Table 2. Gene pairs sets with higher GO similarity or PubMed co-citation significance, have more positive LLS values, and vice versa. Note that gene pairs with negative LLS means they are less likely to be functionally linked than random pairs, which is expected if they share low GO similarity or co-citation. The results suggest that PubMed Co-citation and GO similarity are efficient at discriminating functionally linked gene pairs from not linked ones.
We found that there is a marginal dependence between the GO similarity and PubMed co-citation (Fisher's Z test, p~0.1). Theoretically naïve Bayesian classifier is optimal when the attributes are independent given class. However, empirical studies have shown that the classifier still performs well in many domains when there is moderate attribute dependences [40]. The weak dependence between them indicates that the naïve Bayesian Network is an appropriate choice to integrate their information [41]. Interestingly, the GO and MIPS categories, which are both functional annotations, also only depend weakly on each other. This may be because the present annotations are far from being perfect and complete [42].
Utility of functional linkage information to interaction network modeling
The distribution of p link for yeast gene pairs is given in Figure 2. Note that only a small proportion of gene pairs share high values of p link , for about 92% of the gene pairs this value is less than 0.2. This indicates that most gene pairs share no functional linkage, consistent with the fact that gene networks are usually sparse. The candidate edge reservoir is constructed according to equation 3, and the MCMC samples this distribution to propose new candidate network structure at each iteration. In Figure 2 we have also included the distribution for gene pairs predicted to be interacting to each other, with and without the prior knowledge. Among all possible gene pairs, only ~8% with p link 0.6. In contrast, this proportion increases to 28% among the predicted interactions. It indicates that the prior knowledge did affect the outcome of the BN learning. The results from the other data sets are similar.

Distribution of functional linkage probability for all possible gene pairs, and for predicted interactions with and without prior knowledge.
The assumption of incorporating prior knowledge of functional linkage is that they can help network modeling. Existing data from yeast revealed that genes sharing the same GO attribute interact genetically more often than expected by chance (p < 0.05) [43, 44]. In a very conservative estimate, over ~12% of the genetic interactions are comprised of genes with identical GO annotation (a 12 fold enhancement over what expected by chance, p < 10-12); and over 27% are between genes with similar or identical GO annotations (an 8 fold enhancement, p < 10-10).
We examined whether p link can potentially discriminate interacting gene pairs from non-interacting ones, using the receiver operating characteristic (ROC) curve. ROC is a graphical plot of the sensitivity versus (1-specificity), namely the fraction of true positives versus the fraction of false positives, as the discrimination threshold of a classifier is varied. The area under curve (AUC) reflects the performance. The ROC of a random classifier would be a 45° line with AUC = 0.5. Figure 3 presents the ROC plot for the nine yeast cell cycle regulating transcription factors (TF): Fkh1, Fkh2, Ndd1, Mcm1, Ace2, Swi5, Mbp1, Swi4, and Swi6, and their targets identified using the ChIP-chip technology [45]. The AUC of 0.6064 indicating that is positively correlated with interaction and therefore useful in interaction inference.

ROC curve indicating that functional linkage contains information for interaction. Plotted is the performance of p link as a classifier to identify yeast TF-target pairs defined by ChIP-chip.
Convergence of simulation
In Figure 4A we plot the acceptance ratio versus number of MCMC steps in the yeast cell cycle dataset. Obviously in the later steps the probability to accept the new proposed DAG is small and flattens. The results from the other datasets are similar. In addition, the MCMC simulation was repeated 20 times with independent initializations, and consistency in the marginal posterior probabilities was examined. We found that they correlated well between different runs: 0.83 ± 0.11 for the simulated dataset, 0.68 ± 0.10 for the yeast data set, and 0.51 ± 0.26 for the mouse pancreas dataset. Figure 4B presents the scatter plot of the edge posterior probability from two typical runs that simulate the yeast dataset.

Convergence of simulation. (A) Acceptance ratio versus the number of MCMC steps (B) scatter plot of the marginal posterior probabilities of the edges, obtained from two separate MCMC simulations of the yeast cell cycle data.
Validation using simulated data
In our network inference, the MCMC learning simulation is repeated 20 times with independent initializations and an interaction will be considered in the final network if it is observed more than 15 times. Our new BN algorithm was first tested in a simulated time course (50 time points) gene expression dataset of an artificial network generated using SynTReN [46]. This network contains 76 genes, of which 24 act as regulators with a total of 124 regulatory relationships (i.e. 124 edges). The results are summarized in Table 3, 2nd column. It demonstrates that incorporating the functional linkage as prior knowledge allows the identification of a significantly higher number, 21 versus 14, of the true gene-gene relationships compared with the plain BN modeling of gene expression data only. A random network of the same number of edges was also created for the 76 genes [47]. The improvement of BN with prior knowledge over random is significant (p < 0.01, Table 3), while without prior knowledge it is not (p~0.2, Table 3).
Validation using the yeast cell cycle data
Next the new algorithm was applied to one of the Stanford yeast cell cycle data http://genome-www.stanford.edu/cellcycle/, where the cells from a cdc15 temperature sensitive mutant were studied [48]. To evaluate the performance, we compared the predicted interactions from our algorithm to the annotated interactions in BIND http://bind.ca[49], and the transcription regulation predicted by the ChIP-chip data [45]. Tables 4, 5 list the benchmark interactions for the 107 yeast cell cycle genes that were recovered by the BN modeling. The statistical results are summarized in Table 3, columns 3-4.
Evidently, our method is capable of identifying a higher number of the positive benchmarks compared with the plain BN without prior knowledge. When evaluated with the BIND annotation, the number of correctly identified interactions doubled from 13 to 26 (p~0.13, χ2~2.28). The plain BN actually did not perform better than random selection (p~0.11). In contrast, BN with prior knowledge performed significantly better than random selection with χ2 = 24.5, p < 0.001. When evaluated with the ChIP-chip data, the story is similar. The number of correctly identified gene regulatory relationships increased from 11 to 23 with the addition of prior knowledge (p < 0.01, χ2 = 6.71). Without the prior knowledge, the plain BN is not different from random selection (p~0.1).
Figure 5A-5C shows the ROC curves that give a more quantitative view of the performance of BN with/without prior knowledge, and of the Werhli and Husmeier's algorithm [22, 23], in detecting TF-target gene interactions. Incorporation of prior knowledge significantly improved the performance with higher AUC. Our algorithm performed slightly better than Werhli and Husmeier's.

ROC curves for the network modeling of the yeast cell cycle data using plain BN (A), Werhli and Husmeier's (B), and our algorithm (C). ChIP-chip binding data were used as benchmark. Adding prior knowledge significantly improved BN performance at identifying the TF-target pairs.
Validation using mouse pancreas development data
We also validated our algorithm using a mammal dataset. The experiment profiled gene expression changes in pancreas during embryonic development or during compensatory growth after partial pancreatectomy. Elucidating the networks is key to understand the complex nature of pancreas development and function [50, 51]. A number of efforts have been made to manually annotate the key transcription factors and the gene networks they regulate based on low-throughput data, nicely reviewed by Servitja and Ferrer [52]. In Table 6, we list the 24 experimentally confirmed gene-gene regulatory relationships [52], and their network is depicted in Figure 6A. With prior knowledge BN modeling of the expression data is able to recover half of them (12), as shown in Figure 6C and Table 6. In contrast, the plain BN is only able to identify 6 of them (Figure 6B). This is again a ~two-fold enhancement. In Figures 7A-7C the ROC curves are presented. Incorporation of prior knowledge significantly improved the ability to detect known interactions. Our algorithm performed comparably to Werhli and Husmeier's.

The pancreas development network already established by existing experiments (A), predicted by the plain BN (B), and by BN + prior knowledge (C). The bold edges in (B) and (C) are those that overlap with the edges in (A).

ROC curves for the pancreas development data with plain BN (A), Werhli and Husmeier's algorithm (B), and our approach (C). 24 experimentally confirmed interactions were used as benchmark.
In Additional file 1, we listed the GO similarity and PubMed co-citation of the gene pairs with known regulatory relationships that were missed by plain BN. Clearly, almost all of them have high GO similarity and share a significant number of co-citations. Adding the functional linkage as prior knowledge helped to recover them.
Discussion
In this study we proposed a new algorithm to quantitatively utilize prior biological knowledge in the network modeling of gene expression data. First the functional linkage of gene pairs was assessed based on multiple data sources using the naïve Bayesian classifier. The result was then utilized to construct a candidate network edge reservoir, where the number of replicate edges between each gene pair was proportional to their function linkage probability. During simulation new candidate network structure was formed by sampling from this reservoir at each iteration. Since the edges of gene pairs with stronger functional linkage had more representations in the reservoir, these biologically meaningful edges enjoyed a preferential treatment in network simulation. With both the simulated and real gene expression data, we demonstrated that incorporating the prior knowledge significantly improved the network modeling performance. More information of the gene interaction network could be extracted from the microarray data with higher accuracy. In contrast, in all datasets, without the prior knowledge, though the number of benchmark regulations recovered is more than a random selection, the improvement is not statistically significant, demonstrating the necessity to supplement the gene expression data with additional information. This finding that plain BN did not perform better than random selection was not unexpected, similar observations was recently reported for a number of publically available reverse-engineering algorithms when gene expression data is the sole source of information [47].
Our algorithm provides a practical way to integrate the probabilistic biological knowledge that is different from previous efforts by others [2]. The quantitative nature makes it capable to handle soft constraints. Using the approach by Werhli and Husmeier for instance [22, 23], we differ in several key steps. First, they encode multiple sources of prior knowledge in a weighted sum via an energy function; we integrate information from multiple sources through a Bayesian classifier. Furthermore, in our approach the MCMC samples from a candidate edge distribution defined by the prior knowledge, rather than from the network posterior distribution where the network prior is defined by the prior knowledge. Our algorithm utilizes the prior knowledge at interaction level, while theirs at the network level. Finally the Werhli and Husmeier approach is more computational intensive. To reduce the computational complexity, they sum over all parent configurations of each node and limit the number of parents of each node to 3 or less; the complexity of this operation is (where N is size of the network, and m the maximum FanIn) [23]. We find that it is still memory consuming for networks of moderate or large sizes. For instance, a Dell Optiplex 755 with 2GHZ DUO CPU, 3.25 GB RAM ran out of memory when simulating the 107-gene yeast network. Our algorithm does not have this problem.
We used two sources of prior evidence of functional linkage to assist network modeling: the PubMed co-citation and GO schematic similarity. However, our framework by design allows the integration of other types of data or knowledge, for instance, high throughput genomic data including PPI and ChIP-chip; gene-gene relationships derived from advanced methods including text mining [53], database curation, and computational modeling of sequence information; and many other sources. It has been demonstrated that the degree of improvement brought in by prior knowledge highly depends on the quality of the information being added [54]. Low quality prior knowledge could even lower the performance of BN [54]. Presently, most of the available prior knowledge each on its own suffers from high false positive rate and being incomplete, which can limit their efficacy in network modeling. Integration of data from different sources and utilizing their consensus provides an effective means to deal with this issue [1, 2]. A caveat here is, when considering more sources of data, the inter-dependency among them need to be scrutinized more carefully, and maybe a more sophisticated integration method than the naïve Bayesian classifier is needed.
A number of different approaches have been developed to integrate multiple sources of prior information in the BN modeling of gene expression data, at the different steps of the simulation process [4, 11–14]. It would be of interest to compare the efficiency of the different approaches, investigate whether the optimal approach depends on the types of prior knowledge, and if the different approaches can be combined for a most efficient utilization of prior knowledge in network modeling.
Conclusion
In this paper we proposed a new algorithm to integrate and utilize the prior biological knowledge in the BN modeling of gene expression data. Our study demonstrated that incorporating prior knowledge at the step of network structure simulation is an efficient way to preserve the quantitative information in it, and to improve the performance of network modeling.
Methods
Preparation of gene expression data for algorithm validation
Simulated data
The simulated time course gene expression dataset was generated using SynTReN [46] for a artificial network with 76 genes, of which 24 act as regulators with a total of 124 regulatory relationships (i.e. 124 edges). The total number of time points is 50. All parameters of SynTReN were set to default values [46], except number of correlated inputs, which was set to 50%. The topological structure and inner interacting relationships are sampled from the characteristics of the yeast transcriptional network, therefore the results will be indicative of the algorithm performance on real data.
Yeast cell cycle study
Yeast cell cycle gene expression data were downloaded from http://genome-www.stanford.edu/cellcycle/. These studies [48, 55] profiled expression changes in 6178 genes at ~20 time points under each condition following alpha factor arrest (18 time points from 0-119 minutes), elutriation ELU (14 time points from 0-390 minutes), and arrest of a cdc15 (24 time points from 10-290 minutes) and a cdc28 (28 time points from 0-160 minutes) temperature sensitive mutant. Many genes have missing data points. The cdc28 data is the most severely affected, ~80% of genes contains at least 1 missing values. For the remaining three datasets, it ranged 6-27%. In this study, we chose the cdc15 dataset, as it contains the most number of time points out of the three [56]. Network modeling was performed on the 107 known cell cycle genes [57]. The list is given in Table 7. These are the genes that most likely to have interesting interactions during the time course being studied.
Mouse pancreas development and regeneration after damage
The pancreas development and growth expression data was downloaded from the RNA Abundance Database http://www.cbil.upenn.edu/RAD, with study IDs 2 and 1790. Study 2 profiled mouse pancreas gene expression at six different developmental time points: embryonic day 14.5, 16.5, 18.5, at birth, at postnatal day 7, and at adulthood. 4 samples at E14.5, and 6 at all the following time points, totaling 34 samples. Study 1790 profiled gene expression in mice pancreas following partial pancreatectomy and Exendin-4 treatment. Exendin-4 is a glucagon-like peptide-1 receptor agonist that augments the pancreatic islet beta-cell mass by increasing beta-cell neogenesis and proliferation and by reducing apoptosis. Mice underwent 50% pancreatectomy or sham operation, and received Exendin-4 or vehicle every 24 hours. 3-4 animals from each group were sacrificed at each time point of 12, 24 and 48 hr after operation, together with 4 animals that received no operation, totaling 46 samples. Because the two studies each only contain a few time points, we combined their data for network modeling [58]. Replicate samples under the same condition at the same time point were averaged.
The network modeling was performed on 36 genes manually collected from a recent review by Servitja and Ferrer [52], which are known to be important in pancreas development. They are listed in Table 8.
Digitization of gene expression data
Expression data were further discretized into three levels. In each data set, we calculated the mean (μ) and standard deviation (SD) of expression across all time points for each gene. Each expression value is then assigned to 0, 1 or 2 according to whether the value is less than μ-SD, between μ-SD and μ+SD, or above μ+SD.
Prior data of interaction and transcription binding
Annotations of known yeast gene interaction were downloaded from the Biomolecular Interaction Network Database (BIND, http://bind.ca), a database designed to store full descriptions of interactions, molecular complexes and pathways [49]. BIND includes both directed (such as protein-DNA interaction) and un-directed (such as protein-protein interaction) interactions. Therefore when comparing to BIND annotations, we ignored direction.
Simon et al studied the transcription regulation of yeast genes by 9 cell cycle regulating transcription factors (TF): Fkh1, Fkh2, Ndd1, Mcm1, Ace2, Swi5, Mbp1, Swi4, and Swi6, using the ChIP-chip technology [45]. These nine TFs are among the 107 cell cycle genes that we performed network modeling. The data were downloaded from http://staffa.wi.mit.edu/cgi-bin/young_public/navframe.cgi?s=17%26;f=downloaddata. For each TF, the study derived a binding p-value for each gene which reflects the likelihood that the TF binds to the promoter of this gene. We constructed a positive control target set for each TF that consists of those with p < 0.001, a negative control target set for each TF that consists of those with p > 0.1. Note that the transcription binding data provide directed information.
Abbreviations
- AUC:
-
area under curve
- BN:
-
Bayesian Network
- DAG:
-
directed acyclic graph
- GO:
-
Gene Ontology
- MCMC:
-
Markov Chain Monte Carlo
- PPI:
-
protein-protein interaction
- ROC:
-
receiver operating characteristic
- TF:
-
transcription factor.
References
Fraser AGME: A probabilistic view of gene function. Nat Genet 2004, 6: 559–564.
Lee IDS, Adai AT, Marcotte EM: A Probabilistic Functional Network of Yeast Genes. Science 2004, 306: 1555–1558. 10.1126/science.1099511
Xue-wen Chen GAaXW: An effective structure learning method for constructing gene networks. Bioinformatics 2006, 22: 1367–1374. 10.1093/bioinformatics/btl090
Imoto SHT, Goto T, Tashiro K, Kuhara S, Miyano S: Combining Microarrays and Biological Knowledge for Estimating Gene Networks via Bayesian Networks. J Bioinform Comput Biol 2004, 2: 77–98. 10.1142/S021972000400048X
Wang X, Hessner MJ: Quantitative quality control of microarray experiments: toward accurate gene expression measurements. In Gene expression profiling by microarrays - clinical implications. Edited by: K. HW: Cambridge; 2006.
Troyanskaya OG, Dolinski K, Owen AB, Altman RB, Botstein D: A Bayesian framework for combining heterogeneous data sources for gene function prediction (in Saccharomyces cerevisiae). Proc Natl Acad Sci USA 2003, 100: 8348–8353. 10.1073/pnas.0832373100
Han JJ, McDonald CM: Diagnosis and clinical management of spinal muscular atrophy. Phys Med Rehabil Clin N Am 2008, 19: 661–680. xii xii
Friedman N, Linial M, Nachman I, Pe'er D: Using Bayesian networks to analyze expression data. J Comput Biol 2000, 7: 601–620. 10.1089/106652700750050961
Heckerman D: A tutorial on learning with Bayesian networks. In Learning in Graphical Models. Edited by: Jordan MI. Kluwer, Dordrecht; 1998.
Cooper GF, Herskovits EA: A bayesian method for the induction of probabilistic networks from data. Machine Learning 1992, 9: 309–347.
Le Phillip P, Bahl A, Ungar LH: Using prior knowledge to improve genetic network reconstruction from microarray data. In Silico Biol 2004, 4: 335–353.
Steele E, Tucker A, t Hoen PA, Schuemie MJ: Literature-based priors for gene regulatory networks. Bioinformatics 2009, 25: 1768–1774. 10.1093/bioinformatics/btp277
Gevaert O, Van Vooren S, De Moor B: A framework for elucidating regulatory networks based on prior information and expression data. Ann N Y Acad Sci 2007, 1115: 240–248. 10.1196/annals.1407.002
Le Phillip P, ABA A, Ungar LyleH: Using Prior Knowledge to Improve Genetic Network Reconstruction from Microarray Data. In Silico Biology 2004, 4: 335–353.
Hartemink AJ, Gifford DK, Jaakkola TS, Young RA: Combining location and expression data for principled discovery of genetic regulatory network models. Pac Symp Biocomput 2002, 437–449.
Tamada Y, Kim S, Bannai H, Imoto S, Tashiro K, Kuhara S, Miyano S: Estimating gene networks from gene expression data by combining Bayesian network model with promoter element detection. Bioinformatics 2003, 19(Suppl 2):ii227–236. 10.1093/bioinformatics/btg1082
Larsen P, Almasri E, Chen G, Dai Y: A statistical method to incorporate biological knowledge for generating testable novel gene regulatory interactions from microarray experiments. BMC Bioinformatics 2007, 8: 317. 10.1186/1471-2105-8-317
Eyad Almasri PL, Chen Guanrao, Dai Yang: Incorprating Literature Knowledge in Baysian Network for Inferring Gene Networks with Gene Expression Data. Proceeding of the 4th International Symposium on Bioinformatics Research and Applications 2008, 4983: 184.
Djebbari A, Quackenbush J: Seeded Bayesian Networks: constructing genetic networks from microarray data. BMC Syst Biol 2008, 2: 57. 10.1186/1752-0509-2-57
Imoto S, Goto T, Miyano S: Estimation of genetic networks and functional structures between genes by using Bayesian network and nonparametric regression. Pac Symp Biocomput 2002, 7: 175–186.
Imoto S, Higuchi T, Goto T, Tashiro K, Kuhara S, Miyano S: Combining Microarrays and Biological Knowledge for Estimating Gene Networks via Bayesian Networks. J Bioinform Comput Biol 2004, 2: 77–98. 10.1142/S021972000400048X
Husmeier D, Werhli AV: Bayesian Integration of Biological Prior Knowledge into the Reconstruction of Gene Regulatory Networks with Bayesian Networks. Comput Syst Bioinformatics Conf 2007, 6: 85–95.
Werhli AV, Husmeier D: Reconstructing gene regulatory networks with bayesian networks by combining expression data with multiple sources of prior knowledge. Stat Appl Genet Mol Biol 2007., 6: Article15 Article15
Imoto S, Higuchi T, Goto T, Tashiro K, Kuhara S, Miyano S: Combining microarrays and biological knowledge for estimating gene networks via bayesian networks. J Bioinform Comput Biol 2004, 2: 77–98. 10.1142/S021972000400048X
Ide JS, Cozman FG: Testing MCMC algorithms with randomly generated Bayesian networks. In Workshop de Teses e Dissertações em IA (WTDIA2002). Recife, Pernambuco, Brazil; 2002.
Oti M, Brunner HG: The modular nature of genetic diseases. Clin Genet 2007, 71: 1–11.
Fraser AG, Marcotte EM: A probabilistic view of gene function. Nat Genet 2004, 6: 559–564.
Jansen R, Yu H, Greenbaum D, Kluger Y, Krogan NJ, Chung S, Emili A, Snyder M, Greenblatt JF, Gerstein M: A Bayesian Networks Approach for Predicting Protein-Protein Interactions from Genomic Data. 2003, 302: 449–453.
Madigan D, York J, Allard D: Bayesian Graphical Models for Discrete Data. International Statistical Review 1995, 63: 215–232. 10.2307/1403615
Needham CJ, Bradford JR, Bulpitt AJ, Westhead DR: A primer on learning in Bayesian networks for computational biology. PLoS Comput Biol 2007, 3: e129. 10.1371/journal.pcbi.0030129
Heckerman D, Geiger D, Chickering DM: Learning Bayesian Networks: The Combination of Knowledge and Statistical Data. Machine Learning 1995, 20: 197–243.
Cao SL, Qin L, He WZ, Zhong Y, Zhu YY, Li YX: Semantic search among heterogeneous biological databases based on gene ontology. Acta Biochim Biophys Sin (Shanghai) 2004, 36: 365–370. 10.1093/abbs/36.5.365
Murphy K: The bayes net toolbox for matlab. Computing Science and Statistics 2001., 33:
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T: Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res 2003, 13: 2498–2504. 10.1101/gr.1239303
Lee I, Date SV, Adai AT, Marcotte EM: A Probabilistic Functional Network of Yeast Genes. Science 2004, 306: 1555–1558. 10.1126/science.1099511
Lee I, Li Z, Marcotte EM: An improved, bias-reduced probabilistic functional gene network of baker's yeast, Saccharomyces cerevisiae. PloS one 2007, 2: e988. 10.1371/journal.pone.0000988
Wittig U, De Beuckelaer A: Analysis and comparison of metabolic pathway databases. Briefings in bioinformatics 2001, 2: 126–142. 10.1093/bib/2.2.126
Franke L, Bakel H, Fokkens L, de Jong ED, Egmont-Petersen M, Wijmenga C: Reconstruction of a functional human gene network, with an application for prioritizing positional candidate genes. Am J Hum Genet 2006, 78: 1011–1025. 10.1086/504300
Mewes HW, Frishman D, Guldener U, Mannhaupt G, Mayer K, Mokrejs M, Morgenstern B, Munsterkotter M, Rudd S, Weil B: MIPS: a database for genomes and protein sequences. Nucleic acids research 2002, 30: 31–34. 10.1093/nar/30.1.31
Domingos P, Pazzani M: On the Optimality of the Simple Bayesian Classifier under Zero-One Loss. Machine Learning 1997, 29: 103–130. 10.1023/A:1007413511361
Friedman N, Geiger D, Goldszmidt M: Bayesian Network Classifiers. Machine Learning 1997, 29: 131–163. 10.1023/A:1007465528199
Jansen R, Yu H, Greenbaum D, Kluger Y, Krogan NJ, Chung S, Emili A, Snyder M, Greenblatt JF, Gerstein M: A Bayesian networks approach for predicting protein-protein interactions from genomic data. Science 2003, 302: 449–453. 10.1126/science.1087361
Tong AH, Lesage G, Bader GD, Ding H, Xu H, Xin X, Young J, Berriz GF, Brost RL, Chang M, et al.: Global mapping of the yeast genetic interaction network. Science 2004, 303: 808–813. 10.1126/science.1091317
Consortium GO: Creating the gene ontology resource: design and implementation. Genome Res 2001, 11: 1425–1433. 10.1101/gr.180801
Simon I, Barnett J, Hannett N, Harbison CT, Rinaldi NJ, Volkert TL, Wyrick JJ, Zeitlinger J, Gifford DK, Jaakkola TS, Young RA: Serial regulation of transcriptional regulators in the yeast cell cycle. Cell 2001, 106: 697–708. 10.1016/S0092-8674(01)00494-9
Van den Bulcke T, Van Leemput K, Naudts B, van Remortel P, Ma H, Verschoren A, De Moor B, Marchal K: SynTReN: a generator of synthetic gene expression data for design and analysis of structure learning algorithms. BMC Bioinformatics 2006, 7: 43. 10.1186/1471-2105-7-43
Bansal M, Belcastro V, Ambesi-Impiombato A, di Bernardo D: How to infer gene networks from expression profiles. Mol Syst Biol 2007, 3: 122.
Spellman PT, Sherlock G, Zhang MQ, Iyer VR, Anders K, Eisen MB, Brown PO, Botstein D, Futcher B: Comprehensive identification of cell cycle-regulated genes of the yeast Saccharomyces cerevisiae by microarray hybridization. Mol Biol Cell 1998, 9: 3273–3297.
Alfarano C, Andrade CE, Anthony K, Bahroos N, Bajec M, Bantoft K, Betel D, Bobechko B, Boutilier K, Burgess E, et al.: The Biomolecular Interaction Network Database and related tools 2005 update. Nucleic Acids Res 2005, 33: D418–424.
Lechner A, Habener JF: Stem/progenitor cells derived from adult tissues: potential for the treatment of diabetes mellitus. Am J Physiol Endocrinol Metab 2003, 284: E259–266.
Burns CJ, Persaud SJ, Jones PM: Stem cell therapy for diabetes: do we need to make beta cells? J Endocrinol 2004, 183: 437–443. 10.1677/joe.1.05981
Servitja JM, Ferrer J: Transcriptional networks controlling pancreatic development and beta cell function. Diabetologia 2004, 47: 597–613. 10.1007/s00125-004-1368-9
Jenssen TK, Laegreid A, Komorowski J, Hovig E: A literature network of human genes for high-throughput analysis of gene expression. Nat Genet 2001, 28: 21–28.
Bastos G, Guimaraes KS: Analyzing the Effect of Prior Knowledge in Genetic Regulatory Network Inference. Pattern Recognition and Machine Intelligence, Lecture Notes in Computer Science 2005, 3776: 611–616. 10.1007/11590316_97
Cho RJ, Campbell MJ, Winzeler EA, Steinmetz L, Conway A, Wodicka L, Wolfsberg TG, Gabrielian AE, Landsman D, Lockhart DJ, Davis RW: A genome-wide transcriptional analysis of the mitotic cell cycle. Mol Cell 1998, 2: 65–73. 10.1016/S1097-2765(00)80114-8
Gao S, Hartman J, Carter JL, Hessner MJ, Wang X: Global analysis of phase locking in gene expression during cell cycle: the potential in network modeling. BMC Syst Biol 2010, 4: 167. 10.1186/1752-0509-4-167
Cho RJ, Campbell MJ, Winzeler EA, Steimetz L, Conway A, Wolfsberg TG: A geneome-wide transciptional analysis of the mitotic cell cycle. Mol Cell 1998, 2: 65–73. 10.1016/S1097-2765(00)80114-8
Zhu JCY, Leonardson AS, Wang K, Lamb JR, et al.: Characterizing Dynamic Changes in the Human Blood Transcriptional Network. PLoS Comput Biol 2001., 6:
Acknowledgements
This work was supported in part by National Institute of Diabetes and Digestive and Kidney Diseases Grant R01DK080100 (XW).
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
SG, and XW designed the study. SG wrote the algorithms, performed the analysis, and created the figures and tables. SG and XW wrote the manuscript, read and approved the final version of the manuscript.
Electronic supplementary material
12859_2011_4838_MOESM1_ESM.DOCX
Additional file 1:Predicted regulatory relationships missed by the plain BN. most established regulatory relationships missed by the plain BN involve two genes that share significant GO similarity and PubMed co-citation. (DOCX 16 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Gao, S., Wang, X. Quantitative utilization of prior biological knowledge in the Bayesian network modeling of gene expression data. BMC Bioinformatics 12, 359 (2011). https://doi.org/10.1186/1471-2105-12-359
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2105-12-359