
Abstract
Abstract
The ubiquitin system of protein modification has emerged as a crucial mechanism involved in the regulation of a wide array of cellular processes. As our knowledge of the pathways in this system has grown, so have the ties between the protein ubiquitin and human disease. The power of the ubiquitin system for therapeutic benefit blossomed with the approval of the proteasome inhibitor Velcade in 2003 by the FDA. Current drug discovery activities in the ubiquitin system seek to (i) expand the development of new proteasome inhibitors with distinct mechanisms of action and improved bioavailability, and (ii) validate new targets. This review summarizes our current understanding of the role of the ubiquitin system in various human diseases ranging from cancer, viral infection and neurodegenerative disorders to muscle wasting, diabetes and inflammation. I provide an introduction to the ubiquitin system, highlight some emerging relationships between the ubiquitin system and disease, and discuss current and future efforts to harness aspects of this potentially powerful system for improving human health.
Publication history
Republished from Current BioData's Targeted Proteins database (TPdb; http://www.targetedproteinsdb.com).
Similar content being viewed by others
Broad overview of family
Overview of the ubiquitin system
The ubiquitin system is a hierarchical enzymatic cascade in which a ubiquitin-activating enzyme (E1) activates the 76 amino acid protein UBIQ (ubiquitin) in an ATP-dependent manner and transfers it to the active site cysteine of ubiquitin-conjugating enzymes (E2s) [1]. Ubiquitin ligases (E3s) have a central role in the process of protein modification with UBIQ (known as 'ubiquitination' or 'ubiquitylation'); they recognize specific substrates and facilitate UBIQ transfer from the E2 onto the substrate. Although the precise number of human E3s is unknown, about 500 or more have been proposed to exist [2–5], supportive of the broad role for the ubiquitin system in regulating diverse cellular processes. Ubiquitin-like proteins (UBLs) have also been identified with varying degrees of identity to UBIQ and are conjugated onto proteins through similar enzymatic cascades as UBIQ.
Numerous deubiquitylating enzymes (DUBs) have roles in processing polyubiquitin precursor proteins and may also have regulatory roles, e.g. counteracting the ubiquitylation of a particular protein by its cognate E3 and/or proofreading synthesized UBIQ chains. There are also emerging roles for DUBs in disease (see [6]). Ubiquitin binding proteins also have diverse functions and may represent viable therapeutic targets (see [7]). In a general sense, they act as 'effector' proteins that sense a protein's modification with UBIQ and facilitate downstream signaling.
Two major classes of E3s have been identified and this classification is largely based on how they facilitate UBIQ transfer from E2 onto substrate. HECT (homologous to E6AP C-terminus) domain E3s form a catalytic UBIQ intermediate on a conserved cysteine residue prior to covalent UBIQ transfer (see [8]). The second class of E3s, which contains RING-type and structurally related ligases, facilitates the direct transfer of UBIQ from E2 onto substrate. In general, E3s facilitate covalent UBIQ transfer by properly positioning the site to be modified (i.e. a lysine residue of its recognized substrate) such that it can perform nucleophilic attack of a thioesterified UBIQ molecule either on the active site of the E2 for RING-type E3s or on the conserved cysteine of HECT domain E3s, resulting in isopeptide bond formation [9].
Lysine residues appear to be major sites of UBIQ attachment on proteins, although N-terminal and cysteine modifications have also been reported [10–17]. The type of UBIQ modification could confer distinct encoded protein fate and we are only beginning to understand how this process occurs and how it is recognized and interpreted. Mono-ubiquitylation (i.e. the attachment of a single UBIQ molecule to a single site on a protein) may be involved in histone regulation, receptor endocystosis and signaling [18–22]. UBIQ chains using a lysine residue of one UBIQ molecule attached via an isopeptide bond to the C-terminus of another UBIQ molecule add further complexity to UBIQ-encoded protein fate. Lys48-linked UBIQ chains can trigger degradation by the 26S proteasome [23–26] and Lys63-linked UBIQ chains may regulate signaling pathways [27–30] when attached to a protein. Other types of linkages (including those containing heterogeneous mixtures of linkages or branched chains) could also exist [31–33]; however their roles and physiological significance are currently unclear.
Target validation
Implication of the ubiquitin system in human disease
The basic functions of the UBIQ (ubiquitin) protein were first described in 1980 [34–36], yet its implication in human disease has only recently started to become appreciated. Below, I describe some relationships between the ubiquitin system and various human diseases.
Cancer is associated with alterations in UBIQ-dependent regulation
The ubiquitin system has a widely appreciated role in regulating cellular proliferation. As expected and described in the examples below, alterations in specific pathways involving UBIQ have been associated with cancer.
The stability of P53 (p53) is regulated by ubiquitin ligases and a deubiquitylating enzyme (DUB)
The transcription factor P53 has a crucial role in cellular anticancer mechanisms and it has been estimated that >50% of tumors contain mutations in the P53 gene [37]. MDM2 is a major regulator of P53 function – it binds directly to P53 and targets P53 for degradation through its RING ubiquitin ligase activity [38–42]. MDM2-P53 binding, MDM2-dependent P53 degradation by the proteasome, and P53 ubiquitylation by MDM2 have been demonstrated in cell-based and in vitro systems by a large number of groups.
P53 regulates the stability of the interaction between MDM2 and the DUB known as UBP7 (also known as USP7, HAUSP, herpesvirus-associated ubiquitin-specific protease [43]) [44–47]. Work from the laboratory of Wei Gu demonstrated that partial reduction of UBP7 by RNAi in human cell lines (NHF-1, IMR90, U2OS and H1299 cells were tested) promotes decreased levels of both MDM2 and P53 [45]. By contrast, however, total reduction of UBP7 decreases MDM2 yet stabilizes P53. This observation suggests that the absence of UBP7 promotes MDM2 downregulation, which in turn eliminates MDM2's function as the ubiquitin ligase for P53. This mechanism requires MDM2-dependent function, as depletion of UBP7 in cells inactive for MDM2-dependent P53 turnover (i.e. HeLa cells [48]) results in P53 stabilization.
Cullin-RING ubiquitin ligases and the APC/C regulate cellular proliferation
Cullin-RING ubiquitin ligases (CRLs) and the anaphase-promoting complex/cyclosome (APC/C) are multi-subunit RING ubiquitin ligases that have fundamental roles in controlling the eukaryotic cell cycle (see [49]). CRLs contain a cullin protein (CUL1, CUL2, CUL3, CUL4A, CUL5, CUL7) that binds within the cullin homology domain to the RING protein RBX1 [50–52]. The distinct N-terminal regions of the cullins interact with specific classes of substrate receptors that promote the recruitment of a large number of proteins for ubiquitylation [53]. The APC/C consists of at least 11 subunits and has a mass of 1.5 mDa [54, 55]. One of its subunits, APC2, contains a domain similar to cullins and associates with the RING protein APC11, suggesting that its enzymatic core is similar to CRLs [56–62].
The CUL1-based ubiquitin ligase known as SCF (SKP1-CUL1-F-box) recognizes its substrates through various receptor proteins containing the F-box motif [63]. The SKP2 F-box protein, which functions with SCF in the ubiquitylation of CDN1B (the cyclin-dependent kinase inhibitor p27) at the G1/S transition of the cell cycle [64–66], has garnered attention as a potential oncology target. SKP2, in conjunction with the adapter protein CKS1, recognizes phosphorylated CDN1B late in G1, recruiting it for ubiquitylation by SCF [67, 68]. An inverse correlation between SKP2 overexpression and low CDN1B levels has been found in a variety of human tumors and transgenic mouse models, and has led to the proposal that SKP2 is a proto-oncogene [69–74].
The protein encoded by the tumor suppressor gene VHL (von Hippel-Lindau) serves as a substrate receptor for a CUL2-based ubiquitin ligase [50, 75–80]. Mutations in VHL are associated with lung cancer, sporadic clear cell renal carcinomas and an autosomal dominant familial cancer known as von Hippel-Lindau disease ([81–93] and see [94]). Many of these mutations prevent VHL associating with the other subunits of its ubiquitin ligase, as judged by in vitro binding and co-immunoprecipitation experiments [79, 80, 95]. A substrate for this ubiquitin ligase is a marker of tumor hypoxia, the transcription factor HIF1A (HIF1α, hypoxia-inducible factor 1α), which stimulates angiogenesis [96]. Numerous biochemical and structural studies have determined that HIF1A binds to VHL when hydroxylated on two proline residues through the activity of prolyl hydroxylases, which results in its ubiquitylation and ultimate degradation [77, 78, 97–102]. Transgenic mice overexpressing a stabilized form of HIF1A that cannot be recognized by VHL develop hyper-vascularity without leakage or inflammation [103].
Cervical cancer is linked to HPV infection and involves downregulation of P53 and RB (Rb)
HPVs encode two oncogenic proteins known as E6 and E7, and the sexually transmitted types of HPV have a strong association with cervical cancer (see [104]). Whereas E7 may facilitate the degradation of the tumor suppressor RB through an unclear mechanism, the role of E6 in cellular transformation is more established [105]. E6 binds to a cellular protein known as UBE3A (E6AP), which is a HECT domain ubiquitin ligase [106]. This interaction promotes the recruitment of the tumor suppressor P53 to this complex, resulting in its ubiquitylation and subsequent degradation by the 26S proteasome [107, 108].
Colorectal cancers are associated with defects in the regulation of CTNB1 (β-catenin) stability through mutations in adenomatous polyposis coli
The tumor suppressor gene adenomatous polyposis coli (APC, not to be confused with the ubiquitin ligase APC/C, anaphase-promoting complex/cyclosome, described above) is frequently mutated in colorectal cancers [109–114]. Many of these mutations truncate APC and/or alter its ability to interact with proteins, which may lead to altered regulation of cellular proliferation.
One major target subjected to regulation through APC is CTNB1, a crucial component of Wnt signaling and cell adhesion [115, 116]. The phosphorylation of CTNB1 through APC-associated kinase activity promotes its recognition by the F-box protein β-TRCP [117, 118]. Numerous cell-based and in vitro experiments have demonstrated that the F-box motif of β-TRCP interacts with SKP1 and assembles into an SCF complex with CUL1 [63, 117–119]. Overexpression of β-TRCP containing an F-box deletion results in an increased stability of CTNB1, as demonstrated in pulse chase experiments [117, 119]. Other studies have identified the RING protein SIAH1 (a Drosophila seven in absentia homolog) as a P53-inducible and APC-associated ubiquitin ligase that can also regulate the stability of CTNB1 [120, 121].
Mutations in the BRCA1 ubiquitin ligase complex correlate with breast and ovarian cancer
Germline mutations in the gene encoding the RING protein BRCA1 are associated with the inherited predisposition for breast and ovarian cancer [122–124]. BRCA1 forms a heterodimer with another RING protein known as BARD1 and this complex has E3 activity in vitro [125–128]. These studies, utilizing bacterially expressed proteins and overexpression in mammalian cells, demonstrated that the RING motif of BRCA1 serves as the binding site for the E2 enzyme UB2D3 (UbcH5c) and that BRCA1/BARD1 together have an increased ability to conjugate UBIQ with UB2D3 in vitro than BRCA1 alone. Rachel Klevit's group determined that several of the cancer-predisposing mutations in BRCA1 result in defective E3 activity in vitro by disrupting BRCA1/BARD1 heterodimer formation or by altering the RING domain structure of BRCA1 [129–131]. Cell-based overexpression experiments demonstrated that the abundance of each protein is dependent upon the presence of its binding partner.
Phosphorylated RBBP8 (CtIP) is a reported substrate for ubiquitylation by BRCA1 [132]. Originally identified in a yeast two-hybrid screen for proteins that bind to the BRCA1 C-terminus (BRCT) domain of BRCA1 and confirmed through in vitro binding experiments, RBBP8 interacts with BRCA1 during G2 of the cell cycle [133–135]. Cell-based and in vitro ubiquitylation assays have demonstrated that BRCA1 ubiquitylates phosphorylated RBBP8 [132]. Rather than promoting RBBP8 degradation by the proteasome, UBIQ modification may cause RBBP8 to associate with chromatin following DNA damage and to regulate the G2/M transition of the cell cycle, as determined by cellular localization studies in response to DNA damage [132, 136]. BRCA1 could also be associated with the DNA repair activities of the Fanconi anemia (FA) pathway (see next section), as both physical and functional interactions between BRCA1 and FA complex proteins in response to DNA damage have been described [137].
The Fanconi anemia pathway involves a ubiquitin ligase complex and is associated with increased cancer susceptibility
As described in the [138], studies on the rare autosomal recessive genetic disorder known as Fanconi anemia (FA) have identified a pathway crucial for the cellular response to DNA damage [139–151]. Alterations in this pathway promote increased susceptibility to cancer and have been associated with a wide variety of tumor types, even in non-FA patients [152–166]. Upon DNA damage, two proteins in this pathway are mono-ubiquitylated; FACD2 (FANCD2) and FANCI [137, 141], and recruited to chromatin within nuclear foci. These nuclear foci contain other DNA repair proteins, suggesting that they are sites of DNA damage [137, 147, 167–169].
The role and exact molecular mechanisms underlying the regulation of FACD2 and FANCI mono-ubiquitylation are unclear; however, a protein complex (the FA core complex) contains a subunit known as FANCL that contains a RING motif and likely confers their modification [140]. Work from the D'Andrea and Dutta laboratories identified a ubiquitin-conjugating enzyme, UBE2T, that binds directly to FANCL in a yeast two-hybrid screen and through in vitro pull-down experiments with bacterially expressed proteins [170]. SiRNA depletion of UBE2T in U2OS cells diminished the mono-ubiquitylation of FACD2 in response to DNA damage and promoted the formation of abnormal chromosomes [170]. An siRNA screen for DUBs important for removing UBIQ from FACD2 implicated UBP1 (USP1) as an enzyme that could attenuate the role of FACD2 in DNA damage repair [171].
Viruses exploit the ubiquitin system
Viruses utilize clever mechanisms to exploit their host to facilitate their own propagation. Modification of proteins with UBIQ during infection promotes viral replication and immune response evasion, suggesting potential anti-viral strategies.
HIV encodes proteins that hijack cellular cullin-RING ubiquitin ligases
The rapid evolution of viral subtypes resistant to available treatments suggests that there is still significant need for new anti-HIV therapeutics. Two HIV-encoded proteins, VIF and VPU, interact with distinct cullin-RING ubiquitin ligases to hijack their activity and promote the ubiquitylation of cellular proteins.
VIF interacts directly with a cellular cytidine deaminase, ABC3G (APOBEC3G), and facilitates its proteasome-dependent degradation [172, 173]. In the absence of VIF, ABC3G is packaged into progeny virion particles, which renders them defective in replication [172, 174, 175]. Immunoprecipitation of hemagglutinin (HA)-tagged VIF from H9 cells (human T-cell line) infected with engineered HIV, followed by mass spectrometry, demonstrated that VIF associates with CUL5, ELOB (elongin B) and ELOC (elongin C). Western blotting was used to confirm the presence of all proteins including RBX1 [176]. In vitro ubiquitylation of ABC3G purified from transfected cells has been demonstrated using a reconstituted complex of these proteins from baculovirus-infected insect cells [177, 178].
The CD4 cell surface receptor found on a subclass of T-cells is downregulated through the activity of VPU [179, 180]. As CD4 is a co-receptor for HIV entry into cells, this downregulation could optimize viral replication by blocking further infection, allow for progeny viral particles to be efficiently released, and promote immune response evasion [179]. Co-expression of VPU and CD4 in HeLa cells results in the degradation of CD4, which can be blocked by the proteasome inhibitor MG132 [181]. Work by Margottin et al. originally identified the F-box protein β-TRCP from a yeast two-hybrid screen for VPU-interacting proteins [182]. After demonstrating the formation of the ternary CD4/VPU/β-TRCP complex by overexpressing these proteins in HeLa cells, the authors showed that the F-box motif of β-TRCP is necessary for CD4 degradation in these cells. It was later reported that VPU may block the ubiquitylation of IKBA (Iκbα) and CTNB1, which are phosphorylation-dependent cellular substrates of SCFβ-TRCP, and that VPU itself may also be ubiquitylated by this complex [183–186].
Herpesviruses encode ubiquitin ligases and modulate cellular ubiquitin ligases
Herpesviruses often employ strategies to utilize the host cell's ubiquitin system for their own benefit. The gammaherpesvirus Kaposi's sarcoma herpesvirus (KSHV, alternatively human herpesvirus types 8, HHV8), which is associated with AIDS-related cancer (see [187]), encodes two ubiquitin ligases; K3 (MIR1) and K5 (MIR2), which downregulate a wide range of immunoreceptors (MHC class I, ICAM1, CD86, CD1D (CD1d)) from the surface of infected cells [188–193]. This mechanism could promote immune system evasion by blocking detection by cytotoxic T-lymphocytes.
The molecular details of how K3 and K5 promote immunoreceptor downregulation through the ubiquitin system are beginning to emerge. Laurent Coscoy's group recently reported the unexpected observation that transient expression of K3 in human BJAB cells stably expressing the MHC class I allele HLA.B7 can lead to downregulation of this receptor in the absence of cytoplasmic lysine residues if a cysteine residue is present [17]. Paul Lehner's laboratory used siRNA experiments to identify UBC13 and UB2D2/UB2D3 (UbcH5b/c) as the E2 enzymes important for K3-dependent downregulation of MHC class I [194]. This study suggests that K3-mediated modification of MHC class I occurs through a sequential mechanism in which Lys63-linked UBIQ chains synthesized by UBC13 are added after initial mono-ubiquitylation by UB2D2/UB2D3, thereby promoting receptor endocytosis and lysosomal targeting.
Herpesviruses also encode proteins that modulate cellular ubiquitin ligase activity, such as LMP1 (latent membrane protein 1) – encoded by EBV. LMP1 is required for EBV latency in B-cells and is sufficient to induce transformation [195, 196]. Recent work from Joseph Pagano's laboratory has uncovered differential effects of LMP1 on the SIAH1 ubiquitin ligase, dependent upon cell type [197–199]. EBV-positive B-cells expressing LMP1 or cells transiently transfected to express LMP1 manifest an upregulation of CTNB1, a component of the Wnt signaling pathway whose increased stability has been associated with cancer (see section on Colorectal cancers are associated with defects in the regulation of CTNB1 (β-catenin) stability through mutations in adenomatous polyposis coli) [197]. This observation was attributed to LMP1-mediated downregulation of SIAH1, a component of a RING ubiquitin ligase complex that regulates CTNB1 stability, at the transcriptional level [120, 121]. By contrast, human epithelial cells expressing LMP1 manifest increased SIAH1 protein levels and a resulting decrease in the SIAH1 substrates prolyl hydroxylases 1 and 3 (PHD1, PHD3 [also known as EGLN2 and EGLN3]) [198, 200]. These decreases promote the stability of the transcription factor HIF1A as it cannot be hydroxylated, an event required for its association with the VHL-containing ubiquitin ligase (see section on Cullin-RING ubiquitin ligases and the APC/C regulate cellular proliferation).
Neurodegenerative diseases often have associated impairment of the ubiquitin system
The formation of protein aggregates containing UBIQ has long been associated with neurodegenerative diseases such as Parkinson's, Alzheimer's, Huntington's, and others. For example, polyglutamine repeat expansion in proteins associated with Huntington's disease and the spinocerebellar ataxias could promote the formation of protein aggregates that are resistant to degradation by the proteasome and also impair proteasome function (see [201]). Similarly in Alzheimer's disease, the formation of neurofibrillary tangles and plaques associated with amyloid-β protein aggregation and/or ubiquitylated TAU (tau) accumulation could impair proteasome function (see [202]).
Another example is found with autosomal-recessive juvenile Parkinson's disease, in which mutations in the ubiquitin ligase PRKN2 (parkin) manifest as defects in its ligase activity in vitro [203–205], suggesting that accumulation of its substrates could contribute to disease development.
Metabolic diseases such as diabetes could have associated defects in aspects of the ubiquitin system
The exact relationships between the ubiquitin system and metabolic processes are only beginning to be understood (see [206]). Insulin resistance, associated with diabetes and obesity, manifests as defects in sensing and signaling mechanisms. The ubiquitin system has been associated with insulin signaling through regulating the stability of insulin receptor substrate (IRS) proteins.
IRS proteins serve as adapter molecules, functioning between receptor tyrosine kinases and downstream signaling molecules. IRS2 in particular has a crucial function in controlling the growth and survival of pancreatic β-cells – the body's source of insulin. Irs2 knockout mice are diabetic and exhibit dramatic reduction in β-cell mass, and decreasing IRS2 expression via siRNA in β-cells promotes apoptosis and decreased cell survival [207]. Thus, signaling through IRS2 has a crucial function in regulating the body's response to changes in glucose.
IRS2 function is regulated by phosphorylation and UBIQ-mediated degradation by the proteasome [208, 209]. In pancreatic β-cells, activation of the kinase FRAP (mTOR), as demonstrated by adenoviral delivery of constitutively active FRAP to rat INS-1 cells, promotes IRS2 phosphorylation and degradation by the proteasome [210]. IRS2 interacts with suppressors of cytokine signaling (SOCS) proteins SOCS1 and SOCS3 in human HEK293 cells, mouse 3T3-L1 adipocytes, and mouse hepatocytes [210]. These proteins contain a 'SOCS box' motif, which promotes their interaction with ELOC, a component of a cullin-based ubiquitin ligase, which in turn recruits IRS2 for ubiquitylation and targets it for degradation by the proteasome [211]. Future studies aimed at understanding how IRS2 abundance relates to mechanisms of glucose sensing may lead to novel approaches for combating the growing epidemic of diabetes.
Muscle wasting disorders have increases in ubiquitin system function
Decreases in skeletal muscle mass associated with aging, cancer, disuse and other physiological circumstances occur through proteolytic mechanisms involving calpain proteases and UBIQ-dependent protein degradation (see [212]).
Experimental evidence suggests that numerous genes of the ubiquitin system are upregulated during muscle atrophy, including those encoding a muscle-specific ubiquitin ligase (TRI63, also known as MuRF1) and a potential substrate receptor for SCF (the F-box protein FBX32, also known as MAFbx and Atrogin-1) [213]. Knockout studies in mice performed by Regeneron for each of these genes support their crucial roles in promoting muscle atrophy [213]. Whereas TRI63 deletion results in 36% sparing of muscle mass loss after denervation of the right hindlimb muscle of mice, FBX32 deletion allows for 56% sparing under similar experimental conditions when compared with controls.
Potential substrates for these ubiquitin ligases are only beginning to be identified. TRI63 may target TITIN (titin), a protein implicated in myofibril organization for degradation [214]. The binding between TRI63 and the C-terminal region of TITIN was originally identified by yeast two-hybrid studies [215]. FBX32, as a substrate receptor for SCF, has been proposed to facilitate the ubiquitylation of the calmodulin-dependent phosphatase calcineurin A and MyoD, a transcription factor involved in myogenic differentiation [216, 217]. Cam Patterson's group identified calcineurin A through a yeast two-hybrid screen using FBX32 as bait and validated this interaction through in vitro and cell-based experiments and overexpression studies in mice. Using MyoD as bait in a yeast two-hybrid screen, FBX32 was identified as an interacting protein, and a variety of cell-based and in vitro experiments demonstrated binding, ubiquitylation and turnover dependent upon this interaction [217].
UBIQ is involved in NFκB activation to regulate inflammation and innate immunity
Numerous distinct pathways can promote the activation of NFκB in response to distinct stimuli (such inflammatory cytokines, DNA damaging agents and microbes) and UBIQ has diverse and complex roles in this process. Conventional roles for UBIQ, such as targeting the NFκB inhibitor IKBA for degradation by the proteasome [118, 218–220] and the proteasome-dependent processing of NFκB precursor proteins [221, 222], are mixed with non-proteolytic functions in regulating specific signaling pathways [28, 30]. Also, these signaling mechanisms can be attenuated by specifically associated DUBs [223]. This complexity provides distinct levels of regulation of NFκB activation that could allow for the modulation of this process associated with a wide range of diseases. NFκB activation through TNR11 (receptor activator of NFκB, RANK), for example, has an important role in bone homeostasis and is associated with diseases such as osteoporosis, rheumatoid arthritis and Paget's disease of bone (see [224]).
The importance of regulating NFκB signaling through distinct pathways is highlighted by the tumors associated with the genetic disorder cylindromatosis [225]. Afflicted individuals contain mutations in a DUB known as CYLD. This enzyme functions downstream of the tumor necrosis factor α receptor and is involved in attenuating NFκB activation by deubiquitylating the signaling molecules TRAF2, TRAF6 and TRAF7 [226–228]. The regulation of TRAF2 signaling by CYLD was identified by screening an RNAi library targeting DUBs for ones that attenuate NFκB activation [227] and through a two-hybrid screen for proteins that interact with the regulatory subunit of the IκB kinase complex that could also bind to TRAF2 [228]. Later work studying signaling through the toll-like receptor 2 (TLR2) performed cell-based experiments to demonstrate that CYLD binds to TRAF6 and TRAF7 and that depletion of CYLD increases the ability of transfected TRAF6 or TRAF7 to activate an NFκB-dependent reporter gene [226].
UBIQ-dependent ion channel stability has implications in cardiovascular diseases
Alterations in ion channel stability have been associated with cystic fibrosis and Liddle's syndrome. Cystic fibrosis, one of the most common genetic diseases, is characterized by a wide array of recessive mutations in CFTR (cystic fibrosis transmembrane conductance regulator), a Cl- ion channel protein (see [229]). These mutations promote CFTR misfolding and subsequent clearance through protein quality control pathways of the ubiquitin system. Exactly how CFTR downregulation promotes lung disease is unclear. By contrast, Liddle's syndrome is associated with increased stability of an epithelial Na+ channel (ENaC, see [230]). This autosomal dominant disorder is characterized by mutations in ENaC that block its recognition by the HECT ligase NEDD4 (see [8]), which promotes ENaC accumulation at the cell surface. Alterations of proper ionic balance in the kidney through increases in ENaC may increase blood volume and blood pressure, promoting cardiovascular disease [231, 232].
Lead discovery
Current drug discovery activities focused on the ubiquitin system
The 26S proteasome is the only validated therapeutic target of the ubiquitin system, with a single commercially available drug known as Velcade. Current drug discovery activities related to the ubiquitin system focus on three major areas: (i) expanding indications of proteasome inhibition in therapy, (ii) developing proteasome inhibitors targeting different activities of the proteasome and with improved bio-availability, and (iii) validating the potential of other targets of the ubiquitin system.
Proteasome inhibition is a treatment for cancer
Changes in proteasome function have been implicated in the development of various diseases (see [233]). In 2003, a small molecule proteasome inhibitor known as Velcade (bortezomib by Millennium Pharmaceuticals) was approved by the FDA for the treatment of relapsed or refractory multiple myeloma (see [234]). Velcade is a boronic acid derivative (chemical name [(1R)-3-methyl-1-[[(2S)-1-oxo-3-phenyl-2-[(pyrazinylcarbonyl) amino]propyl]amino]butyl] boronic acid) that is a reversible inhibitor of the chymotrypsin-like activity of the 26S proteasome [235].
Efforts are underway by several other companies to develop inhibitors that target distinct activities of the proteasome. One such company is Proteolix, which has developed PR-171, a synthetic analog of epoxomycin that irreversibly inhibits the chymotryptic site of the proteasome. Treatment of xenograft models over the course of two days demonstrated that PR-171 has a stronger anti-tumor effect than Velcade [236]. Phase I trials are currently underway for PR-171 to evaluate its role in treating multiple myelomas and non-Hodgkin's lymphoma. Another proteasome-targeting drug that is currently in Phase I clinical trials is salinosporamide A (NPI-0052), developed by Nereus Pharmaceuticals. Pre-clinical studies have demonstrated that NPI-0052 achieves a higher and more sustained level of proteasome inhibition when compared with Velcade [237]. It is also well tolerated and improves the response of multi-drug treatment of a colon cancer xenograft model [237]. These two drugs highlight the therapeutic potential of proteasome inhibition when treating various cancerous states. Indeed, Cytomics Systems, Eisai, Novartis AG, Bristol-Myers Squibb, Cell Therapeutics, Cephalon and Ergon Pharmaceuticals are all developing proteasome inhibitors for this purpose.
Small molecules can promote P53 (p53) stability
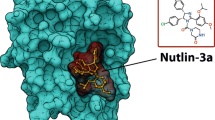
As described in the section Cancer is associated with alterations in UBIQ-dependent regulation, MDM2 is a RING ubiquitin ligase that has a crucial role in regulating P53 stability. The development of inhibitors that disrupt this interaction will play a major role in the regulation of cell cycle progression and potentially the treatment of cancer. One group of small molecules currently in development is the nutlins, by Roche. These represent the first small molecules that can interfere with the ability of MDM2 to mediate P53 ubiquitylation [238]. Nutlins bind to the pocket domain of P53, and inhibit xenograft tumor growth in vivo with no obvious toxicity. They activate the P53 pathway at a range of 1–3 μM, stimulating cell cycle arrest and apoptosis in tumor cells. By contrast, treatment of untransformed cells results in cell cycle arrest, but no apoptosis [239].
RITA [2,5-bis(5-hydroxymethyl-2-thienyl)furan (NSC652287)], isolated from a screen by the National Cancer Institute (NCI) [240, 241], is another compound that has been shown to regulate the interaction between MDM2 and P53. RITA also induces apoptosis in human tumor cells, but has little effect on normal cells. It has been proposed to bind the N-terminus of P53; thereby causing a conformational change that prevents MDM2 binding. However, the sensitivity of human fibroblasts to RITA is varied and depends on the expression level of oncogenes such as MYC (C-MYC) [242]. RITA, as with nutlins, has a significant antitumor effect on mice carrying human tumor xenografts, without apparent toxicity [243], and both are in pre-clinical development. The therapeutic potential of these and other molecules targeting this important regulatory mechanism are currently being explored. Nevertheless, they represent an important class of molecules, which block substrate recognition by a ubiquitin ligase through interfering with a protein-protein interaction interface.
Inhibition of E1 may be a viable therapeutic target
If inhibiting the proteasome has therapeutic utility, then perhaps targeting UBIQ (ubiquitin) activation by E1 might also be beneficial. Both Millennium Pharmaceuticals and Rigel Pharmaceuticals have filed patents disclosing their discovery of E1 inhibitors (Millennium patent: WO2006084281 [244], Rigel patent: WO2005037845 [245], see [246]). These small molecule inhibitors of UBIQ activation may also have potential as cancer therapeutics.
Ubiquitin ligases and deubiquitylating enzymes are emerging therapeutic targets
Proteasome inhibition or E1 inhibition appear to be global approaches to controlling protein activities regulated by UBIQ. As a result, efforts are ongoing to selectively target enzymes involved in specific ubiquitylation pathways. Ubiquitin ligases and deubiquitylating enzymes (DUBs) have gained the most attention due to their direct roles in regulating their recognized protein's stability. However, the complexity of protein ubiquitylation coupled with the absence of catalytic pockets for small molecule binding has made targeting ubiquitin ligases challenging. Nevertheless, Rigel Pharmaceuticals is currently characterizing ubiquitin ligases as potential therapeutic targets. The company has a broad, on-going program to explore the role of specific ubiquitin ligases in oncology, inflammation, virology and metabolism – with Merck and Daiichi as collaborators. Other companies such as Celgene, Amgen and Genentech also have programs exploring ubiquitin ligases for clinical indications. Thus, the flurry of activity surrounding ubiquitin ligases supports their potential therapeutic importance for developing new treatments for a wide variety of conditions.
In contrast to ubiquitin ligases, DUBs have a more simple mechanism of action and a catalytic pocket for targeted binding of small molecules. Hybrigenics recently disclosed the discovery of several small molecules that can inhibit UBP7 (USP7) (HBX 41108) and UBP8 (USP8) (HBX 90397, HBX 90659), which may have oncology applications.
New frontiers in drug discovery for the ubiquitin system
Our understanding of the complexities of the ubiquitin system and its involvement in aspects of physiology is still developing and (perhaps justifiably) so are efforts to discover drugs targeting its enzymes. Here, I summarize some of the current challenges and opportunities that could allow for the potential of the enzymes of the ubiquitin system to be realized as therapeutic targets.
How can ubiquitin ligase activity be inhibited?
Ubiquitin ligases may represent the best target class of the ubiquitin system due to their intrinsic specificity for particular protein substrates. The vast majority of ubiquitin ligases are RING-type. These appear to function primarily as scaffolds to position the substrate to be modified in close juxtaposition with the ubiquitin-conjugating enzyme to promote covalent UBIQ (ubiquitin) transfer [247]. Thus, means to modulate ubiquitin ligase activity may necessarily focus on targeting protein-protein interfaces. Whereas the MDM2-P53 (p53) interface can be disrupted by small molecules, is this possible for other E3-substrate interactions? Do different RING motifs (the binding site for the ubiquitin-conjugating enzyme) look different enough to be considered as targets? If so, how can they be targeted? HECT (homologous to E6AP C-terminus) domain ligases, by contrast, have a catalytic function and may undergo conformational changes [248], suggesting that, at least superficially, they could be more amenable.
Can we harness ubiquitin ligases to artificially target proteins for ubiquitylation?
Increased protein stability correlates with several disease phenotypes. For some of these cases, defects in upstream signaling results in impaired substrate targeting to ubiquitin ligases. In other cases, the ubiquitin ligase itself has functional defects. These observations suggest that artificially recruiting a protein to a ubiquitin ligase could be an approach to restoring proper protein homeostasis. Indeed, there are several reports supporting the potential of this approach [249–251]. One such strategy has involved the development of a chimeric protein or 'ProTaC' (proteolysis targeting chimeric molecule), which targets the protein to the SCFβ-TRCP ubiquitin ligase. SCFβ-TRCP then targets the protein for ubiquitylation, followed by proteasomal degradation. ProTaC consists of a SCFβ-TRCP-binding IKBA (IκBα) phosphopeptide linked to a domain that binds the targeted protein [252]. Further development of this technology could have the potential to realize a powerful and specific tool for the treatment of diseases such as cancer.
Are ubiquitin-conjugating enzymes good therapeutic targets?
Whereas there are considerably fewer ubiquitin-conjugating enzymes than ubiquitin ligases, they may function in specific ubiquitylation pathways. As they represent an essential part of the ubiquitylation process, conferred through their enzymatic activity, they may be reasonable targets. However, these enzymes have a very highly conserved enzymatic core and do not possess defined catalytic pockets [253].
What other approaches should be considered?
Recent work identified small molecules known as ubistatins that bind to Lys48-linked UBIQ chains in vitro and block UBIQ-dependent protein degradation by the proteasome [254]. Randy King's group at Harvard identified ubistatins in a chemical genetic screen for small molecules that stabilize Cyclin B in Xenopus extracts, and they were subsequently shown to inhibit the degradation of the yeast cyclin-dependent kinase inhibitor SIC1 (Sic1) in a reconstituted in vitro system. NMR and in vitro binding studies determined that these molecules bind specifically to Lys48-linked UBIQ chains. Whilst these molecules are not cell permeable and have unclear therapeutic potential, they do represent a new approach for blocking protein degradation.
Another direction could be to target mechanisms that regulate ubiquitin ligase assembly. The SCF ubiquitin ligase, for example, is comprised of multiple subunits in which substrate recognition is conferred by a variable receptor subunit (F-box proteins) [53]. The assembly of specific SCF complexes appears to be regulated at multiple levels ranging from receptor abundance to post-translational modification of its enzymatic core. As our knowledge of the intricacies of the ubiquitin system continues to grow, it is likely that we will uncover new regulatory mechanisms associated with protein ubiquitylation.
Abbreviations
- APC:
-
adenomatous polyposis coli
- APC/C:
-
anaphase promoting complex/cyclosome
- BRCT:
-
BRCA1 C-terminus
- CFTR:
-
cystic fibrosis transmembrane conductance regulator
- CRLs:
-
Cullin-RING ligases
- DUB:
-
deubiquitylating enzyme
- E1:
-
ubiquitin-activating enzyme
- E2:
-
ubiquitin-conjugating enzyme
- E3:
-
ubiquitin ligase
- ENaC:
-
epithelial Na+ channel
- FA:
-
Fanconi anemia
- HECT:
-
homologous to E6AP C-terminus
- HHV8:
-
human herpesvirus type 8
- HIF1α:
-
hypoxia-inducible factor 1α
- IRS:
-
insulin receptor substrate
- KSHV:
-
Kaposi's sarcoma herpesvirus
- ProTaC:
-
proteolysis targeting chimeric molecule
- RANK:
-
receptor activator of NFκB
- SCF:
-
SKP1-CUL1-F-box ubiquitin ligase
- UBLs:
-
ubiquitin-like proteins.
References
Pickart CM, Eddins MJ: Ubiquitin: structures, functions, mechanisms. Biochim Biophys Acta. 2004, 1695: 55-72. 10.1016/j.bbamcr.2004.09.019.
Lorick KL, Tsai Y-C, Yang Y, Weissman AM: RING fingers and relatives: Determination of protein fate. 2005, Weinheim, Germany: VCH Verlag
Semple CA: The comparative proteomics of ubiquitination in mouse. Genome Res. 2003, 13: 1389-1394. 10.1101/gr.980303.
von Arnim AG: A hitchhiker's guide to the proteasome. Sci STKE. 2001, 2001: PE2-10.1126/stke.2001.97.pe2.
Wong BR, Parlati F, Qu K, Demo S, Pray T, Huang J, Payan DG, Bennett MK: Drug discovery in the ubiquitin regulatory pathway. Drug Discov Today. 2003, 8: 746-754. 10.1016/S1359-6446(03)02780-6.
Singhal S, Taylor MC, Baker RT: Deubiquitylating enzymes and disease. BMC Biochemistry. 2008, 9 (Suppl 1): S3-
Madsen L, Shulze A, Seeger M, Hartmann-Petersen R: Ubiquitin domain proteins in disease. BMC Biochemistry. 2007, 8 (Suppl 1): S1-10.1186/1471-2091-8-S1-S1.
Scheffner M, Staub O: HECT E3s and human disease. BMC Biochemistry. 2007, 8 (Suppl 1): S6-10.1186/1471-2091-8-S1-S6.
Hershko A, Heller H, Elias S, Ciechanover A: Components of ubiquitin-protein ligase system. Resolution, affinity purification, and role in protein breakdown. J Biol Chem. 1983, 258: 8206-8214.
Aviel S, Winberg G, Massucci M, Ciechanover A: Degradation of the epstein-barr virus latent membrane protein 1 (LMP1) by the ubiquitin-proteasome pathway. Targeting via ubiquitination of the N-terminal residue. J Biol Chem. 2000, 275: 23491-23499. 10.1074/jbc.M002052200.
Ben-Saadon R, Fajerman I, Ziv T, Hellman U, Schwartz AL, Ciechanover A: The tumor suppressor protein p16(INK4a) and the human papillomavirus oncoprotein-58 E7 are naturally occurring lysine-less proteins that are degraded by the ubiquitin system. Direct evidence for ubiquitination at the N-terminal residue. J Biol Chem. 2004, 279: 41414-41421. 10.1074/jbc.M407201200.
Breitschopf K, Bengal E, Ziv T, Admon A, Ciechanover A: A novel site for ubiquitination: the N-terminal residue, and not internal lysines of MyoD, is essential for conjugation and degradation of the protein. Embo J. 1998, 17: 5964-5973. 10.1093/emboj/17.20.5964.
Coulombe P, Rodier G, Bonneil E, Thibault P, Meloche S: N-Terminal ubiquitination of extracellular signal-regulated kinase 3 and p21 directs their degradation by the proteasome. Mol Cell Biol. 2004, 24: 6140-6150. 10.1128/MCB.24.14.6140-6150.2004.
Reinstein E, Scheffner M, Oren M, Ciechanover A, Schwartz A: Degradation of the E7 human papillomavirus oncoprotein by the ubiquitin-proteasome system: targeting via ubiquitination of the N-terminal residue. Oncogene. 2000, 19: 5944-5950. 10.1038/sj.onc.1203989.
Senga T, Sivaprasad U, Zhu W, Park JH, Arias EE, Walter JC, Dutta A: PCNA is a cofactor for Cdt1 degradation by CUL4/DDB1-mediated N-terminal ubiquitination. J Biol Chem. 2006, 281: 6246-6252. 10.1074/jbc.M512705200.
Bloom J, Amador V, Bartolini F, DeMartino G, Pagano M: Proteasome-mediated degradation of p21 via N-terminal ubiquitinylation. Cell. 2003, 115: 71-82. 10.1016/S0092-8674(03)00755-4.
Cadwell K, Coscoy L: Ubiquitination on nonlysine residues by a viral E3 ubiquitin ligase. Science. 2005, 309: 127-130. 10.1126/science.1110340.
Lucero P, Penalver E, Vela L, Lagunas R: Monoubiquitination is sufficient to signal internalization of the maltose transporter in Saccharomyces cerevisiae. J Bacteriol. 2000, 182: 241-243.
Nakatsu F, Sakuma M, Matsuo Y, Arase H, Yamasaki S, Nakamura N, Saito T, Ohno H: A Di-leucine signal in the ubiquitin moiety. Possible involvement in ubiquitination-mediated endocytosis. J Biol Chem. 2000, 275: 26213-26219. 10.1074/jbc.M907720199.
Robzyk K, Recht J, Osley MA: Rad6-dependent ubiquitination of histone H2B in yeast. Science. 2000, 287: 501-504. 10.1126/science.287.5452.501.
Roth AF, Davis NG: Ubiquitination of the PEST-like endocytosis signal of the yeast a-factor receptor. J Biol Chem. 2000, 275: 8143-8153. 10.1074/jbc.275.11.8143.
Terrell J, Shih S, Dunn R, Hicke L: A function for monoubiquitination in the internalization of a G protein-coupled receptor. Mol Cell. 1998, 1: 193-202. 10.1016/S1097-2765(00)80020-9.
Chau V, Tobias JW, Bachmair A, Marriott D, Ecker DJ, Gonda DK, Varshavsky A: A multiubiquitin chain is confined to specific lysine in a targeted short-lived protein. Science. 1989, 243: 1576-1583. 10.1126/science.2538923.
Finley D, Sadis S, Monia BP, Boucher P, Ecker DJ, Crooke ST, Chau V: Inhibition of proteolysis and cell cycle progression in a multiubiquitination-deficient yeast mutant. Mol Cell Biol. 1994, 14: 5501-5509.
Petroski MD, Deshaies RJ: Context of multiubiquitin chain attachment influences the rate of Sic1 degradation. Mol Cell. 2003, 11: 1435-1444. 10.1016/S1097-2765(03)00221-1.
Thrower JS, Hoffman L, Rechsteiner M, Pickart CM: Recognition of the polyubiquitin proteolytic signal. Embo J. 2000, 19: 94-102. 10.1093/emboj/19.1.94.
Hoege C, Pfander B, Moldovan GL, Pyrowolakis G, Jentsch S: RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature. 2002, 419: 135-141. 10.1038/nature00991.
Kanayama A, Seth RB, Sun L, Ea CK, Hong M, Shaito A, Chiu YH, Deng L, Chen ZJ: TAB2 and TAB3 activate the NF-kappaB pathway through binding to polyubiquitin chains. Mol Cell. 2004, 15: 535-548. 10.1016/j.molcel.2004.08.008.
Spence J, Sadis S, Haas AL, Finley D: A ubiquitin mutant with specific defects in DNA repair and multiubiquitination. Mol Cell Biol. 1995, 15: 1265-1273.
Deng L, Wang C, Spencer E, Yang L, Braun A, You J, Slaughter C, Pickart C, Chen ZJ: Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell. 2000, 103: 351-361. 10.1016/S0092-8674(00)00126-4.
Ben-Saadon R, Zaaroor D, Ziv T, Ciechanover A: The polycomb protein Ring1B generates self atypical mixed ubiquitin chains required for its in vitro histone H2A ligase activity. Mol Cell. 2006, 24: 701-711. 10.1016/j.molcel.2006.10.022.
Kim HT, Kim KP, Lledias F, Kisselev AF, Scaglione KM, Skowyra D, Gygi SP, Goldberg AL: Certain Pairs of Ubiquitin-conjugating Enzymes (E2s) and Ubiquitin-Protein Ligases (E3s) Synthesize Nondegradable Forked Ubiquitin Chains Containing All Possible Isopeptide Linkages. J Biol Chem. 2007, 282: 17375-17386. 10.1074/jbc.M609659200.
Kirkpatrick DS, Hathaway NA, Hanna J, Elsasser S, Rush J, Finley D, King RW, Gygi SP: Quantitative analysis of in vitro ubiquitinated cyclin B1 reveals complex chain topology. Nat Cell Biol. 2006, 8: 700-710. 10.1038/ncb1436.
Ciechanover A, Elias S, Heller H, Ferber S, Hershko A: Characterization of the heat-stable polypeptide of the ATP-dependent proteolytic system from reticulocytes. J Biol Chem. 1980, 255: 7525-7528.
Ciechanover A, Heller H, Elias S, Haas AL, Hershko A: ATP-dependent conjugation of reticulocyte proteins with the polypeptide required for protein degradation. Proc Natl Acad Sci USA. 1980, 77: 1365-1368. 10.1073/pnas.77.3.1365.
Hershko A, Ciechanover A, Heller H, Haas AL, Rose IA: Proposed role of ATP in protein breakdown: conjugation of protein with multiple chains of the polypeptide of ATP-dependent proteolysis. Proc Natl Acad Sci USA. 1980, 77: 1783-1786. 10.1073/pnas.77.4.1783.
Pavletich NP, Chambers KA, Pabo CO: The DNA-binding domain of p53 contains the four conserved regions and the major mutation hot spots. Genes Dev. 1993, 7: 2556-2564. 10.1101/gad.7.12b.2556.
Fang S, Jensen JP, Ludwig RL, Vousden KH, Weissman AM: Mdm2 is a RING finger-dependent ubiquitin protein ligase for itself and p53. J Biol Chem. 2000, 275: 8945-8951. 10.1074/jbc.275.12.8945.
Haupt Y, Maya R, Kazaz A, Oren M: Mdm2 promotes the rapid degradation of p53. Nature. 1997, 387: 296-299. 10.1038/387296a0.
Honda R, Tanaka H, Yasuda H: Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997, 420: 25-27. 10.1016/S0014-5793(97)01480-4.
Honda R, Yasuda H: Association of p19(ARF) with Mdm2 inhibits ubiquitin ligase activity of Mdm2 for tumor suppressor p53. Embo J. 1999, 18: 22-27. 10.1093/emboj/18.1.22.
Kubbutat MH, Jones SN, Vousden KH: Regulation of p53 stability by Mdm2. Nature. 1997, 387: 299-303. 10.1038/387299a0.
Everett RD, Meredith M, Orr A, Cross A, Kathoria M, Parkinson J: A novel ubiquitin-specific protease is dynamically associated with the PML nuclear domain and binds to a herpesvirus regulatory protein. Embo J. 1997, 16: 1519-1530. 10.1093/emboj/16.7.1519.
Cummins JM, Rago C, Kohli M, Kinzler KW, Lengauer C, Vogelstein B: Tumour suppression: disruption of HAUSP gene stabilizes p53. Nature. 2004, 428: 1 p following 486
Li M, Brooks CL, Kon N, Gu W: A dynamic role of HAUSP in the p53-Mdm2 pathway. Mol Cell. 2004, 13: 879-886. 10.1016/S1097-2765(04)00157-1.
Li M, Chen D, Shiloh A, Luo J, Nikolaev AY, Qin J, Gu W: Deubiquitination of p53 by HAUSP is an important pathway for p53 stabilization. Nature. 2002, 416: 648-653. 10.1038/nature737.
Lim SK, Shin JM, Kim YS, Baek KH: Identification and characterization of murine mHAUSP encoding a deubiquitinating enzyme that regulates the status of p53 ubiquitination. Int J Oncol. 2004, 24: 357-364.
Hengstermann A, Linares LK, Ciechanover A, Whitaker NJ, Scheffner M: Complete switch from Mdm2 to human papillomavirus E6-mediated degradation of p53 in cervical cancer cells. Proc Natl Acad Sci USA. 2001, 98: 1218-1223. 10.1073/pnas.031470698.
Cardozo T, Pagano M: Wrenches in the works: drug discovery targeting the SCF ubiquitin ligase and APC/C complexes. BMC Biochemistry. 2007, 8 (Suppl 1): S9-10.1186/1471-2091-8-S1-S9.
Kamura T, Koepp DM, Conrad MN, Skowyra D, Moreland RJ, Iliopoulos O, Lane WS, Kaelin WG, Elledge SJ, Conaway RC, et al.: Rbx1, a component of the VHL tumor suppressor complex and SCF ubiquitin ligase. Science. 1999, 284: 657-661. 10.1126/science.284.5414.657.
Seol JH, Feldman RM, Zachariae W, Shevchenko A, Correll CC, Lyapina S, Chi Y, Galova M, Claypool J, Sandmeyer S, et al.: Cdc53/cullin and the essential Hrt1 RING-H2 subunit of SCF define a ubiquitin ligase module that activates the E2 enzyme Cdc34. Genes Dev. 1999, 13: 1614-1626. 10.1101/gad.13.12.1614.
Skowyra D, Koepp DM, Kamura T, Conrad MN, Conaway RC, Conaway JW, Elledge SJ, Harper JW: Reconstitution of G1 cyclin ubiquitination with complexes containing SCFGrr1 and Rbx1. Science. 1999, 284: 662-665. 10.1126/science.284.5414.662.
Petroski MD, Deshaies RJ: Function and regulation of cullin-RING ubiquitin ligases. Nat Rev Mol Cell Biol. 2005, 6: 9-20. 10.1038/nrm1547.
Dube P, Herzog F, Gieffers C, Sander B, Riedel D, Muller SA, Engel A, Peters JM, Stark H: Localization of the coactivator Cdh1 and the cullin subunit Apc2 in a cryo-electron microscopy model of vertebrate APC/C. Mol Cell. 2005, 20: 867-879. 10.1016/j.molcel.2005.11.008.
Sudakin V, Ganoth D, Dahan A, Heller H, Hershko J, Luca FC, Ruderman JV, Hershko A: The cyclosome, a large complex containing cyclin-selective ubiquitin ligase activity, targets cyclins for destruction at the end of mitosis. Mol Biol Cell. 1995, 6: 185-197.
Gmachl M, Gieffers C, Podtelejnikov AV, Mann M, Peters JM: The RING-H2 finger protein APC11 and the E2 enzyme UBC4 are sufficient to ubiquitinate substrates of the anaphase-promoting complex. Proc Natl Acad Sci USA. 2000, 97: 8973-8978. 10.1073/pnas.97.16.8973.
Kramer KM, Fesquet D, Johnson AL, Johnston LH: Budding yeast RSI1/APC2, a novel gene necessary for initiation of anaphase, encodes an APC subunit. Embo J. 1998, 17: 498-506. 10.1093/emboj/17.2.498.
Leverson JD, Joazeiro CA, Page AM, Huang H, Hieter P, Hunter T: The APC11 RING-H2 finger mediates E2-dependent ubiquitination. Mol Biol Cell. 2000, 11: 2315-2325.
Tang Z, Li B, Bharadwaj R, Zhu H, Ozkan E, Hakala K, Deisenhofer J, Yu H: APC2 Cullin protein and APC11 RING protein comprise the minimal ubiquitin ligase module of the anaphase-promoting complex. Mol Biol Cell. 2001, 12: 3839-3851.
Vodermaier HC, Gieffers C, Maurer-Stroh S, Eisenhaber F, Peters JM: TPR subunits of the anaphase-promoting complex mediate binding to the activator protein CDH1. Curr Biol. 2003, 13: 1459-1468. 10.1016/S0960-9822(03)00581-5.
Yu H, Peters JM, King RW, Page AM, Hieter P, Kirschner MW: Identification of a cullin homology region in a subunit of the anaphase-promoting complex. Science. 1998, 279: 1219-1222. 10.1126/science.279.5354.1219.
Zachariae W, Shevchenko A, Andrews PD, Ciosk R, Galova M, Stark MJ, Mann M, Nasmyth K: Mass spectrometric analysis of the anaphase-promoting complex from yeast: identification of a subunit related to cullins. Science. 1998, 279: 1216-1219. 10.1126/science.279.5354.1216.
Bai C, Sen P, Hofmann K, Ma L, Goebl M, Harper JW, Elledge SJ: SKP1 connects cell cycle regulators to the ubiquitin proteolysis machinery through a novel motif, the F-box. Cell. 1996, 86: 263-274. 10.1016/S0092-8674(00)80098-7.
Carrano AC, Eytan E, Hershko A, Pagano M: SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat Cell Biol. 1999, 1: 193-199. 10.1038/12013.
Sutterluty H, Chatelain E, Marti A, Wirbelauer C, Senften M, Muller U, Krek W: p45SKP2 promotes p27Kip1 degradation and induces S phase in quiescent cells. Nat Cell Biol. 1999, 1: 207-214. 10.1038/12027.
Tsvetkov LM, Yeh KH, Lee SJ, Sun H, Zhang H: p27(Kip1) ubiquitination and degradation is regulated by the SCF(Skp2) complex through phosphorylated Thr187 in p27. Curr Biol. 1999, 9: 661-664. 10.1016/S0960-9822(99)80290-5.
Ganoth D, Bornstein G, Ko TK, Larsen B, Tyers M, Pagano M, Hershko A: The cell-cycle regulatory protein Cks1 is required for SCF(Skp2)-mediated ubiquitinylation of p27. Nat Cell Biol. 2001, 3: 321-324. 10.1038/35060126.
Spruck C, Strohmaier H, Watson M, Smith AP, Ryan A, Krek TW, Reed SI: A CDK-independent function of mammalian Cks1: targeting of SCF(Skp2) to the CDK inhibitor p27Kip1. Mol Cell. 2001, 7: 639-650. 10.1016/S1097-2765(01)00210-6.
Hershko D, Bornstein G, Ben-Izhak O, Carrano A, Pagano M, Krausz MM, Hershko A: Inverse relation between levels of p27(Kip1) and of its ubiquitin ligase subunit Skp2 in colorectal carcinomas. Cancer. 2001, 91: 1745-1751. 10.1002/1097-0142(20010501)91:9<1745::AID-CNCR1193>3.0.CO;2-H.
Latres E, Chiarle R, Schulman BA, Pavletich NP, Pellicer A, Inghirami G, Pagano M: Role of the F-box protein Skp2 in lymphomagenesis. Proc Natl Acad Sci USA. 2001, 98: 2515-2520. 10.1073/pnas.041475098.
Nakayama K, Nagahama H, Minamishima YA, Miyake S, Ishida N, Hatakeyama S, Kitagawa M, Iemura S, Natsume T, Nakayama KI: Skp2-mediated degradation of p27 regulates progression into mitosis. Dev Cell. 2004, 6: 661-672. 10.1016/S1534-5807(04)00131-5.
Shim EH, Johnson L, Noh HL, Kim YJ, Sun H, Zeiss C, Zhang H: Expression of the F-box protein SKP2 induces hyperplasia, dysplasia, and low-grade carcinoma in the mouse prostate. Cancer Res. 2003, 63: 1583-1588.
Signoretti S, Di Marcotullio L, Richardson A, Ramaswamy S, Isaac B, Rue M, Monti F, Loda M, Pagano M: Oncogenic role of the ubiquitin ligase subunit Skp2 in human breast cancer. J Clin Invest. 2002, 110: 633-641.
Singh SP, Lipman J, Goldman H, Ellis FH, Aizenman L, Cangi MG, Signoretti S, Chiaur DS, Pagano M, Loda M: Loss or altered subcellular localization of p27 in Barrett's associated adenocarcinoma. Cancer Res. 1998, 58: 1730-1735.
Duan DR, Pause A, Burgess WH, Aso T, Chen DY, Garrett KP, Conaway RC, Conaway JW, Linehan WM, Klausner RD: Inhibition of transcription elongation by the VHL tumor suppressor protein. Science. 1995, 269: 1402-1406. 10.1126/science.7660122.
Kibel A, Iliopoulos O, DeCaprio JA, Kaelin WG: Binding of the von Hippel-Lindau tumor suppressor protein to Elongin B and C. Science. 1995, 269: 1444-1446. 10.1126/science.7660130.
Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ: The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999, 399: 271-275. 10.1038/20459.
Ohh M, Park CW, Ivan M, Hoffman MA, Kim TY, Huang LE, Pavletich N, Chau V, Kaelin WG: Ubiquitination of hypoxia-inducible factor requires direct binding to the beta-domain of the von Hippel-Lindau protein. Nat Cell Biol. 2000, 2: 423-427. 10.1038/35017054.
Pause A, Lee S, Worrell RA, Chen DY, Burgess WH, Linehan WM, Klausner RD: The von Hippel-Lindau tumor-suppressor gene product forms a stable complex with human CUL-2, a member of the Cdc53 family of proteins. Proc Natl Acad Sci USA. 1997, 94: 2156-2161. 10.1073/pnas.94.6.2156.
Lonergan KM, Iliopoulos O, Ohh M, Kamura T, Conaway RC, Conaway JW, Kaelin WG: Regulation of hypoxia-inducible mRNAs by the von Hippel-Lindau tumor suppressor protein requires binding to complexes containing elongins B/C and Cul2. Mol Cell Biol. 1998, 18: 732-741.
Chen F, Kishida T, Yao M, Hustad T, Glavac D, Dean M, Gnarra JR, Orcutt ML, Duh FM, Glenn G, et al.: Germline mutations in the von Hippel-Lindau disease tumor suppressor gene: correlations with phenotype. Hum Mutat. 1995, 5: 66-75. 10.1002/humu.1380050109.
Crossey PA, Foster K, Richards FM, Phipps ME, Latif F, Tory K, Jones MH, Bentley E, Kumar R, Lerman MI, et al.: Molecular genetic investigations of the mechanism of tumourigenesis in von Hippel-Lindau disease: analysis of allele loss in VHL tumours. Hum Genet. 1994, 93: 53-58. 10.1007/BF00218913.
Crossey PA, Maher ER, Jones MH, Richards FM, Latif F, Phipps ME, Lush M, Foster K, Tory K, Green JS, et al.: Genetic linkage between von Hippel-Lindau disease and three microsatellite polymorphisms refines the localisation of the VHL locus. Hum Mol Genet. 1993, 2: 279-282. 10.1093/hmg/2.3.279.
Crossey PA, Richards FM, Foster K, Green JS, Prowse A, Latif F, Lerman MI, Zbar B, Affara NA, Ferguson-Smith MA, et al.: Identification of intragenic mutations in the von Hippel-Lindau disease tumour suppressor gene and correlation with disease phenotype. Hum Mol Genet. 1994, 3: 1303-1308. 10.1093/hmg/3.8.1303.
Eng C, Crossey PA, Mulligan LM, Healey CS, Houghton C, Prowse A, Chew SL, Dahia PL, O'Riordan JL, Toledo SP, et al.: Mutations in the RET proto-oncogene and the von Hippel-Lindau disease tumour suppressor gene in sporadic and syndromic phaeochromocytomas. J Med Genet. 1995, 32: 934-937.
Foster K, Crossey PA, Cairns P, Hetherington JW, Richards FM, Jones MH, Bentley E, Affara NA, Ferguson-Smith MA, Maher ER: Molecular genetic investigation of sporadic renal cell carcinoma: analysis of allele loss on chromosomes 3p, 5q, 11p, 17 and 22. Br J Cancer. 1994, 69: 230-234.
Foster K, Prowse A, Berg van den A, Fleming S, Hulsbeek MM, Crossey PA, Richards FM, Cairns P, Affara NA, Ferguson-Smith MA, et al.: Somatic mutations of the von Hippel-Lindau disease tumour suppressor gene in non-familial clear cell renal carcinoma. Hum Mol Genet. 1994, 3: 2169-2173. 10.1093/hmg/3.12.2169.
Gallou C, Chauveau D, Richard S, Joly D, Giraud S, Olschwang S, Martin N, Saquet C, Chretien Y, Mejean A, et al.: Genotype-phenotype correlation in von Hippel-Lindau families with renal lesions. Hum Mutat. 2004, 24: 215-224. 10.1002/humu.20082.
Gallou C, Joly D, Mejean A, Staroz F, Martin N, Tarlet G, Orfanelli MT, Bouvier R, Droz D, Chretien Y, et al.: Mutations of the VHL gene in sporadic renal cell carcinoma: of a risk factor for VHL patients to develop an RCC. Hum Mutat. 1999, 13: 464-475. 10.1002/(SICI)1098-1004(1999)13:6<464::AID-HUMU6>3.0.CO;2-A.
Maher ER, Webster AR, Richards FM, Green JS, Crossey PA, Payne SJ, Moore AT: Phenotypic expression in von Hippel-Lindau disease: correlations with germline VHL gene mutations. J Med Genet. 1996, 33: 328-332.
Sekido Y, Bader S, Latif F, Gnarra JR, Gazdar AF, Linehan WM, Zbar B, Lerman MI, Minna JD: Molecular analysis of the von Hippel-Lindau disease tumor suppressor gene in human lung cancer cell lines. Oncogene. 1994, 9: 1599-1604.
Stolle C, Glenn G, Zbar B, Humphrey JS, Choyke P, Walther M, Pack S, Hurley K, Andrey C, Klausner R, Linehan WM: Improved detection of germline mutations in the von Hippel-Lindau disease tumor suppressor gene. Hum Mutat. 1998, 12: 417-423. 10.1002/(SICI)1098-1004(1998)12:6<417::AID-HUMU8>3.0.CO;2-K.
Zbar B, Kishida T, Chen F, Schmidt L, Maher ER, Richards FM, Crossey PA, Webster AR, Affara NA, Ferguson-Smith MA, et al.: Germline mutations in the Von Hippel-Lindau disease (VHL) gene in families from North America, Europe, and Japan. Hum Mutat. 1996, 8: 348-357. 10.1002/(SICI)1098-1004(1996)8:4<348::AID-HUMU8>3.0.CO;2-3.
Corn PG: Role of the ubiquitin proteasome system in renal cell carcinoma. BMC Biochemistry. 2007, 8 (Suppl 1): S4-10.1186/1471-2091-8-S1-S4.
Ohh M, Yauch RL, Lonergan KM, Whaley JM, Stemmer-Rachamimov AO, Louis DN, Gavin BJ, Kley N, Kaelin WG, Iliopoulos O: The von Hippel-Lindau tumor suppressor protein is required for proper assembly of an extracellular fibronectin matrix. Mol Cell. 1998, 1: 959-968. 10.1016/S1097-2765(00)80096-9.
Wang GL, Jiang BH, Rue EA, Semenza GL: Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA. 1995, 92: 5510-5514. 10.1073/pnas.92.12.5510.
Bruick RK, McKnight SL: A conserved family of prolyl-4-hydroxylases that modify HIF. Science. 2001, 294: 1337-1340. 10.1126/science.1066373.
Hon WC, Wilson MI, Harlos K, Claridge TD, Schofield CJ, Pugh CW, Maxwell PH, Ratcliffe PJ, Stuart DI, Jones EY: Structural basis for the recognition of hydroxyproline in HIF-1 alpha by pVHL. Nature. 2002, 417: 975-978. 10.1038/nature00767.
Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG: HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001, 292: 464-468. 10.1126/science.1059817.
Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ, et al.: Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001, 292: 468-472. 10.1126/science.1059796.
Masson N, Willam C, Maxwell PH, Pugh CW, Ratcliffe PJ: Independent function of two destruction domains in hypoxia-inducible factor-alpha chains activated by prolyl hydroxylation. Embo J. 2001, 20: 5197-5206. 10.1093/emboj/20.18.5197.
Yu F, White SB, Zhao Q, Lee FS: HIF-1alpha binding to VHL is regulated by stimulus-sensitive proline hydroxylation. Proc Natl Acad Sci USA. 2001, 98: 9630-9635. 10.1073/pnas.181341498.
Elson DA, Thurston G, Huang LE, Ginzinger DG, McDonald DM, Johnson RS, Arbeit JM: Induction of hypervascularity without leakage or inflammation in transgenic mice overexpressing hypoxia-inducible factor-1alpha. Genes Dev. 2001, 15: 2520-2532. 10.1101/gad.914801.
Beaudenon S, Huibregtse JM: HPV E6, E6AP and cervical cancer. BMC Biochemistry. 2008, 9 (Suppl 1): S4-
Wang J, Sampath A, Raychaudhuri P, Bagchi S: Both Rb and E7 are regulated by the ubiquitin proteasome pathway in HPV-containing cervical tumor cells. Oncogene. 2001, 20: 4740-4749. 10.1038/sj.onc.1204655.
Huibregtse JM, Scheffner M, Howley PM: A cellular protein mediates association of p53 with the E6 oncoprotein of human papillomavirus types 16 or 18. Embo J. 1991, 10: 4129-4135.
Scheffner M, Huibregtse JM, Vierstra RD, Howley PM: The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell. 1993, 75: 495-505. 10.1016/0092-8674(93)90384-3.
Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM: The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell. 1990, 63: 1129-1136. 10.1016/0092-8674(90)90409-8.
Joslyn G, Carlson M, Thliveris A, Albertsen H, Gelbert L, Samowitz W, Groden J, Stevens J, Spirio L, Robertson M, et al.: Identification of deletion mutations and three new genes at the familial polyposis locus. Cell. 1991, 66: 601-613. 10.1016/0092-8674(81)90022-2.
Kinzler KW, Nilbert MC, Su LK, Vogelstein B, Bryan TM, Levy DB, Smith KJ, Preisinger AC, Hedge P, McKechnie D, et al.: Identification of FAP locus genes from chromosome 5q21. Science. 1991, 253: 661-665. 10.1126/science.1651562.
Miki Y, Nishisho I, Horii A, Miyoshi Y, Utsunomiya J, Kinzler KW, Vogelstein B, Nakamura Y: Disruption of the APC gene by a retrotransposal insertion of L1 sequence in a colon cancer. Cancer Res. 1992, 52: 643-645.
Miyoshi Y, Nagase H, Ando H, Horii A, Ichii S, Nakatsuru S, Aoki T, Miki Y, Mori T, Nakamura Y: Somatic mutations of the APC gene in colorectal tumors: mutation cluster region in the APC gene. Hum Mol Genet. 1992, 1: 229-233. 10.1093/hmg/1.4.229.
Nagase H, Miyoshi Y, Horii A, Aoki T, Petersen GM, Vogelstein B, Maher E, Ogawa M, Maruyama M, Utsunomiya J, et al.: Screening for germ-line mutations in familial adenomatous polyposis patients: 61 new patients and a summary of 150 unrelated patients. Hum Mutat. 1992, 1: 467-473. 10.1002/humu.1380010603.
Nishisho I, Nakamura Y, Miyoshi Y, Miki Y, Ando H, Horii A, Koyama K, Utsunomiya J, Baba S, Hedge P: Mutations of chromosome 5q21 genes in FAP and colorectal cancer patients. Science. 1991, 253: 665-669. 10.1126/science.1651563.
Rubinfeld B, Souza B, Albert I, Muller O, Chamberlain SH, Masiarz FR, Munemitsu S, Polakis P: Association of the APC gene product with beta-catenin. Science. 1993, 262: 1731-1734. 10.1126/science.8259518.
Su LK, Vogelstein B, Kinzler KW: Association of the APC tumor suppressor protein with catenins. Science. 1993, 262: 1734-1737. 10.1126/science.8259519.
Hart M, Concordet JP, Lassot I, Albert I, del los Santos R, Durand H, Perret C, Rubinfeld B, Margottin F, Benarous R, Polakis P: The F-box protein beta-TrCP associates with phosphorylated beta-catenin and regulates its activity in the cell. Curr Biol. 1999, 9: 207-210. 10.1016/S0960-9822(99)80091-8.
Winston JT, Strack P, Beer-Romero P, Chu CY, Elledge SJ, Harper JW: The SCFbeta-TRCP-ubiquitin ligase complex associates specifically with phosphorylated destruction motifs in IkappaBalpha and beta-catenin and stimulates IkappaBalpha ubiquitination in vitro. Genes Dev. 1999, 13: 270-283. 10.1101/gad.13.3.270.
Latres E, Chiaur DS, Pagano M: The human F box protein beta-Trcp associates with the Cul1/Skp1 complex and regulates the stability of beta-catenin. Oncogene. 1999, 18: 849-854. 10.1038/sj.onc.1202653.
Liu J, Stevens J, Rote CA, Yost HJ, Hu Y, Neufeld KL, White RL, Matsunami N: Siah-1 mediates a novel beta-catenin degradation pathway linking p53 to the adenomatous polyposis coli protein. Mol Cell. 2001, 7: 927-936. 10.1016/S1097-2765(01)00241-6.
Matsuzawa SI, Reed JC: Siah-1, SIP, and Ebi collaborate in a novel pathway for beta-catenin degradation linked to p53 responses. Mol Cell. 2001, 7: 915-926. 10.1016/S1097-2765(01)00242-8.
Hall JM, Lee MK, Newman B, Morrow JE, Anderson LA, Huey B, King MC: Linkage of early-onset familial breast cancer to chromosome 17q21. Science. 1990, 250: 1684-1689. 10.1126/science.2270482.
Futreal PA, Liu Q, Shattuck-Eidens D, Cochran C, Harshman K, Tavtigian S, Bennett LM, Haugen-Strano A, Swensen J, Miki Y, et al.: BRCA1 mutations in primary breast and ovarian carcinomas. Science. 1994, 266: 120-122. 10.1126/science.7939630.
Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, Liu Q, Cochran C, Bennett LM, Ding W, et al.: A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994, 266: 66-71. 10.1126/science.7545954.
Brzovic PS, Keeffe JR, Nishikawa H, Miyamoto K, Fox D, Fukuda M, Ohta T, Klevit R: Binding and recognition in the assembly of an active BRCA1/BARD1 ubiquitin-ligase complex. Proc Natl Acad Sci USA. 2003, 100: 5646-5651. 10.1073/pnas.0836054100.
Chen A, Kleiman FE, Manley JL, Ouchi T, Pan ZQ: Autoubiquitination of the BRCA1*BARD1 RING ubiquitin ligase. J Biol Chem. 2002, 277: 22085-22092. 10.1074/jbc.M201252200.
Hashizume R, Fukuda M, Maeda I, Nishikawa H, Oyake D, Yabuki Y, Ogata H, Ohta T: The RING heterodimer BRCA1-BARD1 is a ubiquitin ligase inactivated by a breast cancer-derived mutation. J Biol Chem. 2001, 276: 14537-14540. 10.1074/jbc.C000881200.
Xia Y, Pao GM, Chen HW, Verma IM, Hunter T: Enhancement of BRCA1 E3 ubiquitin ligase activity through direct interaction with the BARD1 protein. J Biol Chem. 2003, 278: 5255-5263. 10.1074/jbc.M204591200.
Brzovic PS, Meza J, King MC, Klevit RE: The cancer-predisposing mutation C61G disrupts homodimer formation in the NH2-terminal BRCA1 RING finger domain. J Biol Chem. 1998, 273: 7795-7799. 10.1074/jbc.273.14.7795.
Brzovic PS, Meza JE, King MC, Klevit RE: BRCA1 RING domain cancer-predisposing mutations. Structural consequences and effects on protein-protein interactions. J Biol Chem. 2001, 276: 41399-41406. 10.1074/jbc.M106551200.
Brzovic PS, Rajagopal P, Hoyt DW, King MC, Klevit RE: Structure of a BRCA1-BARD1 heterodimeric RING-RING complex. Nat Struct Biol. 2001, 8: 833-837. 10.1038/nsb1001-833.
Yu X, Fu S, Lai M, Baer R, Chen J: BRCA1 ubiquitinates its phosphorylation-dependent binding partner CtIP. Genes Dev. 2006, 20: 1721-1726. 10.1101/gad.1431006.
Wong AK, Ormonde PA, Pero R, Chen Y, Lian L, Salada G, Berry S, Lawrence Q, Dayananth P, Ha P, et al.: Characterization of a carboxy-terminal BRCA1 interacting protein. Oncogene. 1998, 17: 2279-2285. 10.1038/sj.onc.1202150.
Yu X, Wu LC, Bowcock AM, Aronheim A, Baer R: The C-terminal (BRCT) domains of BRCA1 interact in vivo with CtIP, a protein implicated in the CtBP pathway of transcriptional repression. J Biol Chem. 1998, 273: 25388-25392. 10.1074/jbc.273.39.25388.
Yu X, Chen J: DNA damage-induced cell cycle checkpoint control requires CtIP, a phosphorylation-dependent binding partner of BRCA1 C-terminal domains. Mol Cell Biol. 2004, 24: 9478-9486. 10.1128/MCB.24.21.9478-9486.2004.
Varma AK, Brown RS, Birrane G, Ladias JA: Structural basis for cell cycle checkpoint control by the BRCA1-CtIP complex. Biochemistry. 2005, 44: 10941-10946. 10.1021/bi0509651.
Garcia-Higuera I, Taniguchi T, Ganesan S, Meyn MS, Timmers C, Hejna J, Grompe M, D'Andrea AD: Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol Cell. 2001, 7: 249-262. 10.1016/S1097-2765(01)00173-3.
Jacquemont C, Taniguchi T: The Fanconi anemia pathway and ubiquitin. BMC Biochemistry. 2007, 8 (Suppl 1): S10-10.1186/1471-2091-8-S1-S10.
Lo Ten Foe JR, Rooimans MA, Bosnoyan-Collins L, Alon N, Wijker M, Parker L, Lightfoot J, Carreau M, Callen DF, Savoia A, et al.: Expression cloning of a cDNA for the major Fanconi anaemia gene, FAA. Nat Genet. 1996, 14: 320-323. 10.1038/ng1196-320.
Meetei AR, de Winter JP, Medhurst AL, Wallisch M, Waisfisz Q, Vrugt van de HJ, Oostra AB, Yan Z, Ling C, Bishop CE, et al.: A novel ubiquitin ligase is deficient in Fanconi anemia. Nat Genet. 2003, 35: 165-170. 10.1038/ng1241.
Smogorzewska A, Matsuoka S, Vinciguerra P, McDonald ER, Hurov KE, Luo J, Ballif BA, Gygi SP, Hofmann K, D'Andrea AD, Elledge SJ: Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell. 2007, 129: 289-301. 10.1016/j.cell.2007.03.009.
Dorsman JC, Levitus M, Rockx D, Rooimans MA, Oostra AB, Haitjema A, Bakker ST, Steltenpool J, Schuler D, Mohan S, et al.: Identification of the Fanconi anemia complementation group I gene, FANCI. Cell Oncol. 2007, 29: 211-218.
Levitus M, Waisfisz Q, Godthelp BC, de Vries Y, Hussain S, Wiegant WW, Elghalbzouri-Maghrani E, Steltenpool J, Rooimans MA, Pals G, et al.: The DNA helicase BRIP1 is defective in Fanconi anemia complementation group J. Nat Genet. 2005, 37: 934-935. 10.1038/ng1625.
de Winter JP, Leveille F, van Berkel CG, Rooimans MA, Weel van Der L, Steltenpool J, Demuth I, Morgan NV, Alon N, Bosnoyan-Collins L, et al.: Isolation of a cDNA representing the Fanconi anemia complementation group E gene. Am J Hum Genet. 2000, 67: 1306-1308. 10.1086/321200.
de Winter JP, Rooimans MA, Weel van Der L, van Berkel CG, Alon N, Bosnoyan-Collins L, de Groot J, Zhi Y, Waisfisz Q, Pronk JC, et al.: The Fanconi anaemia gene FANCF encodes a novel protein with homology to ROM. Nat Genet. 2000, 24: 15-16. 10.1038/71626.
de Winter JP, Waisfisz Q, Rooimans MA, van Berkel CG, Bosnoyan-Collins L, Alon N, Carreau M, Bender O, Demuth I, Schindler D, et al.: The Fanconi anaemia group G gene FANCG is identical with XRCC9. Nat Genet. 1998, 20: 281-283. 10.1038/3093.
Howlett NG, Taniguchi T, Olson S, Cox B, Waisfisz Q, De Die-Smulders C, Persky N, Grompe M, Joenje H, Pals G, et al.: Biallelic inactivation of BRCA2 in Fanconi anemia. Science. 2002, 297: 606-609. 10.1126/science.1073834.
Meetei AR, Levitus M, Xue Y, Medhurst AL, Zwaan M, Ling C, Rooimans MA, Bier P, Hoatlin M, Pals G, et al.: X-linked inheritance of Fanconi anemia complementation group B. Nat Genet. 2004, 36: 1219-1224. 10.1038/ng1458.
Meetei AR, Medhurst AL, Ling C, Xue Y, Singh TR, Bier P, Steltenpool J, Stone S, Dokal I, Mathew CG, et al.: A human ortholog of archaeal DNA repair protein Hef is defective in Fanconi anemia complementation group M. Nat Genet. 2005, 37: 958-963. 10.1038/ng1626.
Strathdee CA, Gavish H, Shannon WR, Buchwald M: Cloning of cDNAs for Fanconi's anaemia by functional complementation. Nature. 1992, 358: 434-
Timmers C, Taniguchi T, Hejna J, Reifsteck C, Lucas L, Bruun D, Thayer M, Cox B, Olson S, D'Andrea AD, et al.: Positional cloning of a novel Fanconi anemia gene, FANCD2. Mol Cell. 2001, 7: 241-248. 10.1016/S1097-2765(01)00172-1.
Condie A, Powles RL, Hudson CD, Shepherd V, Bevan S, Yuille MR, Houlston RS: Analysis of the Fanconi anaemia complementation group A gene in acute myeloid leukaemia. Leuk Lymphoma. 2002, 43: 1849-1853. 10.1080/1042819021000009274.
Dhillon VS, Shahid M, Husain SA: CpG methylation of the FHIT, FANCF, cyclin-D2, BRCA2 and RUNX3 genes in Granulosa cell tumors (GCTs) of ovarian origin. Mol Cancer. 2004, 3: 33-10.1186/1476-4598-3-33.
Koul S, McKiernan JM, Narayan G, Houldsworth J, Bacik J, Dobrzynski DL, Assaad AM, Mansukhani M, Reuter VE, Bosl GJ, et al.: Role of promoter hypermethylation in Cisplatin treatment response of male germ cell tumors. Mol Cancer. 2004, 3: 16-10.1186/1476-4598-3-16.
Lensch MW, Tischkowitz M, Christianson TA, Reifsteck CA, Speckhart SA, Jakobs PM, O'Dwyer ME, Olson SB, Le Beau MM, Hodgson SV, et al.: Acquired FANCA dysfunction and cytogenetic instability in adult acute myelogenous leukemia. Blood. 2003, 102: 7-16. 10.1182/blood-2002-09-2781.
Marsit CJ, Liu M, Nelson HH, Posner M, Suzuki M, Kelsey KT: Inactivation of the Fanconi anemia/BRCA pathway in lung and oral cancers: implications for treatment and survival. Oncogene. 2004, 23: 1000-1004. 10.1038/sj.onc.1207256.
Narayan G, Arias-Pulido H, Nandula SV, Basso K, Sugirtharaj DD, Vargas H, Mansukhani M, Villella J, Meyer L, Schneider A, et al.: Promoter hypermethylation of FANCF: disruption of Fanconi Anemia-BRCA pathway in cervical cancer. Cancer Res. 2004, 64: 2994-2997. 10.1158/0008-5472.CAN-04-0245.
Olopade OI, Wei M: FANCF methylation contributes to chemoselectivity in ovarian cancer. Cancer Cell. 2003, 3: 417-420. 10.1016/S1535-6108(03)00111-9.
Rogers CD, Heijden van der MS, Brune K, Yeo CJ, Hruban RH, Kern SE, Goggins M: The genetics of FANCC and FANCG in familial pancreatic cancer. Cancer Biol Ther. 2004, 3: 167-169.
Taniguchi T, Tischkowitz M, Ameziane N, Hodgson SV, Mathew CG, Joenje H, Mok SC, D'Andrea AD: Disruption of the Fanconi anemia-BRCA pathway in cisplatin-sensitive ovarian tumors. Nat Med. 2003, 9: 568-574. 10.1038/nm852.
Tischkowitz M, Ameziane N, Waisfisz Q, De Winter JP, Harris R, Taniguchi T, D'Andrea A, Hodgson SV, Mathew CG, Joenje H: Bi-allelic silencing of the Fanconi anaemia gene FANCF in acute myeloid leukaemia. Br J Haematol. 2003, 123: 469-471. 10.1046/j.1365-2141.2003.04640.x.
Tischkowitz MD, Morgan NV, Grimwade D, Eddy C, Ball S, Vorechovsky I, Langabeer S, Stoger R, Hodgson SV, Mathew CG: Deletion and reduced expression of the Fanconi anemia FANCA gene in sporadic acute myeloid leukemia. Leukemia. 2004, 18: 420-425. 10.1038/sj.leu.2403280.
Heijden van der MS, Brody JR, Gallmeier E, Cunningham SC, Dezentje DA, Shen D, Hruban RH, Kern SE: Functional defects in the fanconi anemia pathway in pancreatic cancer cells. Am J Pathol. 2004, 165: 651-657.
Heijden Van Der MS, Brody JR, Kern SE: Functional screen of the fanconi anemia pathway in cancer cells by Fancd2 immunoblot. Cancer Biol Ther. 2004, 3: 534-537.
Heijden van der MS, Yeo CJ, Hruban RH, Kern SE: Fanconi anemia gene mutations in young-onset pancreatic cancer. Cancer Res. 2003, 63: 2585-2588.
Wang Z, Li M, Lu S, Zhang Y, Wang H: Promoter hypermethylation of FANCF plays an important role in the occurrence of ovarian cancer through disrupting Fanconi anemia-BRCA pathway. Cancer Biol Ther. 2006, 5: 256-260.
Hussain S, Wilson JB, Medhurst AL, Hejna J, Witt E, Ananth S, Davies A, Masson JY, Moses R, West SC, et al.: Direct interaction of FANCD2 with BRCA2 in DNA damage response pathways. Hum Mol Genet. 2004, 13: 1241-1248. 10.1093/hmg/ddh135.
Nakanishi K, Taniguchi T, Ranganathan V, New HV, Moreau LA, Stotsky M, Mathew CG, Kastan MB, Weaver DT, D'Andrea AD: Interaction of FANCD2 and NBS1 in the DNA damage response. Nat Cell Biol. 2002, 4: 913-920. 10.1038/ncb879.
Wang X, Andreassen PR, D'Andrea AD: Functional interaction of monoubiquitinated FANCD2 and BRCA2/FANCD1 in chromatin. Mol Cell Biol. 2004, 24: 5850-5862. 10.1128/MCB.24.13.5850-5862.2004.
Machida YJ, Machida Y, Chen Y, Gurtan AM, Kupfer GM, D'Andrea AD, Dutta A: UBE2T is the E2 in the Fanconi anemia pathway and undergoes negative autoregulation. Mol Cell. 2006, 23: 589-596. 10.1016/j.molcel.2006.06.024.
Nijman SM, Huang TT, Dirac AM, Brummelkamp TR, Kerkhoven RM, D'Andrea AD, Bernards R: The deubiquitinating enzyme USP1 regulates the Fanconi anemia pathway. Mol Cell. 2005, 17: 331-339. 10.1016/j.molcel.2005.01.008.
Sheehy AM, Gaddis NC, Choi JD, Malim MH: Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature. 2002, 418: 646-650. 10.1038/nature00939.
Sheehy AM, Gaddis NC, Malim MH: The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nat Med. 2003, 9: 1404-1407. 10.1038/nm945.
Harris RS, Bishop KN, Sheehy AM, Craig HM, Petersen-Mahrt SK, Watt IN, Neuberger MS, Malim MH: DNA deamination mediates innate immunity to retroviral infection. Cell. 2003, 113: 803-809. 10.1016/S0092-8674(03)00423-9.
Mangeat B, Turelli P, Caron G, Friedli M, Perrin L, Trono D: Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature. 2003, 424: 99-103. 10.1038/nature01709.
Yu X, Yu Y, Liu B, Luo K, Kong W, Mao P, Yu XF: Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science. 2003, 302: 1056-1060. 10.1126/science.1089591.
Kobayashi M, Takaori-Kondo A, Miyauchi Y, Iwai K, Uchiyama T: Ubiquitination of APOBEC3G by an HIV-1 Vif-Cullin5-Elongin B-Elongin C complex is essential for Vif function. J Biol Chem. 2005, 280: 18573-18578. 10.1074/jbc.C500082200.
Shirakawa K, Takaori-Kondo A, Kobayashi M, Tomonaga M, Izumi T, Fukunaga K, Sasada A, Abudu A, Miyauchi Y, Akari H, et al.: Ubiquitination of APOBEC3 proteins by the Vif-Cullin5-ElonginB-ElonginC complex. Virology. 2006, 344: 263-266. 10.1016/j.virol.2005.10.028.
Willey RL, Maldarelli F, Martin MA, Strebel K: Human immunodeficiency virus type 1 Vpu protein induces rapid degradation of CD4. J Virol. 1992, 66: 7193-7200.
Willey RL, Maldarelli F, Martin MA, Strebel K: Human immunodeficiency virus type 1 Vpu protein regulates the formation of intracellular gp160-CD4 complexes. J Virol. 1992, 66: 226-234.
Schubert U, Anton LC, Bacik I, Cox JH, Bour S, Bennink JR, Orlowski M, Strebel K, Yewdell JW: CD4 glycoprotein degradation induced by human immunodeficiency virus type 1 Vpu protein requires the function of proteasomes and the ubiquitin-conjugating pathway. J Virol. 1998, 72: 2280-2288.
Margottin F, Bour SP, Durand H, Selig L, Benichou S, Richard V, Thomas D, Strebel K, Benarous R: A novel human WD protein, h-beta TrCp, that interacts with HIV-1 Vpu connects CD4 to the ER degradation pathway through an F-box motif. Mol Cell. 1998, 1: 565-574. 10.1016/S1097-2765(00)80056-8.
Akari H, Bour S, Kao S, Adachi A, Strebel K: The human immunodeficiency virus type 1 accessory protein Vpu induces apoptosis by suppressing the nuclear factor kappaB-dependent expression of antiapoptotic factors. J Exp Med. 2001, 194: 1299-1311. 10.1084/jem.194.9.1299.
Belaidouni N, Marchal C, Benarous R, Besnard-Guerin C: Involvement of the betaTrCP in the ubiquitination and stability of the HIV-1 Vpu protein. Biochem Biophys Res Commun. 2007, 357: 688-693. 10.1016/j.bbrc.2007.03.195.
Besnard-Guerin C, Belaidouni N, Lassot I, Segeral E, Jobart A, Marchal C, Benarous R: HIV-1 Vpu sequesters beta-transducin repeat-containing protein (betaTrCP) in the cytoplasm and provokes the accumulation of beta-catenin and other SCFbetaTrCP substrates. J Biol Chem. 2004, 279: 788-795. 10.1074/jbc.M308068200.
Bour S, Perrin C, Akari H, Strebel K: The human immunodeficiency virus type 1 Vpu protein inhibits NF-kappa B activation by interfering with beta TrCP-mediated degradation of Ikappa B. J Biol Chem. 2001, 276: 15920-15928. 10.1074/jbc.M010533200.
Shackelford J, Pagano JS: Role of the ubiquitin system and tumor viruses in AIDS-related cancer. BMC Biochemistry. 2007, 8 (Suppl 1): S8-10.1186/1471-2091-8-S1-S8.
Coscoy L, Ganem D: Kaposi's sarcoma-associated herpesvirus encodes two proteins that block cell surface display of MHC class I chains by enhancing their endocytosis. Proc Natl Acad Sci USA. 2000, 97: 8051-8056. 10.1073/pnas.140129797.
Coscoy L, Sanchez DJ, Ganem D: A novel class of herpesvirus-encoded membrane-bound E3 ubiquitin ligases regulates endocytosis of proteins involved in immune recognition. J Cell Biol. 2001, 155: 1265-1273. 10.1083/jcb.200111010.
Hewitt EW, Duncan L, Mufti D, Baker J, Stevenson PG, Lehner PJ: Ubiquitylation of MHC class I by the K3 viral protein signals internalization and TSG101-dependent degradation. Embo J. 2002, 21: 2418-2429. 10.1093/emboj/21.10.2418.
Ishido S, Choi JK, Lee BS, Wang C, DeMaria M, Johnson RP, Cohen GB, Jung JU: Inhibition of natural killer cell-mediated cytotoxicity by Kaposi's sarcoma-associated herpesvirus K5 protein. Immunity. 2000, 13: 365-374. 10.1016/S1074-7613(00)00036-4.
Ishido S, Wang C, Lee BS, Cohen GB, Jung JU: Downregulation of major histocompatibility complex class I molecules by Kaposi's sarcoma-associated herpesvirus K3 and K5 proteins. J Virol. 2000, 74: 5300-5309. 10.1128/JVI.74.11.5300-5309.2000.
Sanchez DJ, Coscoy L, Ganem D: Functional organization of MIR2, a novel viral regulator of selective endocytosis. J Biol Chem. 2002, 277: 6124-6130. 10.1074/jbc.M110265200.
Duncan LM, Piper S, Dodd RB, Saville MK, Sanderson CM, Luzio JP, Lehner PJ: Lysine-63-linked ubiquitination is required for endolysosomal degradation of class I molecules. Embo J. 2006, 25: 1635-1645. 10.1038/sj.emboj.7601056.
Baichwal VR, Sugden B: Transformation of Balb 3T3 cells by the BNLF-1 gene of Epstein-Barr virus. Oncogene. 1988, 2: 461-467.
Wang D, Liebowitz D, Kieff E: An EBV membrane protein expressed in immortalized lymphocytes transforms established rodent cells. Cell. 1985, 43: 831-840. 10.1016/0092-8674(85)90256-9.
Jang KL, Shackelford J, Seo SY, Pagano JS: Up-regulation of beta-catenin by a viral oncogene correlates with inhibition of the seven in absentia homolog 1 in B lymphoma cells. Proc Natl Acad Sci USA. 2005, 102: 18431-18436. 10.1073/pnas.0504054102.
Kondo S, Seo SY, Yoshizaki T, Wakisaka N, Furukawa M, Joab I, Jang KL, Pagano JS: EBV latent membrane protein 1 up-regulates hypoxia-inducible factor 1alpha through Siah1-mediated down-regulation of prolyl hydroxylases 1 and 3 in nasopharyngeal epithelial cells. Cancer Res. 2006, 66: 9870-9877. 10.1158/0008-5472.CAN-06-1679.
Shackelford J, Maier C, Pagano JS: Epstein-Barr virus activates beta-catenin in type III latently infected B lymphocyte lines: association with deubiquitinating enzymes. Proc Natl Acad Sci USA. 2003, 100: 15572-15576. 10.1073/pnas.2636947100.
Nakayama K, Frew IJ, Hagensen M, Skals M, Habelhah H, Bhoumik A, Kadoya T, Erdjument-Bromage H, Tempst P, Frappell PB, et al.: Siah2 regulates stability of prolyl-hydroxylases, controls HIF1alpha abundance, and modulates physiological responses to hypoxia. Cell. 2004, 117: 941-952. 10.1016/j.cell.2004.06.001.
Davies JE, Sarkar S, Rubinsztein DC: The ubiquitin proteasome system in Huntingdon's disease and the spinocerebellar ataxias. BMC Biochemistry. 2007, 8 (Suppl 1): S2-10.1186/1471-2091-8-S1-S2.
Upadhya SC, Hegde AN: Role of the ubiquitin proteasome system in Alzheimer's disease. BMC Biochemistry. 2007, 8 (Suppl 1): S12-10.1186/1471-2091-8-S1-S12.
Imai Y, Soda M, Takahashi R: Parkin suppresses unfolded protein stress-induced cell death through its E3 ubiquitin-protein ligase activity. J Biol Chem. 2000, 275: 35661-35664. 10.1074/jbc.C000447200.
Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N: Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998, 392: 605-608. 10.1038/33416.
Shimura H, Hattori N, Kubo S, Mizuno Y, Asakawa S, Minoshima S, Shimizu N, Iwai K, Chiba T, Tanaka K, Suzuki T: Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet. 2000, 25: 302-305. 10.1038/77060.
Wing SS: The UPS in diabetes and obesity. BMC Biochemistry. 2008, 9 (Suppl 1): S6-
Cosaceanu D, Carapancea M, Alexandru O, Budiu R, Martinsson HS, Starborg M, Vrabete M, Kanter L, Lewensohn R, Dricu A: Comparison of three approaches for inhibiting insulin-like growth factor I receptor and their effects on NSCLC cell lines in vitro. Growth Factors. 2007, 25: 1-8. 10.1080/08977190600702865.
Sun XJ, Goldberg JL, Qiao LY, Mitchell JJ: Insulin-induced insulin receptor substrate-1 degradation is mediated by the proteasome degradation pathway. Diabetes. 1999, 48: 1359-1364. 10.2337/diabetes.48.7.1359.
Zhande R, Mitchell JJ, Wu J, Sun XJ: Molecular mechanism of insulin-induced degradation of insulin receptor substrate 1. Mol Cell Biol. 2002, 22: 1016-1026. 10.1128/MCB.22.4.1016-1026.2002.
Potashnik R, Bloch-Damti A, Bashan N, Rudich A: IRS1 degradation and increased serine phosphorylation cannot predict the degree of metabolic insulin resistance induced by oxidative stress. Diabetologia. 2003, 46: 639-648.
Rui L, Yuan M, Frantz D, Shoelson S, White MF: SOCS-1 and SOCS-3 block insulin signaling by ubiquitin-mediated degradation of IRS1 and IRS2. J Biol Chem. 2002, 277: 42394-42398. 10.1074/jbc.C200444200.
Nury D, Doucet C, Coux O: Roles and potential therapeutic targets of the ubiquitin proteasome system in muscle wasting. BMC Biochemistry. 2007, 8 (Suppl 1): S7-10.1186/1471-2091-8-S1-S7.
Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, Poueymirou WT, Panaro FJ, Na E, Dharmarajan K, et al.: Identification of ubiquitin ligases required for skeletal muscle atrophy. Science. 2001, 294: 1704-1708. 10.1126/science.1065874.
McElhinny AS, Kakinuma K, Sorimachi H, Labeit S, Gregorio CC: Muscle-specific RING finger-1 interacts with titin to regulate sarcomeric M-line and thick filament structure and may have nuclear functions via its interaction with glucocorticoid modulatory element binding protein-1. J Cell Biol. 2002, 157: 125-136. 10.1083/jcb.200108089.
Centner T, Yano J, Kimura E, McElhinny AS, Pelin K, Witt CC, Bang ML, Trombitas K, Granzier H, Gregorio CC, et al.: Identification of muscle specific ring finger proteins as potential regulators of the titin kinase domain. J Mol Biol. 2001, 306: 717-726. 10.1006/jmbi.2001.4448.
Li HH, Kedar V, Zhang C, McDonough H, Arya R, Wang DZ, Patterson C: Atrogin-1/muscle atrophy F-box inhibits calcineurin-dependent cardiac hypertrophy by participating in an SCF ubiquitin ligase complex. J Clin Invest. 2004, 114: 1058-1071.
Tintignac LA, Lagirand J, Batonnet S, Sirri V, Leibovitch MP, Leibovitch SA: Degradation of MyoD mediated by the SCF (MAFbx) ubiquitin ligase. J Biol Chem. 2005, 280: 2847-2856. 10.1074/jbc.M411346200.
Alkalay I, Yaron A, Hatzubai A, Orian A, Ciechanover A, Ben-Neriah Y: Stimulation-dependent I kappa B alpha phosphorylation marks the NF-kappa B inhibitor for degradation via the ubiquitin-proteasome pathway. Proc Natl Acad Sci USA. 1995, 92: 10599-10603. 10.1073/pnas.92.23.10599.
Spencer E, Jiang J, Chen ZJ: Signal-induced ubiquitination of IkappaBalpha by the F-box protein Slimb/beta-TrCP. Genes Dev. 1999, 13: 284-294. 10.1101/gad.13.3.284.
Yaron A, Hatzubai A, Davis M, Lavon I, Amit S, Manning AM, Andersen JS, Mann M, Mercurio F, Ben-Neriah Y: Identification of the receptor component of the IkappaBalpha-ubiquitin ligase. Nature. 1998, 396: 590-594. 10.1038/25159.
Senftleben U, Cao Y, Xiao G, Greten FR, Krahn G, Bonizzi G, Chen Y, Hu Y, Fong A, Sun SC, Karin M: Activation by IKKalpha of a second, evolutionary conserved, NF-kappa B signaling pathway. Science. 2001, 293: 1495-1499. 10.1126/science.1062677.
Xiao G, Harhaj EW, Sun SC: NF-kappaB-inducing kinase regulates the processing of NF-kappaB2 p100. Mol Cell. 2001, 7: 401-409. 10.1016/S1097-2765(01)00187-3.
Wertz IE, O'Rourke KM, Zhou H, Eby M, Aravind L, Seshagiri S, Wu P, Wiesmann C, Baker R, Boone DL, et al.: De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature. 2004, 430: 694-699. 10.1038/nature02794.
Layfield R, Shaw B: Ubiqutin-mediated signaling and Paget's disease of bone. BMC Biochemistry. 2007, 8 (Suppl 1): S5-10.1186/1471-2091-8-S1-S5.
Bignell GR, Warren W, Seal S, Takahashi M, Rapley E, Barfoot R, Green H, Brown C, Biggs PJ, Lakhani SR, et al.: Identification of the familial cylindromatosis tumour-suppressor gene. Nat Genet. 2000, 25: 160-165. 10.1038/76006.
Yoshida H, Jono H, Kai H, Li JD: The tumor suppressor cylindromatosis (CYLD) acts as a negative regulator for toll-like receptor 2 signaling via negative cross-talk with TRAF6 AND TRAF7. J Biol Chem. 2005, 280: 41111-41121. 10.1074/jbc.M509526200.
Brummelkamp TR, Nijman SM, Dirac AM, Bernards R: Loss of the cylindromatosis tumour suppressor inhibits apoptosis by activating NF-kappaB. Nature. 2003, 424: 797-801. 10.1038/nature01811.
Kovalenko A, Chable-Bessia C, Cantarella G, Israel A, Wallach D, Courtois G: The tumour suppressor CYLD negatively regulates NF-kappaB signalling by deubiquitination. Nature. 2003, 424: 801-805. 10.1038/nature01802.
Turnball EL, Rosser MFN, Cyr DM: The role of the UPS in cystic fibrosis. BMC Biochemistry. 2007, 8 (Suppl 1): S11-10.1186/1471-2091-8-S1-S11.
Rotin D: Role of the UPS in Liddle syndrome. BMC Biochemistry. 2008, 9 (Suppl 1): S5-
Hansson JH, Nelson-Williams C, Suzuki H, Schild L, Shimkets R, Lu Y, Canessa C, Iwasaki T, Rossier B, Lifton RP: Hypertension caused by a truncated epithelial sodium channel gamma subunit: genetic heterogeneity of Liddle syndrome. Nat Genet. 1995, 11: 76-82. 10.1038/ng0995-76.
Snyder PM, Price MP, McDonald FJ, Adams CM, Volk KA, Zeiher BG, Stokes JB, Welsh MJ: Mechanism by which Liddle's syndrome mutations increase activity of a human epithelial Na+ channel. Cell. 1995, 83: 969-978. 10.1016/0092-8674(95)90212-0.
Dahlmann B: Role of proteasomes in disease. BMC Biochemistry. 2007, 8 (Suppl 1): S3-10.1186/1471-2091-8-S1-S3.
Chauhan D, Bianchi G, Anderson KC: Targeting the UPS as therapy in multiple myeloma. BMC Biochemistry. 2008, 9 (Suppl 1): S1-
Nalepa G, Rolfe M, Harper JW: Drug discovery in the ubiquitin-proteasome system. Nat Rev Drug Discov. 2006, 5: 596-613. 10.1038/nrd2056.
Demo SD, Kirk CJ, Aujay MA, Buchholz TJ, Dajee M, Ho MN, Jiang J, Laidig GJ, Lewis ER, Parlati F, Shenk KD, Smyth MS, Sun CM, Vallone MK, Woo TM, Molineaux CJ, Bennett MK: Antitumor activity of PR-171, a novel irreversible inhibitor of the proteasome. Cancer Res. 2007, 67: 6383-6391. 10.1158/0008-5472.CAN-06-4086.
Cusack JC, Liu R, Xia L, Chao TH, Pien C, Niu W, Palombella VJ, Neuteboom ST, Palladino MA: NPI-0052 enhances tumoricidal response to conventional cancer therapy in a colon cancer model. Clin Cancer Res. 2006, 12: 6758-6759. 10.1158/1078-0432.CCR-06-1151.
Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, et al.: In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004, 303: 844-848. 10.1126/science.1092472.
Kussie PH, Gorina S, Marechal V, Elenbaas B, Moreau J, Levine AJ, Pavletich NP: Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science. 1996, 274: 948-953. 10.1126/science.274.5289.948.
Lane DP, Crawford LV: T antigen is bound to a host protein in SV40-transformed cells. Nature. 1979, 278: 261-263. 10.1038/278261a0.
Gatz SA, Wiesmüller L: p53 in recombination and repair. Cell Death Differ. 2006, 13: 1003-1016. 10.1038/sj.cdd.4401903.
Issaeva N, Bozko P, Enge M, Protopopova M, Verhoef LG, Masucci M, Pramanik A, Selivanova G: Small molecule RITA binds to p53, blocks p53-HDM-2 interaction and activates p53 function in tumors. Nat Med. 2004, 10: 1321-1328. 10.1038/nm1146.
Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA: In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004, 303: 844-848. 10.1126/science.1092472.
Critchley S, Gant TG, Langston SP, Olhava EJ, Peluso S, [Millennium Pharmaceuticals Inc.]: Inhibitors of E1 activating enzymes. WO2006084281.
Parlati F, Ramesh UV, Singh R, Payan DG, Lowe R, Look GC, [Rigel Pharmaceuticals Inc.]: Benzothiazole and Thiazole'5,5-B!Pyridine compositions and their use as Ubiquitin ligase Inhibitors. WO2005037845.
Gudeat P, Colland F: Patented small molecule inhibitors in the ubiquitin proteasome system. BMC Biochemistry. 2007, 8 (Suppl 1): S14-10.1186/1471-2091-8-S1-S14.
Passmore LA, Barford D: Getting into position: the catalytic mechanisms of protein ubiquitylation. Biochem J. 2004, 379: 513-525. 10.1042/BJ20040198.
Verdecia MA, Joazeiro CA, Wells NJ, Ferrer JL, Bowman ME, Hunter T, Noel JP: Conformational flexibility underlies ubiquitin ligation mediated by the WWP1 HECT domain E3 ligase. Mol Cell. 2003, 11: 249-259. 10.1016/S1097-2765(02)00774-8.
Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, Deshaies RJ: Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc Natl Acad Sci USA. 2001, 98: 8554-8559. 10.1073/pnas.141230798.
Sakamoto KM, Kim KB, Verma R, Ransick A, Stein B, Crews CM, Deshaies RJ: Development of Protacs to target cancer-promoting proteins for ubiquitination and degradation. Mol Cell Proteomics. 2003, 2: 1350-1358. 10.1074/mcp.T300009-MCP200.
Schneekloth JS, Fonseca FN, Koldobskiy M, Mandal A, Deshaies R, Sakamoto K, Crews CM: Chemical genetic control of protein levels: selective in vivo targeted degradation. J Am Chem Soc. 2004, 126: 3748-3754. 10.1021/ja039025z.
Sakamoto KM, Kim KB, Verma R, Ransick A, Stein B, Crews CM, Deshaies RJ: Development of Protacs to target cancer-promoting proteins for ubiquitination and degradation. Mol Cell Proteomics. 2003, 2: 1350-8. 10.1074/mcp.T300009-MCP200.
Winn PJ, Religa TL, Battey JN, Banerjee A, Wade RC: Determinants of functionality in the ubiquitin conjugating enzyme family. Structure. 2004, 12: 1563-1574. 10.1016/j.str.2004.06.017.
Verma R, Peters NR, D'Onofrio M, Tochtrop GP, Sakamoto KM, Varadan R, Zhang M, Coffino P, Fushman D, Deshaies RJ, King RW: Ubistatins inhibit proteasome-dependent degradation by binding the ubiquitin chain. Science. 2004, 306: 117-120. 10.1126/science.1100946.
Publication history
Republished from Current BioData's Targeted Proteins database (TPdb; http://www.targetedproteinsdb.com).
Acknowledgements
I thank Julia Toth, Jianing Huang, Lyuben Tsvetkov, Sarkiz Daniel-Issakani, and Donald Payan for critically reading the manuscript, Amelia Cervantes and Carine Richards for technical assistance, the staff of Current BioData Limited for editorial input, and my colleagues at Rigel Pharmaceuticals, Inc. for helpful discussions and suggestions.
This article has been published as part of BMC Biochemistry Volume 9 Supplement 1, 2008: Ubiquitin-Proteasome System in Disease Part 2. The full contents of the supplement are available online at http://www.biomedcentral.com/1471-2091/9?issue=S1.
Additional TPdb reviews on the ubiquitin-proteasome system are also available in BMC Biochemistry – see Volume 8 Suppl 1 http://www.biomedcentral.com/1471-2091/8?issue=S1.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The author was employed by a pharmaceutical company with an interest in the ubiquitin system during the writing of this review.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an open access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Petroski, M.D. The ubiquitin system, disease, and drug discovery. BMC Biochem 9 (Suppl 1), S7 (2008). https://doi.org/10.1186/1471-2091-9-S1-S7
Published:
DOI: https://doi.org/10.1186/1471-2091-9-S1-S7