
Abstract
Abstract
Hypertension is a serious medical problem affecting a large population worldwide. Liddle syndrome is a hereditary form of early onset hypertension caused by mutations in the epithelial Na+ channel (ENaC). The mutated region, called the PY (Pro-Pro-x-Tyr) motif, serves as a binding site for Nedd4-2, an E3 ubiquitin ligase from the HECT family. Nedd4-2 binds the ENaC PY motif via its WW domains, normally leading to ENaC ubiquitylation and endocytosis, reducing the number of active channels at the plasma membrane. In Liddle syndrome, this endocytosis is impaired due to the inability of the mutated PY motif in ENaC to properly bind Nedd4-2. This leads to accumulation of active channels at the cell surface and increased Na+ (and fluid) absorption in the distal nephron, resulting in elevated blood volume and blood pressure. Small molecules/compounds that destabilize cell surface ENaC, or enhance Nedd4-2 activity in the kidney, could potentially serve to alleviate hypertension.
Publication history
Republished from Current BioData's Targeted Proteins database (TPdb; http://www.targetedproteinsdb.com).
Similar content being viewed by others

Protein pathway involvement in disease
Introduction
Elevated arterial blood pressure, or hypertension, poses a serious public health problem, affecting approximately 25% of the adult population in the industrial world [1], and becoming, along with obesity, a serious health problem in the developing world as well. In recent years, the causes of several genetic disorders leading to hypertension or hypotension have been identified, and deleterious mutations have been mapped to components of the aldosterone pathway, as well as to key ion channels and transporters expressed along the nephron. Prominent examples include Bartter syndrome type I, II or III, Gitelman syndrome, pseudohypoaldosteronism I (PHAI) and Liddle syndrome [2]; the latter two are associated with mutations in the epithelial Na+ channel (ENaC) and are discussed in this review, with a particular focus on Liddle syndrome, a hereditary form of hypertension.
The epithelial Na+ channel
The amiloride-sensitive ENaC is an ion channel expressed in Na+-transporting epithelia such as those present in the distal nephron, respiratory epithelium, distal colon and taste buds [3]. In the kidney, it is primarily expressed in the distal connecting tubules (CNTs) and cortical collecting tubules (CCTs) of the nephron [4, 5], where it provides the rate limiting step for Na+ (and fluid) reabsorption into the blood stream [3, 6, 7]. This regulation of Na+ and fluid absorption is tightly controlled by the hormones aldosterone (i.e. the renin-angiotensin-aldosterone pathway) and vasopressin (antidiuretic hormone, ADH), which stimulate channel activity [6, 8]. The single channel characteristics of ENaC reveal high selectivity for Na+ over K+, low single channel conductance (~5 pS), high sensitivity to amiloride (~100 nM) and slow gating [6]. ENaC activity is primarily regulated by control of its opening (Po) and numbers at the plasma membrane [8].
ENaC is comprised of three subunits, α, β and γ [9], each consisting of two transmembrane domains flanked by a large extracellular loop and two intracellular N- and C-termini, and is preferentially assembled at a stoichiometry of α2βγ [10, 11] (although other configurations have been proposed [12, 13]). Maximal channel activity is obtained when all three subunits are expressed together, but expression of α alone, or a combination of αβ, or αγ, results in low or moderate channel activity, respectively [9].
Genetic disease-causing mutations in ENaC, as well as mouse models, have shed important light on ENaC function and the pathology of ENaC-related diseases. For example, loss of function mutations in either α, β, or γ ENaC cause PHAI [14, 15], a salt-wasting disease leading to hypotension, which is also mimicked in knockout mouse models lacking β or γ ENaC [16, 17], or models expressing reduced levels of α ENaC [18], all of which exhibit reduced levels of channel expression and activity [9, 19]. In contrast to the ENaC loss of function mutations causing PHAI, gain of function mutations in this channel cause Liddle syndrome.
Liddle syndrome
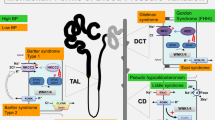
Liddle syndrome (pseudoaldosteronism, OMIM 177200) is an autosomal dominant disease leading to early onset of hypertension. It is associated with hypokalemic alkalosis, reduced plasma rennin activity and low plasma aldosterone levels [20]. Over the past 12 years, work from Lifton's group (Yale University) and others has identified several deletions/mutations that cause Liddle syndrome, all of which map to β or γ ENaC and lead to elevated channel numbers and activity at the plasma membrane, as assessed by heterologously expressing these mutant ENaCs in Xenopus oocytes or cultured mammalian cells [2, 21, 22]. These genetic defects either delete the C-terminus of β or γ ENaC [23, 24], or mutate a proline or a tyrosine within a short sequence, called the PY (Pro-Pro-x-Tyr) motif [25–28]. The PY motif, or extended PY motif (PPxYxxL [21, 29]), is highly conserved in the C-termini of all ENaC subunits [26], and serves as a binding site for the Nedd4 family of ubiquitin ligases [30], as assessed by in vitro binding, yeast two-hybrid and co-immunoprecipitation assays, as well as by structural analysis (e.g. [29, 30]).
Regulation of ENaC by the ubiquitin system and its impairment in Liddle syndrome
Ubiquitylation, carried out by the sequential activity of E1, E2 and E3 (ubiquitin ligase) enzymes, usually regulates stability of target proteins that are tagged with ubiquitin by the E3 ligases [31]. Most of these proteins are degraded by the proteasome [32]. Recent studies have demonstrated, however, that ubiquitylation of transmembrane proteins can tag them for endocytosis and/or vesicular sorting, often resulting in their degradation in the lysosome [33, 34]. This is usually achieved by the presence of ubiquitin binding motifs or domains (e.g. UIM, UBA, CUE, GAT, UEV, VHS) within proteins such as epsin/Eps15, Hrs and GGA, which function to recognize the ubiquitylated transmembrane proteins and facilitate their endocytosis or sorting [35].
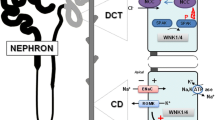
Nedd4 family members are E3 ubiquitin ligases that comprise a C2 domain responsible for membrane targeting [36, 37], three to four WW domains that bind the PY motifs of ENaC [29, 30, 38–42], and a ubiquitin ligase HECT (homologous to E6AP carboxyl-terminus) domain [43, 44] (Figures 1 and 2). Of the two closely related Nedd4 members, Nedd4-1 and Nedd4-2, the latter binds ENaC more strongly due to the presence of an additional, high affinity WW domain (WW3, out of four WW domains) [41, 42]. Accordingly, Nedd4-2 was shown to effectively suppress ENaC activity by enhancing removal of the channel from the plasma membrane [45–47], and ubiquitylation of ENaC was demonstrated to destabilize cell surface ENaC [48] (Figure 2). Indeed, our recent work has demonstrated that Nedd4-2 can ubiquitylate ENaC present at the apical membrane of cultured kidney epithelial cells [49]. The few Nedd4-1 proteins that also contain this high affinity WW3 domain (e.g. human and Drosophila Nedd4-1) are also able to suppress ENaC activity when heterologously expressed in Xenopus oocytes or cultured cells, although in some cases this is prevented in the presence of the C2 domain (for example, in the case of human Nedd4-1 [41, 47, 50]), possibly (albeit speculatively) due to inhibitory interactions between the C2 and HECT domains.

Regulation of ENaC by Nedd4-2 in homeostasis. The ubiquitin ligase Nedd4-2 binds (via its WW domains) to the PY motifs of ENaC, in turn ubiquitylating and targeting ENaC for endocytosis and lysosomal degradation. This process can be inhibited by Sgk1- or Akt-mediated phosphorylation of Nedd4-2, which leads to binding of 14-3-3 proteins to phosphorylated Nedd4-2, thus preventing Nedd4-2 from associating with ENaC and thus increasing ENaC levels at the plasma membrane. Sgk1 can also upregulate ENaC independently of Nedd4-2.

Regulation of ENaC by Nedd4-2 and its impairment in Liddle syndrome. In Liddle syndrome, deletion/mutation of the PY motif in βENaC (or γENaC, not shown) impairs the ability of Nedd4-2 to bind (and thus ubiquitylate) ENaC, leading to accumulation of ENaC channels at the plasma membrane and increased channel activity. (Modified with permission from Staub and Rotin).
Experiments performed in Xenopus oocytes or mammalian cultured cells that ectopically express ENaC reveal that unlike the ability of Nedd4-2 to induce removal of wild-type ENaC from the plasma membrane by ubiquitylation (likely linked to subsequent clathrin-mediated endocytosis [51]), Liddle syndrome mutations in the PY motif of β or γ ENaC severely attenuate this removal, leading to increased retention of mutant ENaC at the cell surface and to elevated channel activity [45, 46, 49] (Figure 2). Accordingly, conditional knockout mice bearing a deletion of the PY motif in βENaC (a mouse model for Liddle syndrome) develop hypertension that is induced by high salt diet [52]. Moreover, channel feedback inhibition by elevated intracellular Na+ concentrations exhibited by wild-type ENaC (demonstrated ex vivo in the CCTs of rats [53]) is defective in ENaC bearing the Liddle syndrome mutations in the PY motif (as shown by ectopic expression of PY motif-mutated ENaC in Xenopus oocytes [54]), further exacerbating Na+ loading. Together, this results in increased Na+ and fluid reabsorption in the distal nephron, and increased blood volume and blood pressure, which are hallmarks of Liddle syndrome.
As indicated in the section The epithelial Na+channel, ENaC is tightly (positively) regulated in the kidney by the mineralocorticoid hormone aldosterone. One of the recently discovered aldosterone targets is Sgk1, a Ser/Thr kinase from the Akt (PKB) family that was found to elevate ENaC levels/activity in response to aldosterone in rat or mouse kidney and in A6 cells (a Xenopus cell line endogenously expressing ENaC and responsive to aldosterone) [55, 56]. This effect can be mediated either without (see below) or via regulation of Nedd4-2 (Figure 1). Nedd4-2 (but not Nedd4-1) possesses Sgk1 phosphorylation sites and, when phosphorylated by Sgk1 (in cultured cells), is prevented from downregulating ENaC, leading to increased ENaC retention at the cell surface and thus increased ENaC activity [57, 58]. This effect is believed to be mediated by association of the adaptor protein 14-3-3, known to bind phosphoSer/Thr [59], with Ser-phosphorylated Nedd4-2, thus preventing Nedd4-2 from binding ENaC [60, 61] (by as yet unknown mechanism(s)). In support, the expression of 14-3-3β and Nedd4-2, as well as Nedd4-2 phosphorylation, were recently shown to be induced in CCT cells by dietary salt and by aldosterone [62–64]. However, aldosterone and Sgk1 can stimulate ENaC independently of Nedd4-2 and their role in Liddle syndrome is controversial: aldosterone and Sgk1 were found to increase cell surface abundance of ENaC channels bearing Liddle syndrome deletions/mutations (which cannot bind Nedd4-2) [65–68] and, importantly, CCTs harvested from mutant mice bearing a Liddle syndrome deletion [52] revealed a normal response to aldosterone [69].
In addition to aldosterone, the hormone vasopressin also increases ENaC activity (as well as water absorption) in the distal nephron by binding to V2 receptors and stimulating activation of adenylate cyclase and the release of cAMP [6]. cAMP increases the density of ENaC channels (endogenously or ectopically expressed in epithelial cells) at the plasma membrane [70, 71], an effect suggested to be impaired in channels bearing the Liddle syndrome PY motif mutations due to defective trafficking to the cell surface [72], or mobilization from a sub-apical pool [49]. Recent studies also suggested that Nedd4-2 phosphorylation by PKA (which is activated by cAMP) provides inhibitory function much like Sgk1, thus inhibiting the ability of Nedd4-2 to suppress ENaC, leading to increased cell surface abundance of this channel [73].
Among the other factors that regulate ENaC (aside from hormones, ions and Nedd4-2), are proteases such as CAP proteins and TMPRSS3, which activate ENaC by proteolytic cleavage of its ectodomains [3, 74–77]. A recent paper suggests that Liddle syndrome mutations increase the number of cleaved (active) ENaCs at the cell surface (thus further increasing Na+ absorption), and that Nedd4-2 and ENaC ubiquitylation regulate the number of cleaved channels at the plasma membrane [78].
Disease models, knockouts and assays
To date, only one mouse model for Liddle syndrome has been generated. These mice, created by the Rossier/Hummler groups (Institut de Pharmacologie et de Toxicologie, Switzerland) in 1999, bear a deletion of the PY motif in βENaC (a stop codon is inserted at a residue corresponding to residue Arg566 in human βENaC, as found in the original pedigree described by R. Lifton [23]). The mice have no phenotype under a normal salt diet, but develop hypertension when fed a high salt diet [52]. To date, no other relevant knockout mice of Liddle syndrome have been developed.
Disease targets and ligands
Liddle syndrome patients are treated with the ENaC antagonist amiloride-triamterene and a low salt diet to stabilize their high blood pressure. While Liddle syndrome is a rare disorder, as are several genetic forms of hypertension [2], other forms of hypertension are very common in the population and have no known genetic components. Inhibiting ENaC activity, the rate limiting step in the regulation of Na+ and fluid reabsorption in the nephron, could provide an attractive target to treat hypertension. With the advent of high throughput technology it is possible to test for inhibition of ENaC activity by screening with small molecules/compound libraries, with the hope of identifying inhibitory compounds that may be superior to amiloride and its analogs. In that regard, we have recently developed a high throughput assay that allows quantification of the amounts of cell surface ENaC (Chen and Rotin, unpublished). Given the key role played by the ubiquitin system/Nedd4-2 in regulating ENaC cell surface stability and ENaC function, identifying compounds that destabilize/decrease ENaC levels at the plasma membrane could have potential therapeutic benefits for the treatment of hypertension. Stimulating Nedd4-2 activity, which leads to ENaC endocytosis/degradation, could also be a possibility. However, since Nedd4-2 likely has other targets in other tissues/cells, this approach needs to be scrutinized to ensure it is targeted specifically to ENaC in the kidney. It is likely that use of putative compounds that aim to enhance ENaC internalization or Nedd4-2 activity would be more effective towards other forms of hypertension and not Liddle syndrome, since the latter carries mutations that already inhibit ENaC internalization and are insensitive to Nedd4-2.
New frontiers in drug discovery
Despite significant recent advances, several key questions remain to be answered regarding the regulation of ENaC by Nedd4-2 and the ubiquitin system. These include:
(i) How is sensing of elevation of intracellular concentrations of Na+ (that normally shuts down ENaC) related to the Liddle syndrome mutations? If this is regulated via Nedd4-2, how is Nedd4-2 (directly or indirectly) able to sense Na+?
(ii) How does phosphorylation of Nedd4-2 by Sgk1 (on sites not within the WW domains), which leads to binding of 14-3-3 to Nedd4-2, inhibit the association of Nedd4-2 with ENaC, which is mediated via the WW domains?
(iii) What is the exact stoichiometry of Nedd4-2-ENaC interactions, and how is it that loss of only one PY motif is sufficient to cause Liddle syndrome?
(iv) How is the activity of Nedd4-2 itself regulated in the cell?
(v) What is the physiological function of Nedd4 proteins in vivo in mammals, especially in the kidney? The latter should be answered with the generation of knockout murine models for these proteins (not yet published). Future work will undoubtedly address these and other important questions that investigate the relationship between ENaC and the ubiquitin system.
Abbreviations
- CCT:
-
cortical collecting tubule
- CNT:
-
distal connecting tubule
- ENaC:
-
epithelial Na+ channel
- HECT:
-
homologous to E6AP carboxyl-terminus
- PHAI:
-
pseudohypoaldosteronism I
- PY:
-
Pro-Pro-x-Tyr.
References
Burt VL, Whelton P, Roccella EJ, Brown C, Cutler JA, Higgins M, Horan MJ, Labarthe D: Prevalence of hypertension in the US adult population. Results from the Third National Health and Nutrition Examination Survey, 1988–1991. Hypertension. 1995, 25 (3): 305-313.
Lifton RP, Gharavi AG, Geller DS: Molecular mechanisms of human hypertension. Cell. 2001, 104 (4): 545-556.
Rossier BC: The epithelial sodium channel: activation by membrane-bound serine proteases. Proc Am Thorac Soc. 2004, 1 (1): 4-9.
Duc C, Farman N, Canessa CM, Bonvalet JP, Rossier BC: Cell-specific expression of epithelial sodium channel alpha, beta, and gamma subunits in aldosterone-responsive epithelia from the rat: localization by in situ hybridization and immunocytochemistry. J Cell Biol. 1994, 127 (6 Pt 2): 1907-1921.
Frindt G, Palmer LG: Na channels in the rat connecting tubule. Am J Physiol Renal Physiol. 2004, 286 (4): F669-674.
Garty H, Palmer LG: Epithelial sodium channels: function, structure, and regulation. Physiol Rev. 1997, 77 (2): 359-396.
Kellenberger S, Schild L: Epithelial sodium channel/degenerin family of ion channels: a variety of functions for a shared structure. Physiol Rev. 2002, 82 (3): 735-767.
Schild L: The epithelial sodium channel: from molecule to disease. Rev Physiol Biochem Pharmacol. 2004, 151: 93-107.
Canessa CM, Schild L, Buell G, Thorens B, Gautschi I, Horisberger JD, Rossier BC: Amiloride-sensitive epithelial Na+ channel is made of three homologous subunits. Nature. 1994, 367 (6462): 463-467.
Firsov D, Gautschi I, Merillat AM, Rossier BC, Schild L: The heterotetrameric architecture of the epithelial sodium channel (ENaC). Embo J. 1998, 17 (2): 344-352.
Kosari F, Sheng S, Li J, Mak DO, Foskett JK, Kleyman TR: Subunit stoichiometry of the epithelial sodium channel. J Biol Chem. 1998, 273 (22): 13469-13474.
Snyder PM, Cheng C, Prince LS, Rogers JC, Welsh MJ: Electrophysiological and biochemical evidence that DEG/ENaC cation channels are composed of nine subunits. J Biol Chem. 1998, 273 (2): 681-684.
Staruschenko A, Medina JL, Patel P, Shapiro MS, Booth RE, Stockand JD: Fluorescence resonance energy transfer analysis of subunit stoichiometry of the epithelial Na+ channel. J Biol Chem. 2004, 279 (26): 27729-27734.
Strautnieks SS, Thompson RJ, Gardiner RM, Chung E: A novel splice-site mutation in the gamma subunit of the epithelial sodium channel gene in three pseudohypoaldosteronism type 1 families. Nat Genet. 1996, 13 (2): 248-250.
Chang SS, Grunder S, Hanukoglu A, Rosler A, Mathew PM, Hanukoglu I, Schild L, Lu Y, Shimkets RA, Nelson-Williams C, et al.: Mutations in subunits of the epithelial sodium channel cause salt wasting with hyperkalaemic acidosis, pseudohypoaldosteronism type 1. Nat Genet. 1996, 12 (3): 248-253.
Barker PM, Nguyen MS, Gatzy JT, Grubb B, Norman H, Hummler E, Rossier B, Boucher RC, Koller B: Role of gammaENaC subunit in lung liquid clearance and electrolyte balance in newborn mice. Insights into perinatal adaptation and pseudohypoaldosteronism. J Clin Invest. 1998, 102 (8): 1634-1640.
McDonald FJ, Yang B, Hrstka RF, Drummond HA, Tarr DE, McCray PB, Stokes JB, Welsh MJ, Williamson RA: Disruption of the beta subunit of the epithelial Na+ channel in mice: hyperkalemia and neonatal death associated with a pseudohypoaldosteronism phenotype. Proc Natl Acad Sci USA. 1999, 96 (4): 1727-1731.
Hummler E, Barker P, Talbot C, Wang Q, Verdumo C, Grubb B, Gatzy J, Burnier M, Horisberger JD, Beermann F, et al.: A mouse model for the renal salt-wasting syndrome pseudohypoaldosteronism. Proc Natl Acad Sci USA. 1997, 94 (21): 11710-11715.
Grunder S, Firsov D, Chang SS, Jaeger NF, Gautschi I, Schild L, Lifton RP, Rossier BC: A mutation causing pseudohypoaldosteronism type 1 identifies a conserved glycine that is involved in the gating of the epithelial sodium channel. Embo J. 1997, 16 (5): 899-907.
Liddle GW, Bledsoe T, Coppage WS: Hypertension reviews. J Tenn Med Assoc. 1974, 67 (8): 669-
Snyder PM, Price MP, McDonald FJ, Adams CM, Volk KA, Zeiher BG, Stokes JB, Welsh MJ: Mechanism by which Liddle's syndrome mutations increase activity of a human epithelial Na+ channel. Cell. 1995, 83 (6): 969-978.
Firsov D, Schild L, Gautschi I, Merillat AM, Schneeberger E, Rossier BC: Cell surface expression of the epithelial Na channel and a mutant causing Liddle syndrome: a quantitative approach. Proc Natl Acad Sci USA. 1996, 93 (26): 15370-15375.
Shimkets RA, Warnock DG, Bositis CM, Nelson-Williams C, Hansson JH, Schambelan M, Gill JR, Ulick S, Milora RV, Findling JW, et al.: Liddle's syndrome: heritable human hypertension caused by mutations in the beta subunit of the epithelial sodium channel. Cell. 1994, 79 (3): 407-414.
Hansson JH, Nelson-Williams C, Suzuki H, Schild L, Shimkets R, Lu Y, Canessa C, Iwasaki T, Rossier B, Lifton RP: Hypertension caused by a truncated epithelial sodium channel gamma subunit: genetic heterogeneity of Liddle syndrome. Nat Genet. 1995, 11 (1): 76-82.
Hansson JH, Schild L, Lu Y, Wilson TA, Gautschi I, Shimkets R, Nelson-Williams C, Rossier BC, Lifton RP: A de novo missense mutation of the beta subunit of the epithelial sodium channel causes hypertension and Liddle syndrome, identifying a proline-rich segment critical for regulation of channel activity. Proc Natl Acad Sci USA. 1995, 92 (25): 11495-11499.
Schild L, Lu Y, Gautschi I, Schneeberger E, Lifton RP, Rossier BC: Identification of a PY motif in the epithelial Na channel subunits as a target sequence for mutations causing channel activation found in Liddle syndrome. Embo J. 1996, 15 (10): 2381-2387.
Tamura H, Schild L, Enomoto N, Matsui N, Marumo F, Rossier BC: Liddle disease caused by a missense mutation of beta subunit of the epithelial sodium channel gene. J Clin Invest. 1996, 97 (7): 1780-1784.
Inoue J, Iwaoka T, Tokunaga H, Takamune K, Naomi S, Araki M, Takahama K, Yamaguchi K, Tomita K: A family with Liddle's syndrome caused by a new missense mutation in the beta subunit of the epithelial sodium channel. J Clin Endocrinol Metab. 1998, 83 (6): 2210-2213.
Kanelis V, Rotin D, Forman-Kay JD: Solution structure of a Nedd4 WW domain – ENaC peptide complex. Nature Structure Biol. 2001, 8 (5): 407-412.
Staub O, Dho S, Henry P, Correa J, Ishikawa T, McGlade J, Rotin D: WW domains of Nedd4 bind to the proline-rich PY motifs in the epithelial Na+ channel deleted in Liddle's syndrome. Embo J. 1996, 15 (10): 2371-2380.
Hershko A, Ciechanover A: The ubiquitin system. Annu Rev Biochem. 1998, 67: 425-479.
Glickman MH, Ciechanover A: The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev. 2002, 82 (2): 373-428.
Hicke L: A new ticket for entry into budding vesicles-ubiquitin. Cell. 2001, 106 (5): 527-530.
Staub O, Rotin D: Role of ubiquitylation in cellular membrane transport. Physiol Rev. 2006, 86 (2): 669-707.
Hicke L, Schubert HL, Hill CP: Ubiquitin-binding domains. Nat Rev Mol Cell Biol. 2005, 6 (8): 610-621.
Plant PJ, Lafont F, Lecat S, Verkade P, Simons K, Rotin D: Apical membrane targeting of Nedd4 is mediated by an association of its C2 domain with annexin XIIIb. J Cell Biol. 2000, 149 (7): 1473-1484.
Plant PJ, Yeger H, Staub O, Howard P, Rotin D: The C2 domain of the ubiquitin protein ligase Nedd4 mediates Ca2+-dependent plasma membrane localization. J Biol Chem. 1997, 272 (51): 32329-32336.
Goulet CC, Volk KA, Adams CM, Prince LS, Stokes JB, Snyder PM: Inhibition of the epithelial Na+ channel by interaction of Nedd4 with a PY motif deleted in Liddle's syndrome. J Biol Chem. 1998, 273 (45): 30012-30017.
Harvey KF, Dinudom A, Komwatana P, Jolliffe CN, Day ML, Parasivam G, Cook DI, Kumar S: All three WW domains of murine Nedd4 are involved in the regulation of epithelial sodium channels by intracellular Na+. J Biol Chem. 1999, 274 (18): 12525-12530.
Farr TJ, Coddington-Lawson SJ, Snyder PM, McDonald FJ: Human Nedd4 interacts with the human epithelial Na+ channel: WW3 but not WW1 binds to Na+-channel subunits. Biochem J. 2000, 345 (Pt 3): 503-509.
Henry PC, Kanelis V, O'Brien MC, Kim B, Gautschi I, Forman-Kay J, Schild L, Rotin D: Affinity and specificity of interactions between Nedd4 isoforms and the epithelial Na+ channel. J Biol Chem. 2003, 278 (22): 20019-20028.
Kanelis V, Bruce MC, Skrynnikov NR, Rotin D, Forman-Kay JD: Structural determinants for high-affinity binding in a Nedd4 WW3* domain-Comm PY motif complex. Structure. 2006, 14 (3): 543-553.
Rotin D, Staub O, Haguenauer-Tsapis R: Ubiquitination and endocytosis of plasma membrane proteins: role of Nedd4/Rsp5p family of ubiquitin-protein ligases. J Membr Biol. 2000, 176 (1): 1-17.
Ingham RJ, Gish G, Pawson T: The Nedd4 family of E3 ubiquitin ligases: functional diversity within a common modular architecture. Oncogene. 2004, 23 (11): 1972-1984.
Abriel H, Loffing J, Rebhun JF, Pratt JH, Schild L, Horisberger JD, Rotin D, Staub O: Defective regulation of the epithelial Na+ channel by Nedd4 in Liddle's syndrome. J Clin Invest. 1999, 103 (5): 667-673.
Kamynina E, Debonneville C, Bens M, Vandewalle A, Staub O: A novel mouse Nedd4 protein suppresses the activity of the epithelial Na+ channel. Faseb J. 2001, 15 (1): 204-214.
Snyder PM, Olson DR, McDonald FJ, Bucher DB: Multiple WW domains, but not the C2 domain, are required for inhibition of the epithelial Na+ channel by human Nedd4. J Biol Chem. 2001, 276 (30): 28321-28326.
Staub O, Gautschi I, Ishikawa T, Breitschopf K, Ciechanover A, Schild L, Rotin D: Regulation of stability and function of the epithelial Na+ channel (ENaC) by ubiquitination. Embo J. 1997, 16 (21): 6325-6336.
Lu C, Pribanic S, Debonneville A, Jiang C, Rotin D: The PY motif of ENaC, mutated in Liddle syndrome, regulates channel internalization, sorting and mobilization from subapical pool. Traffic. 2007,
Kamynina E, Tauxe C, Staub O: Distinct characteristics of two human Nedd4 proteins with respect to epithelial Na(+) channel regulation. Am J Physiol Renal Physiol. 2001, 281 (3): F469-477.
Shimkets RA, Lifton RP, Canessa CM: The activity of the epithelial sodium channel is regulated by clathrin-mediated endocytosis. J Biol Chem. 1997, 272 (41): 25537-25541.
Pradervand S, Wang Q, Burnier M, Beermann F, Horisberger JD, Hummler E, Rossier BC: A mouse model for Liddle's syndrome. J Am Soc Nephrol. 1999, 10 (12): 2527-2533.
Palmer LG, Sackin H, Frindt G: Regulation of Na+ channels by luminal Na+ in rat cortical collecting tubule. J Physiol. 1998, 509 (Pt 1): 151-162.
Kellenberger S, Gautschi I, Rossier BC, Schild L: Mutations causing Liddle syndrome reduce sodium-dependent downregulation of the epithelial sodium channel in the Xenopus oocyte expression system. J Clin Invest. 1998, 101 (12): 2741-2750.
Chen SY, Bhargava A, Mastroberardino L, Meijer OC, Wang J, Buse P, Firestone GL, Verrey F, Pearce D: Epithelial sodium channel regulated by aldosterone-induced protein sgk. Proc Natl Acad Sci USA. 1999, 96 (5): 2514-2519.
Naray-Fejes-Toth A, Canessa C, Cleaveland ES, Aldrich G, Fejes-Toth G: sgk is an aldosterone-induced kinase in the renal collecting duct. Effects on epithelial na+ channels. J Biol Chem. 1999, 274 (24): 16973-16978.
Debonneville C, Flores SY, Kamynina E, Plant PJ, Tauxe C, Thomas MA, Munster C, Chraibi A, Pratt JH, Horisberger JD, et al.: Phosphorylation of Nedd4-2 by Sgk1 regulates epithelial Na(+) channel cell surface expression. Embo J. 2001, 20 (24): 7052-7059.
Snyder PM, Olson DR, Thomas BC: Serum and glucocorticoid-regulated kinase modulates Nedd4-2-mediated inhibition of the epithelial Na+ channel. J Biol Chem. 2002, 277 (1): 5-8.
Kjarland E, Keen TJ, Kleppe R: Does isoform diversity explain functional differences in the 14-3-3 protein family?. Curr Pharm Biotechnol. 2006, 7 (3): 217-223.
Ichimura T, Yamamura H, Sasamoto K, Tominaga Y, Taoka M, Kakiuchi K, Shinkawa T, Takahashi N, Shimada S, Isobe T: 14-3-3 proteins modulate the expression of epithelial Na+ channels by phosphorylation-dependent interaction with Nedd4-2 ubiquitin ligase. J Biol Chem. 2005, 280 (13): 13187-13194.
Bhalla V, Daidie D, Li H, Pao AC, LaGrange LP, Wang J, Vandewalle A, Stockand JD, Staub O, Pearce D: Serum- and glucocorticoid-regulated kinase 1 regulates ubiquitin ligase neural precursor cell-expressed, developmentally down-regulated protein 4-2 by inducing interaction with 14-3-3. Mol Endocrinol. 2005, 19 (12): 3073-3084.
Liang X, Peters KW, Butterworth MB, Frizzell RA: 14-3-3 isoforms are induced by aldosterone and participate in its regulation of epithelial sodium channels. J Biol Chem. 2006, 281 (24): 16323-16332.
Flores SY, Loffing-Cueni D, Kamynina E, Daidie D, Gerbex C, Chabanel S, Dudler J, Loffing J, Staub O: Aldosterone-induced serum and glucocorticoid-induced kinase 1 expression is accompanied by Nedd4-2 phosphorylation and increased Na+ transport in cortical collecting duct cells. J Am Soc Nephrol. 2005, 16 (8): 2279-2287.
Loffing-Cueni D, Flores SY, Sauter D, Daidie D, Siegrist N, Meneton P, Staub O, Loffing J: Dietary sodium intake regulates the ubiquitin-protein ligase nedd4-2 in the renal collecting system. J Am Soc Nephrol. 2006, 17 (5): 1264-1274.
Alvarez de la Rosa D, Zhang P, Naray-Fejes-Toth A, Fejes-Toth G, Canessa CM: The serum and glucocorticoid kinase sgk increases the abundance of epithelial sodium channels in the plasma membrane of Xenopus oocytes. J Biol Chem. 1999, 274 (53): 37834-37839.
Shigaev A, Asher C, Latter H, Garty H, Reuveny E: Regulation of sgk by aldosterone and its effects on the epithelial Na(+) channel. Am J Physiol Renal Physiol. 2000, 278 (4): F613-619.
Auberson M, Hoffmann-Pochon N, Vandewalle A, Kellenberger S, Schild L: Epithelial Na+ channel mutants causing Liddle's syndrome retain ability to respond to aldosterone and vasopressin. Am J Physiol Renal Physiol. 2003, 285 (3): F459-471.
Diakov A, Korbmacher C: A novel pathway of epithelial sodium channel activation involves a serum- and glucocorticoid-inducible kinase consensus motif in the C terminus of the channel's alpha-subunit. J Biol Chem. 2004, 279 (37): 38134-38142.
Dahlmann A, Pradervand S, Hummler E, Rossier BC, Frindt G, Palmer LG: Mineralocorticoid regulation of epithelial Na+ channels is maintained in a mouse model of Liddle's syndrome. Am J Physiol Renal Physiol. 2003, 285 (2): F310-318.
Morris RG, Schafer JA: cAMP Increases Density of ENaC Subunits in the Apical Membrane of MDCK Cells in Direct Proportion to Amiloride-sensitive Na(+) Transport. J Gen Physiol. 2002, 120 (1): 71-85.
Butterworth MB, Edinger RS, Johnson JP, Frizzell RA: Acute ENaC stimulation by cAMP in a kidney cell line is mediated by exocytic insertion from a recycling channel pool. J Gen Physiol. 2005, 125 (1): 81-101.
Snyder PM: Liddle's syndrome mutations disrupt cAMP-mediated translocation of the epithelial Na(+) channel to the cell surface. J Clin Invest. 2000, 105 (1): 45-53.
Snyder PM, Olson DR, Kabra R, Zhou R, Steines JC: cAMP and serum and glucocorticoid-inducible kinase (SGK) regulate the epithelial Na(+) channel through convergent phosphorylation of Nedd4-2. J Biol Chem. 2004, 279 (44): 45753-45758.
Vallet V, Chraibi A, Gaeggeler HP, Horisberger JD, Rossier BC: An epithelial serine protease activates the amiloride-sensitive sodium channel. Nature. 1997, 389 (6651): 607-610.
Hughey RP, Bruns JB, Kinlough CL, Harkleroad KL, Tong Q, Carattino MD, Johnson JP, Stockand JD, Kleyman TR: Epithelial sodium channels are activated by furin-dependent proteolysis. J Biol Chem. 2004, 279 (18): 18111-18114.
Hughey RP, Bruns JB, Kinlough CL, Kleyman TR: Distinct pools of epithelial sodium channels are expressed at the plasma membrane. J Biol Chem. 2004, 279 (47): 48491-48494.
Hughey RP, Mueller GM, Bruns JB, Kinlough CL, Poland PA, Harkleroad KL, Carattino MD, Kleyman TR: Maturation of the epithelial Na+ channel involves proteolytic processing of the alpha- and gamma-subunits. J Biol Chem. 2003, 278 (39): 37073-37082.
Knight KK, Olson DR, Zhou R, Snyder PM: Liddle's syndrome mutations increase Na+ transport through dual effects on epithelial Na+ channel surface expression and proteolytic cleavage. Proc Natl Acad Sci USA. 2006, 103 (8): 2805-2808.
Publication history
Republished from Current BioData's Targeted Proteins database (TPdb; http://www.targetedproteinsdb.com).
Acknowledgements
Work from the author's lab described in this review was supported by grants from the Canadian Institute of Health Research and the Canadian CF Foundation.
This article has been published as part of BMC Biochemistry Volume 9 Supplement 1, 2008: Ubiquitin-Proteasome System in Disease Part 2. The full contents of the supplement are available online at http://www.biomedcentral.com/1471-2091/9?issue=S1.
Additional TPdb reviews on the ubiquitin-proteasome system are also available in BMC Biochemistry – see Volume 8 Suppl 1 http://www.biomedcentral.com/1471-2091/8?issue=S1.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The author declares that they have no competing interests.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an open access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Rotin, D. Role of the UPS in Liddle syndrome. BMC Biochem 9 (Suppl 1), S5 (2008). https://doi.org/10.1186/1471-2091-9-S1-S5
Published:
DOI: https://doi.org/10.1186/1471-2091-9-S1-S5