
Abstract
Abstract
Deubiquitylating enzymes (DUBs) can hydrolyze a peptide, amide, ester or thiolester bond at the C-terminus of UBIQ (ubiquitin), including the post-translationally formed branched peptide bonds in mono- or multi-ubiquitylated conjugates. DUBs thus have the potential to regulate any UBIQ-mediated cellular process, the two best characterized being proteolysis and protein trafficking. Mammals contain some 80–90 DUBs in five different subfamilies, only a handful of which have been characterized with respect to the proteins that they interact with and deubiquitylate. Several other DUBs have been implicated in various disease processes in which they are changed by mutation, have altered expression levels, and/or form part of regulatory complexes. Specific examples of DUB involvement in various diseases are presented. While no specific drugs targeting DUBs have yet been described, sufficient functional and structural information has accumulated in some cases to allow their rapid development.
Publication history
Republished from Current BioData's Targeted Proteins database (TPdb; http://www.targetedproteinsdb.com).
Similar content being viewed by others

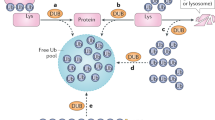
Localization and function
Introduction
In this chapter, the term 'deubiquitylating enzyme' (DUB) is used to describe any enzyme that can hydrolyze a peptide, amide, ester or thiolester bond at the C-terminus of UBIQ (ubiquitin). DUBs can cleave the linear products of UBIQ gene translation [1]; the post-translationally formed branched peptide bonds in mono- or multi-ubiquitylated conjugates [2]; ubiquitylated remnants resulting from proteasome-mediated degradation [3] and other small amide or ester adducts [3, 4]. DUBs thus have the potential to regulate any UBIQ-mediated cellular process, the two best characterized being proteolysis and protein trafficking/endocytosis.
In mammals there are some 80–90 DUBs categorized into five gene families: the ubiquitin C-terminal hydrolases (UCHs); the ubiquitin-specific peptidases (USPs/UBPs); the ovarian tumor (OTU) domain proteins; the Josephin or Machado-Joseph disease (MJD) proteins and the JAMM (Ja b1/M PN domain-associated m etalloisopeptidase) domain proteins. The first four families are cysteine peptidases, while the JAMM proteins are zinc metalloisopeptidases. These DUB families have been the subjects of recent reviews [5–7]. Since linear UBIQ fusion proteins are cleaved very rapidly, perhaps co-translationally [1], it is unclear which DUB(s) cleave these precursors in vivo to provide free UBIQ. However, mutating or inhibiting a DUB(s) that produces free UBIQ would have pleiotropic effects, in that it would deplete free UBIQ levels and inhibit all UBIQ-dependent functions non-specifically. DUBs that cleave branched UBIQ conjugates presumably make up the bulk of the 80–90 DUBs, and provide substrate specificity to deubiquitylation. Most DUBs contain a catalytic domain that has sequence similarity within subfamilies and structural similarity across subfamilies [5–8], and unrelated sequences either N-terminal or C-terminal (or both) to the catalytic domain. These flanking sequences have been shown to mediate substrate binding in a few cases (see DUBs and disease: UBP7/USP7/HAUSP and DUBs and disease: UBP33/USP33/VDU1, UBP20USP20/VDU2) and presumably serve as substrate binding domains in all DUBs. These flanking sequences, along with the catalytic core, could also contribute binding and cleavage specificity for different UBIQ-UBIQ linkages. Some DUBs function at the proteasome to edit and/or remove UBIQ chains; one example that is linked to disease is UBP14 (USP14) (see DUBs and disease: UBP14/USP14).
Since most DUBs have been identified only by means of sequence similarity to catalytic motifs, there is little known functional information on many of these enzymes. However, the relatively few examples where functional insights have been gained (see DUBs and disease) indicate that DUBs can play crucial regulatory roles in the ubiquitin proteasome system (UPS), making them ideal drug target candidates for therapeutic intervention in UPS-related diseases.
DUBs and disease
UBP6/USP6/TRE-17/TRE-2
UBP6 (encoded by USP6/TRE-17), the first DUB to be identified as an oncogene [9, 10], has in recent years been directly linked to human cancers, primarily aneurysmal bone cysts (ABCs), which are locally aggressive bone tumors. ABCs were previously regarded as non-neoplastic, but recent cytogenetic studies have identified clonal rearrangements that often feature chromosome 17p13 – the USP6 or TRE-17 (originally termed TRE-2) locus. There are five known examples of chromosomal rearrangements that have positioned USP6 downstream of a heterologous gene promoter, in turn forcing inappropriate USP6 expression in a bone/mesenchymal context: Osteomodulin; COL1A1 (Collagen 1A1); THRAP3 (TRAP150); CNBP (ZNF9) and CDH11 [11–13]. High-level UBP6 expression was also detected in four other human cancers originating from mesenchymal neoplastic cells in a bone context (one Ewing's sarcoma, two osteoblastomas and one myofibroma), but not in 50 other non-ABC tumors, suggesting that UBP6 could have a broader oncogenic role in mesenchymal tumors [13]. Recent work has also revealed that the USP6 product regulates actin remodeling and vesicular trafficking, and could thus regulate cell motility and invasiveness [14]. In all five cases of UBP6-linked human cancers referred to above, it remains unclear whether the heterologous promoters cause overexpression of normal, full-length UBP6 protein, or whether there have been further mutations, deletions, or alternate splicing within USP6 to produce an altered, oncogenic UBP6 protein.
UBP7/USP7/HAUSP
One well characterized case that illustrates the possible link between DUBs and disease is the mammalian DUB UBP7 (encoded by USP7/HAUSP), a 1102 amino acid member of the USP family. The N-terminal domain of UBP7 (residues 1–208) contains a TRAF (tumor necrosis factor receptor-associated factor) domain [15] that binds to both the P53 (p53) tumor suppressor protein [16, 17] and the MDM2 ubiquitin ligase, which ubiquitylates P53 [18]. The remainder of UBP7 comprises a catalytic core (residues 208–560) that cleaves UBIQ, and a C-terminal domain (residues 560–1102) of unknown function. Since UBP7 can deubiquitylate and stabilize P53 in vitro, it was suggested that the role of UBP7 was to stabilize P53 in vivo [16]. More recent work, however, showed that UBP7 can also deubiquitylate and thus stabilize MDM2 (which autoubiquitylates itself) [19, 20]. UBP7 forms a tighter complex with MDM2 than with P53 [21], consistent with observations that UBP7's primary role could be deubiquitylating and stabilizing MDM2 (and thus increasing ubiquitylation of P53), rather than deubiquitylating and stabilizing P53 [20]. MDM2 is not the only P53 ubiquitin ligase and additional proteins that play a role in P53 ubiquitylation, deubiquitylation and degradation have been recently reviewed [22]. One further member of this complex interplay was recently identified as the protein DAXX (death domain-associated protein). DAXX simultaneously binds to MDM2 and UBP7, and mediates the stabilizing effect of UBP7 on MDM2 [23]. In response to DNA damage, DAXX (and UBP7) dissociate from MDM2, which correlates with MDM2 self-degradation.
Pathogenic mutations within UBP7 itself have not yet been described. However, a recent study that investigated UBP7 expression and TP53 gene status in non-small cell lung carcinomas found that, in 93 of the 131 patients examined, either mutant P53 or reduced UBP7 expression was observed [24]. A statistically significant association between reduced UBP7 levels and reduced P53 protein expression was observed in tumors with wild-type P53, while a more dramatic association was seen in tumors with mutant P53. The authors concluded that the concurrent evaluation of both UBP7 expression and P53 gene status was a significant prognostic indicator in adenocarcinoma patients [24]. MDM2 expression was not investigated in this study though it could be another useful prognostic indicator, given that the protein can regulate P53 levels.
Several disease-causing viruses also target P53 levels by manipulating the interaction between UBP7 and P53. These include the HSV protein ICPO (which is a ubiquitin RING finger ligase that targets several cellular proteins for degradation, including P53 [25]), and the Epstein-Barr nuclear antigen 1 (EBNA1) protein, which displaces UBP7 from P53 [26].
In addition to having multiple targets in the same pathway (i.e. P53, MDM2 and MDM4 [MDMX]), UBP7 has at least one other non-proteolytic target. The transcription factor forkhead box O (FOXO) becomes monoubiquitylated in response to increased cellular oxidative stress, resulting in its re-localization to the nucleus and an increase in its transcriptional activity. UBP7 removes UBIQ from monoubiquitylated FOXO, and negatively regulates FOXO transcriptional activity towards endogenous promoters [27]. Thus, we must keep in mind that DUBs not only regulate protein degradation, but also protein trafficking/localization.
CYLD
Mutations in the cylindromatosis protein (CYLD), a tumor suppressor, are linked to familial cylindromatosis (MIM132700), an autosomal dominant predisposition to multiple neoplasms of skin appendages [28, 29]. The C-terminal 365 amino acids of the 953-residue CYLD protein comprise a variant USP-type DUB catalytic core, and CYLD has been shown to possess deubiquitylating activity both in vitro and in whole cells. This deubiquitylating activity was specific to non-Lys48-linked UBIQ chains [30]. CYLD functions to downregulate NFκB signaling, in which the UPS has several roles [30]. Upon receptor activation, TRAF2, TRAF6 and the NEMO (IKKγ) subunit are polyubiquitylated with Lys63-linked UBIQ chains, which is a necessary process to activate the IKK complex to phosphorylate IκB. IκB in turn becomes polyubiquitylated with Lys48-linked ubiquitin chains and is degraded, releasing NFκB for translocation to the nucleus to enable gene activation [30–32]. CYLD acts to downregulate NFκB signaling by removing these activating Lys63-linked UBIQ chains [30]. Reduced or absent CYLD activity allows prolonged NFκB signaling, increases resistance to apoptosis and hence could lead to tumor formation [31]. CYLD is expressed in a broad range of tissue types, and it remains unclear as to why CYLD mutations only give rise to skin tumors [33].
In a study of 25 cylindromatosis families, 21 had CYLD mutations resulting in truncations or frameshift alterations within the C-terminal two-thirds of the protein that would abolish its deubiquitylating activity, thus indicating a correlation between tumorigenesis and reduced deubiquitylating activity [33]. Recent studies on CYLD-deficient mice demonstrated a deficiency in T-cell development [34], and observed that activation of their immune cells led to increased NFκB signaling, a higher susceptibility to induced colonic inflammation and increased incidence of tumors when compared with controls in a colitis-associated cancer model [35].
TNAP3/A20/TNFAIP3
Another DUB that plays a role in downregulating NFκB signaling is TNAP3 (A20, encoded by TNFAIP), which has an OTU DUB domain. Previous studies of yet another negative regulator of NFκB signaling, OTU7B (Cezanne), determined that the N-terminal OTU domain of this protein conferred its DUB activity [36], which was subsequently shown for TNAP3 [37]. TNA3P can cleave both Lys48- and Lys63-linked UBIQ chains in vitro, but in vivo appears to have specificity for Lys63 chains. However, TNAP3 also possesses a novel zinc finger-type ubiquitin ligase (E3) domain, which can assemble Lys48-linked UBIQ chains in vitro and in transfected cells [37]. Thus, TNAP3 acts as an inhibitor of NFκB signaling by removing the (activating) Lys63 chains on the tumor necrosis factor (TNF) receptor-interacting protein RIP, and then assembling Lys48 linked chains on RIP by virtue of its ubiquitin ligase domain. This results in the degradation of RIP, thus preventing its activation of NFκB via the TNF-mediated pathway [37].
To date, no TNAP3 mutations have been linked to human disease. However, TNAP3-deficient mice develop severe inflammation and cachexia, are hypersensitive to both lipopolysaccharide and TNF, and die prematurely, consistent with failure to terminate TNF-induced NFκB responses [38]. The role of the UBIQ pathway in NFκB signaling has been extensively reviewed [39–41].
UBP33/USP33/VDU1, UBP20/USP20/VDU2 and von Hippel-Lindau disease
Von Hippel-Lindau disease is an autosomal dominant disorder that predisposes affected individuals to a variety of tumors, including hemangioblastomas in the CNS and retina, clear cell carcinomas of the kidney, pheochromocytomas of the adrenal gland, and pancreatic cysts, adenomas and islet cell tumors (reviewed in [42]). UBP33 (encoded by USP33/VDU1) and UBP20 (encoded by USP20/VDU2) are 59% identical USP-type DUBs that interact with the tumor suppressor E3 ubiquitin ligase VHL (pVHL), mutations in which are associated with von Hippel-Lindau disease [43]. UBP33 and UBP20 interact with the β-domain of VHL, leading to their ubiquitylation and degradation by the proteasome [44]. The β-domain of VHL is the region of the protein that harbors the naturally occurring mutations found in von Hippel-Lindau disease. Some of these mutations have been shown to disrupt UBP33/20 interaction with VHL, suggesting an important role for UBP33/20 in this disease [43, 44]. One target of VHL is the α-subunit of the transcription factor hypoxia-inducible factor-1 (HIF-1) [45], which regulates the genes involved in angiogenesis, glucose metabolism, cell proliferation, invasion and metastasis [46]. The inability to degrade HIF1A (HIF-1α) leads to overexpression of HIFA target genes and to a variety of tumors [42]. Recently, it has been shown that UBP20, but not UBP33, interacts with HIFIA and can specifically deubiquitylate and stabilize it, antagonizing VHL-mediated ubiquitylation of the transcription factor [45]. This UBP20/HIF1A/VHL interplay is functionally similar to the UBP7/P53/MDM2 situation described previously (see DUBs and disease: UBP7/USP7/HAUSP).
UBP14
UBP14 (encoded by ubp14/gid6) is a USP-type DUB that is localized to the proteasome by virtue of a ubiquitin-like domain N-terminal to its catalytic core. This DUB has been best studied in yeast, where it was found that UBP14 activity was increased 300-fold upon binding to the proteasome, and it was originally proposed to assist in the release of UBIQ from proteasome-bound multi-ubiquitylated conjugates [47]. In yeast, loss of UBP14 depletes free cellular UBIQ levels due to increased degradation of UBIQ by the proteasome, and renders cells susceptible to stresses that impose extra load on UBIQ-dependent proteolysis [47]. However, more recent studies suggest that UBP14 regulates proteasome activity by actually delaying UBIQ chain removal, and that it is involved in a dynamic remodeling of UBIQ chains at the proteasome in conjunction with a proteasome-bound ubiquitin ligase, HUL5 [48, 49]. In mammals, UBP14 (encoded by USP14) is also associated with the proteasome, and loss of UBP14 in mice also leads to depletion of UBIQ [50], resulting in an ataxia phenotype due to defects in synaptic transmission [51]. However, it is not yet clear whether this is due to a general defect in proteolysis (by analogy to the yeast ubp14 deletion mutant) or to a more specific, perhaps non-proteasomal role for UBP14 in neurons.
UCHL1
Ubiquitin C-terminal hydrolase isozyme L1 (UCHL1) catalyzes the hydrolysis of C-terminal ubiquityl esters and amides, releasing UBIQ from substrates [4]. UCHL1 is a highly abundant neuronal enzyme, comprising up to 2% of total brain protein [52]. Although it has mainly been implicated in deubiquitylation, it has also been shown in vitro to have ubiquitin ligase activity and this activity has been correlated with dimerization of the enzyme. Mutations in the UCHL1 gene have been linked to susceptibility to and protection from Parkinson's disease (PD) [53, 54]. UCHL1 was initially linked to PD in a German family [55] where a point mutation at nucleotide C277G led to the amino acid substitution Ile93Met [56, 57]. The Met93 variant has a severely diminished hydrolase activity and a lower E3 ligase activity compared with wild-type UCHL1. UCHL1 is found in Lewy body protein aggregates associated with PD. These Lewy bodies amass a range of normal and abnormal proteins, many of which are ubiquitylated [58]. However, this polymorphism has only been observed in one PD family. The second polymorphism in UCHL1 results in an amino acid change, Ser18Tyr [59]. This mutation is, however, protective against PD [60] and delays the age of onset of disease. The Tyr18 allele prevents the formation of UCHL1 dimers and therefore lacks ubiquitin ligase activity. In addition, the Tyr18 allele has been shown to increase the hydrolase activity of UCHL1 in vitro [61, 62]. Taken together, these results suggest that increased UCHL1 hydrolase activity is protective against PD, but that the specific substrates of UCHL1 require further investigation.
ATX3
Spinocerebellar ataxia (SCA) type-3 or Machado-Joseph disease (MJD) is a member of the CAG/polyglutamine repeats disease family [63, 64]. Expanded polyglutamine confers a toxic gain of function on the disease protein, presumably through an increased propensity towards aggregation, altered protein expression or both [65]. ATX3 (Ataxin-3), the disease protein associated with SCA3/MJD, is a ubiquitously expressed protein and a member of a novel family of DUBs defined by the Josephin or MJD domain [66–68]. Normally, the protein contains 12–40 glutamines near its C-terminus, whereas in disease the polyglutamine domain expands to ~55–84 glutamines. Expression of mutant ATX3 is widespread, although the neurodegeneration in MJD has been described only in particular regions of the brain such as the cerebellum, substantia nigra and pontine nuclei. It has been proposed that the cellular expression of the disease gene is not in itself sufficient to cause neuronal degeneration, and that other cell-specific factors must be invoked to explain the restricted neuropathology seen in MJD [63].
DUBs linked to other diseases
There are also numerous examples of other DUBs linked to different diseases that have not been covered herein, owing to space constraints. These include (but are not limited to): UBP1 (encoded by USP1), which deubiquitylates a component of the Fanconi anemia DNA repair complex [69]; BAP1, a UCH-type DUB that binds to the BRCA1 breast-cancer susceptibility protein [70]; UBP11 (encoded by USP11), which binds to BRCA2 [71]; UBP4 (encoded by USP4), an oncoprotein linked to lung cancer [72] that interacts with the Retinoblastoma tumor suppressor proteins [73, 74], and also interacts with and deubiquitylates the ubiquitin ligase RO52 [75], an autoantigen associated with the autoimmune disease Sjögren's syndrome [76]; the DUB-1 and DUB-2 family of cytokine-inducible USP-type DUBs, where DUB-2 deubiquitylates the common cytokine receptor subunit gamma(c) [77]; and the UBP2a splice variant of UBP2 (encoded by USP2), which regulates the stability of fatty acid synthase in prostate cancer, protects cells from apoptosis and interacts with MDM2 [78, 79]. Also, while this review has focused on mammalian DUBs, there are also relevant examples from other species, such as the Drosophila DUB fat facets (FAF) and its substrate, the epsin ortholog liquid facets (LQF) [80, 81], which play a clear role in endocytosis and Notch signaling, with implications for human disease.
Disease, mutation, expression, knockout
Earlier biochemical assays of DUBs employed small ubiquitin (UBIQ)-ester adducts [3, 4] and these were followed by gel-based or enzymatic assays based on larger UBIQ protein fusions [6]. More recently, a UBIQ-fluorescent leaving group substrate has been widely used [82, 83]. UBIQ chains of defined lysine linkage have also been developed, mainly through the pioneering work of Cecile Pickart (Johns Hopkins University, Baltimore, USA) [84], and these are useful in determining DUB preference for chain topology.
Recently, RNAi-based knockdown of a large number of DUBs has been successfully employed in cell-based screens aimed at identifying roles for DUBs in regulatory pathways, such as CYLD in NFκB signaling [31] and UBP1 (encoded by USP1) in Fanconi anemia [69]. More recently, mice lacking specific DUBs have been developed, and these will provide useful models for further understanding the roles of DUBs in disease. For example, the CYLD- [34, 35] and TNAP (A20)- [38] deficient mice provide useful models to address the role of these DUBs in inflammatory disease. Similarly, the UBP7 (USP7)-null mouse, which shows increased P53 (p53) levels [20], led investigators to conclude that UBP7's main substrate could be the MDM2 ubiquitin ligase, rather than P53 itself.
In two other cases, mutations in naturally occurring mouse disease models were mapped to DUB genes. The Ataxia mouse, bred for some 50 years by (and available from) the Jackson Laboratories, was found in 2002 to harbor an inactivating insertion in the proteasome-associated DUB UBP14 [51]. While it is clear that the null mutant has a neurological phenotype, it remains unclear whether this is a non-specific (proteasomal) or specific substrate effect. However, the availability of the Usp14 mutant mouse should accelerate discovery in this area.
Secondly, the mutation in the gracile axonal dystrophy (gad) mouse was mapped to a deletion in the Uchl1 gene [85], and subsequent work on this model revealed that UCHL1 regulates the morphology and differentiation of neural progenitor cells [86]. Thus the Uchl1 (gad) mouse provides a useful model for further study.
One other mouse model, the Usp18-null mouse, is useful in the study of interferon signaling and innate immunity against viral and bacterial infection [87]. However, it has not been covered in detail here, because UBP18 cleaves the interferon-stimulated ubiquitin-like protein UCRP (ISG15), rather than UBIQ itself.
Disease targets and ligands
DUBs represent the newest, and least studied, family of enzymes in the ubiquitin proteasome pathway. Potentially, they have the ability to regulate the ubiquitylation status, and thus function, localization, and/or degradation, of any ubiquitylated protein, and therefore should be ideal drug targets for therapeutic intervention. However, there are no specific drugs reported for any DUB to date. Theoretically, there should be two possible routes of intervention: (i) modulation of the DUB's enzymatic activity, or (ii) modulation of the DUB's interaction with its ubiquitylated substrate and/or with the cognate ligase. There are several non-specific inhibitors of whole classes of DUBs, such as ubiquitin-aldehyde [80], which blocks at least the UCH and USP families, as well as other cysteine protease inhibitors. However, these target the catalytic protease domains of DUBs, and would at best have broad specificity. Other screens using branched peptide mimics of UBIQ-UBIQ linkages have also been performed (e.g. [88]); these may allow development of inhibitors of cleavage of specific lysine linkages. Further approaches aim to identify candidate inhibitors of deubiquitylating activity based on the X-ray crystallographic structure of DUB active sites (World Patent WO9901567). It could also be possible to stimulate DUB activity with small molecules that may enhance active site configuration, or that induce DUB gene expression, although such approaches have not been reported.
Given that the interacting regions between DUBs and their substrates (or cognate ubiquitin ligases) are now being defined, it is hoped that more rapid progress towards specific drugs will be made. For example, given the apparent primary role of UBP7 (USP7) in regulating MDM2 levels, and the well-defined binding sites of these proteins [21], UBP7 would be a logical choice for therapeutic intervention. Preventing UBP7-MDM2 interaction would destabilize MDM2, and thus stabilize P53 (p53) (see DUBs and disease: UBP7/USP7/HAUSP). This should have application in any disease where P53 is wild-type, and where stabilizing it would restore normal P53 check-point function. Such possibilities have been theoretically explored as a therapeutic approach to modulate P53 levels in hematopoietic tumors (where P53 is rarely mutated and thus amenable to manipulation [89]), but as yet, no UBP7-targeted drugs have been described. Similarly, once the full in vivo ramifications of the UBP20 (USP20)/HIF1A (HIF-1κ/VHL (pVHL) interplay are understood (see DUBs and disease: UBP33/USP33/VDU1, UBP20/USP20/VDU2 and von Hippel-Lindau disease), modulation of these protein-protein interactions should allow regulation of HIF1A levels in von Hippel-Lindaudisease.
New frontiers in drug discovery
The most important issue to be addressed for most DUBs is to define the proteins that they interact with and deubiquitylate, be they (i) ubiquitylated proteins destined for degradation, trafficking, or some other consequence, or (ii) ubiquitin ligases, as it is becoming apparent that many DUBs interact with, and stabilize, ligases [19, 21, 44, 75, 79]. For the large majority of DUBs, physiological substrates remain unidentified, though it is clear from the examples discussed in this review that many DUBs have clear links to disease. This remains a major barrier to drug development. Current efforts, such as the DUB family-wide RNAi knockdown screens (e.g. [31, 69]), should identify pathways that DUBs regulate, thus allowing focused efforts on substrate identification.
Once these interactions have been defined at the molecular and structural level, they can be exploited as drug targets. The P53 (p53)/MDM2/UBP7 (USP7) example gives a clear indication that we need to understand all aspects of a DUB's role before drugs can be rationally designed. One major unresolved question concerns the numbers of ubiquitin ligases (perhaps 500) compared with the number of DUBs (some 80–90). Does the fewer number of DUBs mean that some substrates do not have a protective DUB, and/or that DUBs are more promiscuous than ligases and can deubiquitylate several substrates, and/or that some ligases do not have a protective DUB? We need to understand the complex interplay between ubiquitin ligases, DUBs and substrates, and whether the DUB's role is to promote degradation, inhibit degradation, or is non-proteolytic, before pharmacological intervention is explored.
Abbreviations
- CYLD:
-
cylindromatosis
- DUB:
-
deubiquitylating enzyme
- EBNA:
-
Epstein-Barr nuclear antigen
- FOXO:
-
forkhead box O
- HAUSP:
-
herpesvirus-associated ubiquitin-specific peptidase
- JAMM:
-
Jab1/MPN domain-associated metalloisopeptidase
- MJD:
-
Machado-Joseph disease
- OUT:
-
ovarian tumor
- PD:
-
Parkinson's disease
- RIP:
-
receptor-interacting protein
- TNF:
-
tumor necrosis factor
- TRAF:
-
tumor necrosis factor receptor-associated factor
- UBP:
-
ubiquitin-processing peptidase
- USP:
-
ubiquitin-specific peptidase
- UCH:
-
ubiquitin C-terminal hydrolase
- VHL:
-
von Hippel-Lindau.
References
Baker RT, Tobias JT, Varshavsky A: Ubiquitin-specific proteases of Saccharomyces cerevisiae. Cloning of UBP2 and UBP3, and functional analysis of the UBP gene family. J Biol Chem. 1992, 267: 23364-23375.
Matsui S, Sandberg AA, Negoro S, Seon BK, Goldstein G: Isopeptidase: a novel eukaryotic enzyme that cleaves isopeptide bonds. Proc Natl Acad Sci USA. 1982, 79: 1535-1539. 10.1073/pnas.79.5.1535.
Pickart CM, Rose IA: Ubiquitin carboxyl-terminal hydrolase acts on ubiquitin carboxyl-terminal amides. J Biol Chem. 1985, 260: 7903-7910.
Mayer AN, Wilkinson KD: Detection, resolution, and nomenclature of multiple ubiquitin carboxyl-terminal esterases from bovine calf thymus. Biochemistry. 1989, 28: 166-172. 10.1021/bi00427a024.
Amerik AY, Hochstrasser M: Mechanism and function of deubiquitinating enzymes. Biochim Biophys Acta. 2004, 1695: 189-207. 10.1016/j.bbamcr.2004.10.003.
Soboleva TA, Baker RT: Deubiquitinating enzymes: their functions and substrate specificity. Curr Protein Pept Sci. 2004, 5: 191-200. 10.2174/1389203043379765.
Nijman SM, Luna-Vargas MP, Velds A, Brummelkamp TR, Dirac AM, Sixma TK, Bernards R: A genomic and functional inventory of deubiquitinating enzymes. Cell. 2005, 123: 773-786. 10.1016/j.cell.2005.11.007.
Wilkinson KD: Regulation of ubiquitin-dependent processes by deubiquitinating enzymes. Faseb J. 1997, 11: 1245-1256.
Onno M, Nakamura T, Mariage-Samson R, Hillova J, Hill M: Human TRE17 oncogene is generated from a family of homologous polymorphic sequences by single-base changes. DNA Cell Biol. 1993, 12: 107-118.
Papa FR, Hochstrasser M: The yeast DOA4 gene encodes a deubiquitinating enzyme related to a product of the human tre-2 oncogene. Nature. 1993, 366: 313-319. 10.1038/366313a0.
Oliveira AM, Hsi BL, Weremowicz S, Rosenberg AE, Dal Cin P, Joseph N, Bridge JA, Perez-Atayde AR, Fletcher JA: USP6 (Tre2) fusion oncogenes in aneurysmal bone cyst. Cancer Res. 2004, 64: 1920-1923. 10.1158/0008-5472.CAN-03-2827.
Oliveira AM, Perez-Atayde AR, Inwards CY, Medeiros F, Derr V, Hsi BL, Gebhardt MC, Rosenberg AE, Fletcher JA: USP6 and CDH11 oncogenes identify the neoplastic cell in primary aneurysmal bone cysts and are absent in so-called secondary aneurysmal bone cysts. Am J Pathol. 2004, 165: 1773-1780.
Oliveira AM, Perez-Atayde AR, Dal Cin P, Gebhardt MC, Chen CJ, Neff JR, Demetri GD, Rosenberg AE, Bridge JA, Fletcher JA: Aneurysmal bone cyst variant translocations upregulate USP6 transcription by promoter swapping with the ZNF9, COL1A1, TRAP150, and OMD genes. Oncogene. 2005, 24: 3419-3426. 10.1038/sj.onc.1208506.
Martinu L, Masuda-Robens JM, Robertson SE, Santy LC, Casanova JE, Chou MM: The TBC (Tre-2/Bub2/Cdc16) domain protein TRE17 regulates plasma membrane-endosomal trafficking through activation of Arf6. Mol Cell Biol. 2004, 24: 9752-9762. 10.1128/MCB.24.22.9752-9762.2004.
Zapata JM, Pawlowski K, Haas E, Ware CF, Godzik A, Reed JC: A diverse family of proteins containing tumor necrosis factor receptor-associated factor domains. J Biol Chem. 2001, 276: 24242-24252. 10.1074/jbc.M100354200.
Li M, Chen D, Shiloh A, Luo J, Nikolaev AY, Qin J, Gu W: Deubiquitination of p53 by HAUSP is an important pathway for p53 stabilization. Nature. 2002, 416: 648-653. 10.1038/nature737.
Hu M, Li P, Li M, Li W, Yao T, Wu JW, Gu W, Cohen RE, Shi Y: Crystal structure of a UBP-family deubiquitinating enzyme in isolation and in complex with ubiquitin aldehyde. Cell. 2002, 111: 1041-1054. 10.1016/S0092-8674(02)01199-6.
Sheng Y, Saridakis V, Sarkari F, Duan S, Wu T, Arrowsmith CH, Frappier L: Molecular recognition of p53 and MDM2 by USP7/HAUSP. Nat Struct Mol Biol. 2006, 13: 285-291. 10.1038/nsmb1067.
Li M, Brooks CL, Kon N, Gu W: A dynamic role of HAUSP in the p53-Mdm2 pathway. Mol Cell. 2004, 13: 879-886. 10.1016/S1097-2765(04)00157-1.
Cummins JM, Vogelstein B: HAUSP is required for p53 destabilization. Cell Cycle. 2004, 3: 689-692.
Hu M, Gu L, Li M, Jeffrey PD, Gu W, Shi Y: Structural basis of competitive recognition of p53 and MDM2 by HAUSP/USP7: implications for the regulation of the p53-MDM2 pathway. PLoS Biol. 2006, 4 (2): e27-10.1371/journal.pbio.0040027.
Brooks CL, Gu W: p53 ubiquitination: Mdm2 and beyond. Mol Cell. 2006, 21: 307-315. 10.1016/j.molcel.2006.01.020.
Tang J, Qu LK, Zhang J, Wang W, Michaelson JS, Degenhardt YY, El-Deiry WS, Yang X: Critical role for Daxx in regulating Mdm2. Nat Cell Biol. 2006, 8: 855-862. 10.1038/ncb1442.
Masuya D, Huang C, Liu D, Nakashima T, Yokomise H, Ueno M, Nakashima N, Sumitomo S: The HAUSP gene plays an important role in non-small cell lung carcinogenesis through p53-dependent pathways. J Pathol. 2006, 208: 724-732. 10.1002/path.1931.
Boutell C, Everett RD: The herpes simplex virus type 1 (HSV-1) regulatory protein ICP0 interacts with and Ubiquitinates p53. J Biol Chem. 2003, 278: 36596-36602. 10.1074/jbc.M300776200.
Saridakis V, Sheng Y, Sarkari F, Holowaty MN, Shire K, Nguyen T, Zhang RG, Liao J, Lee W, Edwards AM, Arrowsmith CH, Frappier L: Structure of the p53 binding domain of HAUSP/USP7 bound to Epstein-Barr nuclear antigen 1 implications for EBV-mediated immortalization. Mol Cell. 2005, 18: 25-36. 10.1016/j.molcel.2005.02.029.
Horst van der A, de Vries-Smits AM, Brenkman AB, van Triest MH, Broek van den N, Colland F, Maurice MM, Burgering BM: FOXO4 transcriptional activity is regulated by monoubiquitination and USP7/HAUSP. Nat Cell Biol. 2006, 8: 1064-1073. 10.1038/ncb1469.
Bignell GR, Warren W, Seal S, Takahashi M, Rapley E, Barfoot R, Green H, Brown C, Biggs PJ, Lakhani SR, et al.: Identification of the familial cylindromatosis tumour-suppressor gene. Nat Genet. 2000, 25: 160-165. 10.1038/76006.
Bowen S, Gill M, Lee DA, Fisher G, Geronemus RG, Vazquez ME, Celebi JT: Mutations in the CYLD gene in Brooke-Spiegler syndrome, familial cylindromatosis, and multiple familial trichoepithelioma: lack of genotype-phenotype correlation. J Invest Dermatol. 2005, 124: 919-920. 10.1111/j.0022-202X.2005.23688.x.
Kovalenko A, Chable-Bessia C, Cantarella G, Israel A, Wallach D, Courtois G: The tumour suppressor CYLD negatively regulates NF-kappaB signalling by deubiquitination. Nature. 2003, 424: 801-805. 10.1038/nature01802.
Brummelkamp TR, Nijman SM, Dirac AM, Bernards R: Loss of the cylindromatosis tumour suppressor inhibits apoptosis by activating NF-kappaB. Nature. 2003, 424: 797-801. 10.1038/nature01811.
Trompouki E, Hatzivassiliou E, Tsichritzis T, Farmer H, Ashworth A, Mosialos G: CYLD is a deubiquitinating enzyme that negatively regulates NF-kappaB activation by TNFR family members. Nature. 2003, 424: 793-796. 10.1038/nature01803.
Ikeda F, Dikic I: CYLD in ubiquitin signaling and tumor pathogenesis. Cell. 2006, 125: 643-645. 10.1016/j.cell.2006.05.003.
Reiley WW, Zhang M, Jin W, Losiewicz M, Donohue KB, Norbury CC, Sun SC: Regulation of T cell development by the deubiquitinating enzyme CYLD. Nat Immunol. 2006, 7: 411-417. 10.1038/ni1315.
Zhang J, Stirling B, Temmerman ST, Ma CA, Fuss IJ, Derry JM, Jain A: Impaired regulation of NF-kappaB and increased susceptibility to colitis-associated tumorigenesis in CYLD-deficient mice. J Clin Invest. 2006, 116: 3042-3049. 10.1172/JCI28746.
Evans PC, Smith TS, Lai MJ, Williams MG, Burke DF, Heyninck K, Kreike MM, Beyaert R, Blundell TL, Kilshaw PJ: A novel type of deubiquitinating enzyme. J Biol Chem. 2003, 278: 23180-23186. 10.1074/jbc.M301863200.
Wertz IE, O'Rourke KM, Zhou H, Eby M, Aravind L, Seshagiri S, Wu P, Wiesmann C, Baker R, Boone DL, Ma A, Koonin EV, Dixit VM: De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature. 2004, 430: 694-699. 10.1038/nature02794.
Lee EG, Boone DL, Chai S, Libby SL, Chien M, Lodolce JP, Ma A: Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science. 2000, 289: 2350-2354. 10.1126/science.289.5488.2350.
Chen ZJ: Ubiquitin signalling in the NF-kappaB pathway. Nat Cell Biol. 2005, 7: 758-765. 10.1038/ncb0805-758.
Krappmann D, Scheidereit C: A pervasive role of ubiquitin conjugation in activation and termination of IkappaB kinase pathways. EMBO Rep. 2005, 6: 321-326. 10.1038/sj.embor.7400380.
Sun L, Chen ZJ: The novel functions of ubiquitination in signaling. Curr Opin Cell Biol. 2004, 16: 119-126. 10.1016/j.ceb.2004.02.005.
Kaelin WG: Molecular basis of the VHL hereditary cancer syndrome. Nat Rev Cancer. 2002, 2: 673-682. 10.1038/nrc885.
Li Z, Na X, Wang D, Schoen SR, Messing EM, Wu G: Ubiquitination of a novel deubiquitinating enzyme requires direct binding to von Hippel-Lindau tumor suppressor protein. J Biol Chem. 2002, 277: 4656-4662. 10.1074/jbc.M108269200.
Li Z, Wang D, Na X, Schoen SR, Messing EM, Wu G: Identification of a deubiquitinating enzyme subfamily as substrates of the von Hippel-Lindau tumor suppressor. Biochem Biophys Res Commun. 2002, 294: 700-709. 10.1016/S0006-291X(02)00534-X.
Li Z, Wang D, Messing EM, Wu G: VHL protein-interacting deubiquitinating enzyme 2 deubiquitinates and stabilizes HIF-1alpha. EMBO Rep. 2005, 6: 373-378. 10.1038/sj.embor.7400377.
Semenza GL: Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003, 3: 721-732. 10.1038/nrc1187.
Leggett DS, Hanna J, Borodovsky A, Crosas B, Schmidt M, Baker RT, Walz T, Ploegh H, Finley D: Multiple associated proteins regulate proteasome structure and function. Mol Cell. 2002, 10: 495-507. 10.1016/S1097-2765(02)00638-X.
Hanna J, Hathaway NA, Tone Y, Crosas B, Elsasser S, Kirkpatrick DS, Leggett DS, Gygi SP, King RW, Finley D: Deubiquitinating enzyme Ubp6 functions noncatalytically to delay proteasomal degradation. Cell. 2006, 127: 99-111. 10.1016/j.cell.2006.07.038.
Crosas B, Hanna J, Kirkpatrick DS, Zhang DP, Tone Y, Hathaway NA, Buecker C, Leggett DS, Schmidt M, King RW, Gygi SP, Finley D: Ubiquitin chains are remodeled at the proteasome by opposing ubiquitin ligase and deubiquitinating activities. Cell. 2006, 127: 1401-1413. 10.1016/j.cell.2006.09.051.
Anderson C, Crimmins S, Wilson JA, Korbel GA, Ploegh HL, Wilson SM: Loss of Usp14 results in reduced levels of ubiquitin in ataxia mice. J Neurochem. 2005, 95: 724-731. 10.1111/j.1471-4159.2005.03409.x.
Wilson SM, Bhattacharyya B, Rachel RA, Coppola V, Tessarollo L, Householder DB, Fletcher CF, Miller RJ, Copeland NG, Jenkins NA: Synaptic defects in ataxia mice result from a mutation in Usp14, encoding a ubiquitin-specific protease. Nat Genet. 2002, 32: 420-425. 10.1038/ng1006.
Jackson P, Thompson RJ: The demonstration of new human brain-specific proteins by high-resolution two-dimensional polyacrylamide gel electrophoresis. J Neurol Sci. 1981, 49: 429-438. 10.1016/0022-510X(81)90032-0.
Ardley HC, Scott GB, Rose SA, Tan NG, Robinson PA: UCH-L1 aggresome formation in response to proteasome impairment indicates a role in inclusion formation in Parkinson's disease. J Neurochem. 2004, 90: 379-391. 10.1111/j.1471-4159.2004.02485.x.
Das C, Hoang QQ, Kreinbring CA, Luchansky SJ, Meray RK, Ray SS, Lansbury PT, Ringe D, Petsko GA: Structural basis for conformational plasticity of the Parkinson's disease-associated ubiquitin hydrolase UCH-L1. Proc Natl Acad Sci USA. 2006, 103: 4675-4680. 10.1073/pnas.0510403103.
Leroy E, Boyer R, Auburger G, Leube B, Ulm G, Mezey E, Harta G, Brownstein MJ, Jonnalagada S, Chernova T, et al.: The ubiquitin pathway in Parkinson's disease. Nature. 1998, 395: 451-452. 10.1038/26652.
McNaught KS, Jenner P: Proteasomal function is impaired in substantia nigra in Parkinson's disease. Neurosci Lett. 2001, 297: 191-194. 10.1016/S0304-3940(00)01701-8.
McNaught KS, Perl DP, Brownell AL, Olanow CW: Systemic exposure to proteasome inhibitors causes a progressive model of Parkinson's disease. Ann Neurol. 2004, 56: 149-162. 10.1002/ana.20186.
Lowe J, McDermott H, Landon M, Mayer RJ, Wilkinson KD: Ubiquitin carboxyl-terminal hydrolase (PGP 9.5) is selectively present in ubiquitinated inclusion bodies characteristic of human neurodegenerative diseases. J Pathol. 1990, 161: 153-160. 10.1002/path.1711610210.
Elbaz A, Levecque C, Clavel J, Vidal JS, Richard F, Correze JR, Delemotte B, Amouyel P, Alperovitch A, Chartier-Harlin MC, et al.: S18Y polymorphism in the UCH-L1 gene and Parkinson's disease: evidence for an age-dependent relationship. Mov Disord. 2003, 18: 130-137. 10.1002/mds.10326.
Lincoln S, Vaughan J, Wood N, Baker M, Adamson J, Gwinn-Hardy K, Lynch T, Hardy J, Farrer M: Low frequency of pathogenic mutations in the ubiquitin carboxy-terminal hydrolase gene in familial Parkinson's disease. Neuroreport. 1999, 10: 427-429. 10.1097/00001756-199902050-00040.
Maraganore DM, Farrer MJ, Hardy JA, Lincoln SJ, McDonnell SK, Rocca WA: Case-control study of the ubiquitin carboxy-terminal hydrolase L1 gene in Parkinson's disease. Neurology. 1999, 53: 1858-1860.
Maraganore DM, Lesnick TG, Elbaz A, Chartier-Harlin MC, Gasser T, Kruger R, Hattori N, Mellick GD, Quattrone A, Satoh J, et al.: UCHL1 is a Parkinson's disease susceptibility gene. Ann Neurol. 2004, 55: 512-521. 10.1002/ana.20017.
Colomer Gould VF: Mouse models of Machado-Joseph disease and other polyglutamine spinocerebellar ataxias. NeuroRx. 2005, 2: 480-483. 10.1602/neurorx.2.3.480.
Perez MK, Paulson HL, Pittman RN: Ataxin-3 with an altered conformation that exposes the polyglutamine domain is associated with the nuclear matrix. Hum Mol Genet. 1999, 8: 2377-2385. 10.1093/hmg/8.13.2377.
Paulson HL, Das SS, Crino PB, Perez MK, Patel SC, Gotsdiner D, Fischbeck KH, Pittman RN: Machado-Joseph disease gene product is a cytoplasmic protein widely expressed in brain. Ann Neurol. 1997, 41: 453-462. 10.1002/ana.410410408.
Fujigasaki H, Uchihara T, Koyano S, Iwabuchi K, Yagishita S, Makifuchi T, Nakamura A, Ishida K, Toru S, Hirai S, et al.: Ataxin-3 is translocated into the nucleus for the formation of intranuclear inclusions in normal and Machado-Joseph disease brains. Exp Neurol. 2000, 165: 248-256. 10.1006/exnr.2000.7479.
Burnett B, Li F, Pittman RN: The polyglutamine neurodegenerative protein ataxin-3 binds polyubiquitylated proteins and has ubiquitin protease activity. Hum Mol Genet. 2003, 12: 3195-3205. 10.1093/hmg/ddg344.
Chow MK, Mackay JP, Whisstock JC, Scanlon MJ, Bottomley SP: Structural and functional analysis of the Josephin domain of the polyglutamine protein ataxin-3. Biochem Biophys Res Commun. 2004, 322: 387-394. 10.1016/j.bbrc.2004.07.131.
Nijman SM, Huang TT, Dirac AM, Brummelkamp TR, Kerkhoven RM, D'Andrea AD, Bernards R: The deubiquitinating enzyme USP1 regulates the Fanconi anemia pathway. Mol Cell. 2005, 17: 331-339. 10.1016/j.molcel.2005.01.008.
Jensen DE, Proctor M, Marquis ST, Gardner HP, Ha SI, Chodosh LA, Ishov AM, Tommerup N, Vissing H, Sekido Y, et al.: BAP1: a novel ubiquitin hydrolase which binds to the BRCA1 RING finger and enhances BRCA1-mediated cell growth suppression. Oncogene. 1998, 16: 1097-1112. 10.1038/sj.onc.1201861.
Schoenfeld AR, Apgar S, Dolios G, Wang R, Aaronson SA: BRCA2 is ubiquitinated in vivo and interacts with USP11, a deubiquitinating enzyme that exhibits prosurvival function in the cellular response to DNA damage. Mol Cell Biol. 2004, 24: 7444-7455. 10.1128/MCB.24.17.7444-7455.2004.
Gray DA, Inazawa J, Gupta K, Wong A, Ueda R, Takahashi T: Elevated expression of Unph, a proto-oncogene at 3p21.3, in human lung tumors. Oncogene. 1995, 10: 2179-2183.
Blanchette P, Gilchrist CA, Baker RT, Gray DA: Association of UNP, a ubiquitin-specific protease, with the pocket proteins pRb, p107 and p130. Oncogene. 2001, 20: 5533-5537. 10.1038/sj.onc.1204823.
DeSalle LM, Latres E, Lin D, Graner E, Montagnoli A, Baker RT, Pagano M, Loda M: The de-ubiquitinating enzyme Unp interacts with the retinoblastoma protein. Oncogene. 2001, 20: 5538-5542. 10.1038/sj.onc.1204824.
Wada K, Kamitani T: UnpEL/Usp4 is ubiquitinated by Ro52 and deubiquitinated by itself. Biochem Biophys Res Commun. 2006, 342: 253-258. 10.1016/j.bbrc.2006.01.144.
Di Donato F, Chan EK, Askanase AD, Miranda-Carus M, Buyon JP: Interaction between 52 kDa SSA/Ro and deubiquitinating enzyme UnpEL: a clue to function. Int J Biochem Cell Biol. 2001, 33: 924-934. 10.1016/S1357-2725(01)00055-3.
Gesbert F, Malarde V, Dautry-Varsat A: Ubiquitination of the common cytokine receptor gammac and regulation of expression by an ubiquitination/deubiquitination machinery. Biochem Biophys Res Commun. 2005, 334: 474-480. 10.1016/j.bbrc.2005.06.121.
Graner E, Tang D, Rossi S, Baron A, Migita T, Weinstein LJ, Lechpammer M, Huesken D, Zimmermann J, Signoretti S, Loda M: The isopeptidase USP2a regulates the stability of fatty acid synthase in prostate cancer. Cancer Cell. 2004, 5: 253-261. 10.1016/S1535-6108(04)00055-8.
Priolo C, Tang D, Brahamandan M, Benassi B, Sicinska E, Ogino S, Farsetti A, Porrello A, Finn S, Zimmermann J, Febbo P, Loda M: The isopeptidase USP2a protects human prostate cancer from apoptosis. Cancer Res. 2006, 66: 8625-8632. 10.1158/0008-5472.CAN-06-1374.
Huang Y, Baker RT, Fischer-Vize JA: Control of cell fate by a deubiquitinating enzyme encoded by the fat facets gene. Science. 1995, 270: 1828-1831. 10.1126/science.270.5243.1828.
Chen X, Zhang B, Fischer JA: A specific protein substrate for a deubiquitinating enzyme: Liquid facets is the substrate of Fat facets. Genes Dev. 2002, 16: 289-294. 10.1101/gad.961502.
Dang LC, Melandri FD, Stein RL: Kinetic and mechanistic studies on the hydrolysis of ubiquitin C-terminal 7-amido-4-methylcoumarin by deubiquitinating enzymes. Biochemistry. 1998, 37: 1868-1879. 10.1021/bi9723360.
Russell NS, Wilkinson KD: Deubiquitinating enzyme purification, assay inhibitors, and characterization. Methods Mol Biol. 2005, 301: 207-219.
Pickart CM, Raasi S: Controlled synthesis of polyubiquitin chains. Methods Enzymol. 2005, 399: 21-36. 10.1016/S0076-6879(05)99002-2.
Saigoh K, Wang YL, Suh JG, Yamanishi T, Sakai Y, Kiyosawa H, Harada T, Ichihara N, Wakana S, Kikuchi T, Wada K: Intragenic deletion in the gene encoding ubiquitin carboxy-terminal hydrolase in gad mice. Nat Genet. 1999, 23: 47-51.
Sakurai M, Ayukawa K, Setsuie R, Nishikawa K, Hara Y, Ohashi H, Nishimoto M, Abe T, Kudo Y, Sekiguchi M, Sato Y, Aoki S, Noda M, Wada K: Ubiquitin C-terminal hydrolase L1 regulates the morphology of neural progenitor cells and modulates their differentiation. J Cell Sci. 2006, 119: 162-71. 10.1242/jcs.02716.
Ritchie KJ, Hahn CS, Kim KI, Yan M, Rosario D, Li L, de la Torre JC, Zhang DE: Role of ISG15 protease UBP43 (USP18) in innate immunity to viral infection. Nat Med. 2004, 10: 1374-1378. 10.1038/nm1133.
Mason DE, Ek J, Peters EC, Harris JL: Substrate profiling of deubiquitin hydrolases with a positional scanning library and mass spectrometry. Biochemistry. 2004, 43: 6535-6544. 10.1021/bi049722j.
Cheon KW, Baek KH: HAUSP as a therapeutic target for hematopoietic tumors (review). Int J Oncol. 2006, 28: 1209-1215.
Publication history
Republished from Current BioData's Targeted Proteins database (TPdb; http://www.targetedproteinsdb.com).
Acknowledgements
This article has been published as part of BMC Biochemistry Volume 9 Supplement 1, 2008: Ubiquitin-Proteasome System in Disease Part 2. The full contents of the supplement are available online at http://www.biomedcentral.com/1471-2091/9?issue=S1.
Additional TPdb reviews on the ubiquitin-proteasome system are also available in BMC Biochemistry – see Volume 8 Suppl 1 http://www.biomedcentral.com/1471-2091/8?issue=S1.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Singhal, S., Taylor, M.C. & Baker, R.T. Deubiquitylating enzymes and disease. BMC Biochem 9 (Suppl 1), S3 (2008). https://doi.org/10.1186/1471-2091-9-S1-S3
Published:
DOI: https://doi.org/10.1186/1471-2091-9-S1-S3