
Abstract
A functional ubiquitin proteasome system is essential for all eukaryotic cells and therefore any alteration to its components has potential pathological consequences. Though the exact underlying mechanism is unclear, an age-related decrease in proteasome activity weakens cellular capacity to remove oxidatively modified proteins and favours the development of neurodegenerative and cardiac diseases. Up-regulation of proteasome activity is characteristic of muscle wasting conditions including sepsis, cachexia and uraemia, but may not be rate limiting. Meanwhile, enhanced presence of immunoproteasomes in aging brain and muscle tissue could reflect a persistent inflammatory defence and anti-stress mechanism, whereas in cancer cells, their down-regulation reflects a means by which to escape immune surveillance. Hence, induction of apoptosis by synthetic proteasome inhibitors is a potential treatment strategy for cancer, whereas for other diseases such as neurodegeneration, the use of proteasome-activating or -modulating compounds could be more effective.
Publication history: Republished from Current BioData's Targeted Proteins database (TPdb; http://www.targetedproteinsdb.com).
Similar content being viewed by others


Proteasome localization and function
As proteins play crucial roles in virtually all biological processes, the finely tuned equilibrium between their synthesis and degradation influences cellular homoeostasis. Protein degradation is predominantly catalysed by the proteasome, a giant protein breakdown enzyme complex, any disturbance to which can result in the onset of disease processes. The present review briefly summarizes some of the major aspects relating to the role of proteasomes in disease.
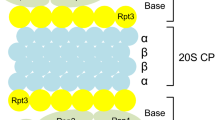
The majority of cellular proteins are degraded by the ubiquitin proteasome system (UPS), which consists of both substrate-recruiting and substrate-degrading machinery. The former is composed of three enzymes, the first of which (E1) activates the polypeptide ubiquitin in an ATP-dependent manner, enabling its transfer onto a ubiquitin carrier enzyme (E2). Activated ubiquitin is further transferred by a ubiquitin protein ligase (E3) to a substrate protein [1]. The substrate-recruiting machinery then catalyses the formation of an isopeptide bond between the C-terminal glycine residue of ubiquitin and the ε-amino group of a substrate protein lysine residue. Repeated addition of ubiquitin moieties onto the first results in a polyubiquitylated substrate protein that is recognized by the proteolytic machinery of the UPS, the 26S proteasome [2]. The 26S proteasome contains a central, barrel-like core particle (the 20S proteasome) composed of four stacked seven-membered rings, with the subunit stoichiometry α1-7β1-7β1-7α1-7[3]. A six-membered ring of AAA ATPase proteins binds to one or both outer α-rings and, together with two non-ATPase subunits, forms the base, while nine other subunits comprise the adjoining lid. In turn, the base and lid comprise the 19S regulator complex (19S REG) [4–6], which functions in the recognition of ubiquitylated substrates and their subsequent binding [7], de-ubiquitylation [8, 9], unfolding and transfer into the central chamber of the 20S proteasome [10, 11]. Within the 20S proteasome, subunits β1, β2, and β5 of both adjacent β-rings expose their proteolytically active sites, exhibiting post-glutamyl peptide hydrolysing (PGPH), trypsin-like and chymotrypsin-like cleavage specificity, respectively [12, 13]. Under conditions of acute immune or stress response, these three β subunits can be substituted during de novo proteasome biosynthesis for the interferon-γ-inducible subunits β1i, β2i, and β5i. This results in the replacement of standard 20S proteasomes with immunoproteasomes, which have different cleavage specificities to those described above [14, 15]. Replacement of only one or two of the active site-containing β subunits results in the formation of intermediate type proteasomes [16]. Alternatively, the proteasome activator PA28 can associate with the 20S proteasome in place of 19S REG [1].
In mammalian cells, proteasomes are located throughout the cytoplasm though most highly concentrated at the centrosome [18]. By contrast, immunoproteasomes specifically concentrate at the endoplasmic reticulum [19]. A nuclear localization signal directs proteasomes to enter the cell nucleus [20, 21] (particularly after induction of cell stress [22]), in which they accumulate in focal subdomains [23]. Circulating 20S proteasomes (probably released from (dying) cells) have been found in human plasma and could potentially be used as diagnostic markers [24–26]. In yeast cells, the majority of proteasomes have been detected in the cell nucleus [27].
Whereas polyubiquitylation of proteins labels them for degradation by proteasomes, monoubiquitylation confers a cellular localization signal that, for example, directs them from the plasma membrane to endosomes and multivesicular bodies. The monoubiquitin localization signal also plays a role in regulating protein activity and function [28, 29].
The role of the proteasome in disease
By catalysing limited or complete degradation of proteins, the UPS functions in many basic cellular processes such as differentiation [30], proliferation [31], apoptosis [31], gene transcription [32], signal transduction [33], metabolic regulation [34], immune surveillance [35] and many others [36]. Thus, the UPS is essential for development and maintenance of all eukaryotic cells [37]. This biological importance implies that the UPS is also inevitably involved in patho-physiological processes resulting in the development of many diseases, including autoimmune, neurodegenerative and rheumatoid diseases, cancer, viral infections and cachexia (Table 1). Many of these pathological states are caused by defects in the E2 and E3 enzymes or by genomic or post-translational alterations to proteins that affect their ubiquitylation and subsequent susceptibility to proteasomal degradation. This in turn leads to disruptions in biochemical reaction sequences. Numerous reviews have summarized the UPS-associated patho-biochemical mechanisms of these disorders [e.g. [38–43].
The present paper summarizes knowledge of the changes in proteasomes that lead to the triggering and development of various diseases.
Changes in proteasome activity – a cause of disease development?
In comparison to most other cellular proteins, the concentration of proteasomes is very high, ranging from 1 to 20 µg/mg of soluble protein [44]. As for all cellular proteins, proteasomes have a limited lifetime and thus are continuously synthesized and degraded. In HeLa cells, the half-life of standard- and immuno-proteasomes is around 5 days and 27 hours, respectively [14].
NLVS (4-hydroxy-5-iodo-3-nitrophenylacetyl-Ala-Ala-Phe-vinyl sulfone) is a compound that covalently binds the threonine residues of all three active site-containing β subunits, thus significantly reducing cellular proteasome activity. When mice thymoma and lymphoma cells are incubated with this compound, the residual proteasome activity (perhaps complemented by other proteases) ensures their survival. However, functions such as MHC class I antigen processing and presentation are clearly impaired by NLVS treatment [45]. This indicates that cells contain no real surplus of proteasome activity and that any change in the function of the complex could affect cellular homoeostasis. This may not necessarily compromise cell survival, but may be important enough to induce pathological consequences, for example, through impaired activation of the transcription factor NFκB, which mediates cellular responses to the many signals received from outside [46].
Decreased proteasomal activity and disease
An age-related decrease in proteasome activity has been observed in different tissues including bovine eye lens [47], rat liver [48, 49], human skin and epidermal cells [50, 51], rat heart [52], human lymphocytes [53], human fibroblasts [54], rat kidney [55], rat lung [55] and rat muscle [56, 57]. In all of these investigations, proteasome activity was measured using fluorigenic peptide substrates and PGPH activity was found to be consistently depressed with increasing age. Only in rat liver extracts [48] was an increase in chymotrypsin- and trypsin-like activity observed (when measured in the presence of SDS) with increasing age. However, when specific activity was measured in purified liver 20S proteasomes, a loss of PGPH activity was observed with no change in chymotrypsin- and trypsin-like activity [49]. Therefore, results obtained by use of tissue extracts [48, 50, 52] may not reflect the intrinsic activity of proteasomes, but in fact be influenced by other components within the homogenate, for example proteasome activator PA28 or 19S REG [57]. Alternatively, decreased activity could be due to reduced amounts of proteasome, as observed in aging keratinocytes [50], but not in liver tissue of aged rats when compared with young rats [48]. However, when the specific activity of 20S proteasomes purified from rat liver [49], rat heart [52], human epidermis [51] and bovine lens [47] was calculated, a loss of PGPH activity [49] or PGPH and chymotrypsin-like activity [51], or PGPH and trypsin-like activity [47, 52] was detected, supporting the link between intrinsic changes in 20S proteasomes and increasing age. In some of the studies mentioned [49–52], a rise in the concentration of oxidatively modified proteins was detected in aged donor tissue samples. Since oxidatively modified proteins have been found to be substrates of 20S proteasomes [58], all of these investigations have been confined to 20S proteasomes. The concentration of ubiquitylated proteins was also found to be increased in aged tissue and cells [52, 53], suggesting that the activity of 26S proteasomes could be affected by aging in addition to 20S proteasomes. Specifically, this was shown in human lymphocytes of elderly donors [53], as well as in late passages of a human fibroblast cell line, which both exhibited a significant reduction in 26S proteasome activity [54]. Accordingly, a cell model for studying aging processes has been developed by Chondrogianni and Gonos (Institute of Biological Research and Biotechnology, Athens, Greece) that uses human embryonic lung fibroblasts in which a senescence-like phenotype is induced by treatment with low doses of commercially available proteasome inhibitors (epoxomicin or MG132) [59]. Epoxomicin, a microbial epoxyketone, is known to react irreversibly with all three active sites but primarily with the β5 subunit, which exhibits chymotrypsin-like activity [60]. Though less specific, MG132, a peptide aldehyde (Z-L-leucyl-L-leucyl-L-leucinal), is probably the most widely used proteasome inhibitor. It reacts reversibly, primarily with proteasome subunit β5 [61].
As aging is a progressive and irreversible (but not pathological) phenomenon, a decline in proteasome activity may be regarded as the natural answer to an age-related decrease in the rate of protein synthesis, a process counterbalanced by protein degradation. Therefore, it is no surprise that experimental inhibition of proteasome activity in primary neuronal cells, for example, induces impairments in protein synthesis [62], especially since many transcription factors and some translation-initiation factors, as well as ribosome biogenesis, are regulated by proteasomal processing [63–65]. However, as previously mentioned, proteasome activity may also be necessary to remove products of other processes that become imbalanced during aging, such as the antioxidant response system. For example, a lowered capacity to eliminate oxidatively damaged proteins due to a decrease in all three proteasome peptidase activities in eye lens nuclei favours cataract formation in elderly individuals [66] and, in heart muscle, reduces tolerance of the aged heart to ischemia/reperfusion [67]. Experimental thoracic aortic constriction was also found to depress proteasome activity in mouse heart and thus favours accumulation of pro-apoptotic proteins that later induce apoptosis and cardiac dysfunction [68].
Using fluorigenic peptide substrates, an age-related decrease in 20S proteasome activity has been detected in certain areas of rat, mice and marmoset brain. These especially include the substantia nigra, striatum, cerebral cortex and spinal cord but not cerebellum [55, 69, 70], although statistical significance of the difference in activity could depend on the interval of age chosen for the investigations [71]. A life-long steady decrease in proteasome activity is proposed to be responsible for accumulation of abnormally folded proteins, formation of inclusion bodies and development of neurodegenerative diseases such as Alzheimer's and Parkinson's disease, amyotrophic lateral sclerosis and Huntington's disease [72–76].
Thus, age-related proteasomal dysfunction could be regarded as a factor in these disease processes, which involve the formation of plaques, filaments and aggregates. Once generated, these protein inclusions have been found to further inhibit proteasome activity and thus amplify the formation of inclusion bodies [77–80]. Additionally, the age-related loss of antioxidant capacity affects the proteasomal system, particularly the 26S proteasome, which appears to be itself sensitive to oxidative inactivation [81]. The latter finding has been tested by measuring proteasome activity in the presence and absence of ATP in neuronal cells exposed to non-toxic doses of hydrogen peroxide (H2O2) [82]. In either case, residual proteasomal capacity appears unable to eliminate the aggregated and highly oxidized proteins [83], leading to irreversible development of neurodegenerative diseases.
Which mechanisms underlie the age-related attenuation of proteasome activity?
Investigations into the mechanisms underlying the change in proteasome activity during aging are usually performed by comparing cells and tissues from individuals (mostly rats) of different ages, since experimental models mimicking the aging process are difficult to obtain. In particular, the effect of aging on proteasome activity cannot be mimicked by proteasome knockout models, as they are non-viable [37].
Therefore, data summarized in this review were obtained from studies utilising many different tissues from various species. In most investigations into age-dependent changes in proteasome activity, a decrease in PGPH activity was detected, while changes in chymotrypsin-like and trypsin-like activity were not as consistent. Thus, a general decrease in 20S/26S proteasome concentration, as found in aging keratinocytes [50] and in a human fibroblast cell line that was experimentally induced to senescence-like phenotype by incubation with low dose proteasome inhibitors [54], cannot be the general underlying mechanism. Certainly, a lower concentration of subunit α4 mRNA was measured in mesencephalon of adult rats when compared with young, suggesting an age-dependent decreased expression of proteasomes [84]. However, if this were the case, a general decrease in all three proteasome activities catalyzed by subunits β1 (PGPH-activity, β2 (trypsin-like activity) and β5 (chymotrypsin-like activity) should be observed during aging.
Glycation and/or conjugation with the lipid peroxidation product 4-hydroxy-2-nonenal of several α and β subunits (including active site-containing β subunits) was found to coincide with a decrease in chymotrypsin-like activity of 26S proteasomes in lymphocytes from healthy 50–63 year old human donors when compared with 20–35 year old donors [53]. Only two subunits from the 19S REG base were glycated without an effect on the stability and abundance of 26S proteasomes [53]. This observation corroborates the finding that aging does not coincide with an enhanced dissociation of 26S proteasomes into core 20S particles and 19S regulators [54]. Similarly, modifications by 4-hydroxy-2-nonenal, a peroxidation product derived from oxidized ω-6 polyunsaturated fatty acids [85], were found to affect neuronal proteasomes [86–88], as well as myocardial proteasomes after coronary occlusion/reperfusion [89, 90].
The chaperone Hsp90 was shown to protect 20S proteasomes from oxidative inactivation in rat hepatoma cells treated with iron/ascorbate [91]. Interestingly, 20S proteasomes were found to be associated with Hsp90 when purified from bovine lens of one month old but not of two year old animals [47]. This suggests an age-dependent loss of the protective agent in bovine lens, thus favouring oxidation of proteins including proteasomes [92], leading to age-related cataract formation [93]. Similar to the proteasome, Hsp90 is essential for cell survival and therefore this protecting function cannot be tested with Hsp90 knockout cells.
Complex alterations in the proteasome system were found in sarcopenia of rat soleus muscle. Specifically, the concentration of 20S proteasomes in the muscle of old rats (29–40 months) was three-fold higher, but of significantly lower specific activity towards fluorigenic peptide substrates, when compared with young rats (5–12 months) [94]. This loss of activity may in part be due to oxidative modification because it was partly rescued by addition of the reducing agent dithiothreitol (DTT) to the test system [95]. The three-fold increase in 20S proteasome concentration led to increased ratios of 20S proteasomes to both PA28 proteasome activator and 19S REG in aged rats when compared with young rats. Additionally, the interferon-γ-inducible proteasome subunits β1i and β5i were detected in 3–6 fold higher amounts in aged muscle compared with young muscle. Interestingly, high expression of β1i and β5i was also found in neurons, astrocytes and endothelial cells of the hippocampus region of elderly humans (mean age 70 years), but only scarcely in that of young donors (mean age 42 years) [96]. These data suggest that during aging, intermediate-type proteasomes and immunoproteasomes may accumulate in muscle and brain, tissues that normally predominantly contain standard proteasomes.
Enhanced proteasome activity and disease
Muscle atrophy due to decreased protein synthesis and accelerated protein degradation is a hallmark of many patho-physiological situations, examples of which are chronic kidney diseases, type I diabetes mellitus, sepsis, cancer cachexia and starvation. Increased expression of proteasomes in muscle tissue has been observed in rats suffering from NH4Cl-induced metabolic acidosis [97], tumors [98], starvation, denervation atrophy [99], sepsis induced by cecal ligation and puncture [100] and other catabolic conditions [reviewed in [101]], as shown by enhanced transcription of genes encoding proteasome subunits and enhanced proteasomal activity towards fluorigenic substrate proteins [100]. mRNA levels of the 19S REG subunit Rpt1 were also found to be increased in starving rat skeletal muscle; however, knowledge of the adaptive mechanisms of this 26S proteasome sub-complex to catabolic situations is still very limited [102]. Accelerated muscle protein breakdown in burn-injured rats can be inhibited by the proteasome inhibitor lactacystin and also by the less specific but commercially available reversible proteasome inhibitor LLnL (N-acetyl-L-leucyl-L-leucyl-L-norleucinal) [103], which binds to all three active sites with different affinities [104]. The same inhibitors [105], as well as the commercially available proteasome inhibitor PSI (N-benzyloxycarbonyl-Ile-Glu-(O-t-butyl)-Ala-leucinal), which blocks the chymotrypsin-like activity of proteasomes [106], prevent the sepsis-induced increase in protein degradation in rat skeletal muscle [107]. These data clearly demonstrate that proteasomes are responsible for enhanced muscle protein degradation under catabolic conditions [108, 109]. However, proteasomes appear not to be the initiating enzymes under these conditions, since they are not able to degrade contractile proteins and their regulators while they are assembled within the myofibrils that constitute the majority of muscle proteins [110]. Rather, the protease caspase-3, the activity and amount of which is increased in rat skeletal muscle under experimental diabetes mellitus, appears to catalyse the disassembly of myofibrils [111, 112]. This leads to the delivery of substrates to muscle-specific E3 ubiquitin protein ligases (such as atrogin-1 and MuRF1), which prepare muscle proteins for subsequent proteasomal degradation [113]. Using microarray hybridization, the mRNAs of both E3 ligases (similar to that of several proteasome subunits) were found to be expressed at a significantly higher level in mice and rat skeletal muscle during fasting, tumor bearing, chronic renal failure and experimentally induced diabetes mellitus [114]. This suggests that in some cases, compounds inhibiting caspase-3 and E3 enzymes could be more useful in the clinical management of these muscle wasting conditions than proteasome inhibitors. However, a diminution of proteasomal activity could still be useful, for example in preventing degradation of IκB and thus activation of NFκB, since this transcription factor appears to be involved in the induction of muscle atrophy [115] by possibly up-regulating MuRF1 [116]. These findings corroborate the data referred to earlier, which showed that treatment of rats with proteasome inhibitors suppresses their muscle atrophy induced by sepsis or burn injury [103, 105, 106].
A complex role for the proteasome in apoptosis is supported by the finding that inhibition of proteasome activity has pro- as well as anti-apoptotic effects [31]. The activation of NFκB by proteasomes (through degradation of IκB) induces the expression of anti-apoptotic members of the BCL2 family that maintain the mitochondrial membrane barrier [117]. Additionally, proteasomes degrade pro-apoptotic proteins such as Bax and Bid [118, 119], the tumor suppressor p53 [120] and the negative cell cycle regulators p21 (Cip1) and p27 (Kip1) [reviewed in [121]]. All of these anti-apoptotic proteasomal effects are predominantly found in neoplastic and rapidly growing cells. Therefore, a main treatment strategy for multiple myeloma involves the induction of apoptosis through the introduction of proteasome inhibitors such as bortezomib and NPI-0052 into these cells [122, 123].
Bortezomib (formally known as MLN341, PS-341, LDP-341) is a dipeptide boronate (pyrazylcarbonyl-phenylalanyl-leucyl-boronate) originally developed at ProScript and now distributed by Millennium Pharmaceuticals. It binds to the N-terminal threonine hydroxyl group of the active site-containing β5 subunit in a similar manner to peptide aldehyde inhibitors but with a slower dissociation rate and higher specificity, since no protease other than the proteasome was found to be inhibited in rat [124]. As this proteasome inhibitor was found to be active against a broad range of tumours, it entered clinical trials and is now used as Velcade® in clinical oncology for treatment of multiple myeloma. NPI-0052 (Salinsporamide-A) is a non-peptide proteasome inhibitor from the marine actinomycete Salinospora containing a β-lacton structure similar to that of lactacystin. It was found to irreversibly inhibit all three proteasomal peptide cleaving activities. This compound is being developed by Nereus Pharmaceuticals and is currently in Phase I clinical trials for the patients resistant to Velcade® treatment [125].
Referring back to the complex role of proteasomes in apoptosis, a clear pro-apoptotic proteasomal activity was observed in human umbilical vein endothelial cells (HUVEC), primary thymocytes and neurons. Specifically, BCL2 and inhibitors of apoptosis proteins (IAP) were shown to be degraded by proteasomes, resulting in a stimulation of apoptosis that could be prevented by treatment of these cells with the proteasome inhibitors MG132 (Z-L-leucyl-L-leucyl-L-leucinal), ALLN (Acetyl-L-Leucyl-L-Leucyl-L-Norleucinal) or lactacystin [126–128]. MG132 is more potent and more selective than ALLN, which was originally used as an inhibitor of calpains, a heterogenous family of Ca-dependent cysteine proteinases [129]. These data indicate that therapeutic use of proteasome inhibitors must therefore be based on a clear knowledge of the underlying molecular mechanism of the disease.
Furthermore, elevated proteolytic and ATPase activity due to increased expression of 20S proteasome and 19S REG ATPase subunits was found to occur during developmentally regulated and endogenously triggered apoptosis [130–132].
Immunoproteasomes and disease
Transcription of the gene encoding the immunoproteasome subunit β1i requires binding of the transcription factors Stat-1 and IRF-1 to the partially overlapping interferon-consensus-sequence-2/γ-interferon-activated sequence (ICS2/GAS) in its promoter region. Due to the presence of interferon-stimulated response elements, transcription and expression of β1i (as well as β5i) is induced by γ-interferon. However, cells professionally involved in immune surveillance, such as splenocytes and dendritic cells, constitutively express immunoproteasomes due to binding of unphosphorylated and non-dimerized Stat-1 to ICS2/GAS [133]. If, as mentioned above, brain and muscle tissue show an age-related increase in the concentration of immunoproteasome subunits, it could reflect a state of constant inflammation or cell stress. Thus, increased concentration of immunoproteasomes in muscle of patients suffering from myofibrillar myopathy and inclusion body myositis [134], as well as in neurons of a mouse model of Huntington's disease [135], can be regarded as a consequence of, rather than the cause of, these diseases. Alternatively, this increased concentration could result from a potential defence mechanism, since induction of immunoproteasome subunits was recently shown to occur in endothelial cells by nitric oxide (NO) via cGMP/cAMP-mediated mechanisms. The NO-induced synthesis of immunoproteasomes protected the cells against transferrin iron-induced oxidative stress by regulating the level of transferrin receptor [136]. Since NO regulates processes such as vasodilatation, neurotransmittance, the immune response and apoptosis, an imbalance in this mediator has many pathological consequences. An NO-dependent change in the ratio of standard- to immuno-proteasomes is thought to contribute to these consequences [137–140]. Similarly, treatment of SH-SY5Y neural cells with non-toxic doses (1–10 µM) of H2O2 induced not only the formation of oxidized proteins but also synthesis of immunoproteasome subunits (detected by western blotting and real-time RT-PCR) [82], indicating the sensitivity of the proteasome system to react and to cope with cell stress. Such an adaptation of the proteasome system was found to be lost in senescent human fibroblasts, which displayed a decreased concentration in standard proteasome subunits but retention in the immunoproteasome subunits β1i, β2i and β5i. In contrast to confluent young fibroblasts, the concentration of immunoproteasome subunits could not be augmented by treatment with γ-interferon [141].
The adverse process, namely down-regulation of immunoproteasomes, has been found to serve as an immune surveillance escape mechanism in several tumor cells [142–145], since generation of certain MHC class I-presented antigenic epitopes appears less efficient when performed by standard proteasomes compared with immunoproteasomes. Another successful oncogenic mechanism relying on insufficient antigen processing was observed in human cervical carcinoma and melanoma cells that express a non-functional variant of the immunoproteasome subunit β5i, designated E1. In contrast to its functional counterpart, designated β5i-E2, variant E1 cannot be recruited and incorporated by the proteasome maturation factor, POMP, into nascent 20S proteasomes [146]. This defect prevents formation of immunoproteasomes and suppresses the generation of epitopes that elicit a cytotoxic immune response. It is therefore not surprising that the human immunodeficiency virus (HIV) interferes with the antigen processing/presentation machinery of its host cell by means of competition between HIV-tat protein and Stat-1 for binding to IRF-1, thus suppressing synthesis of β2i and functional immunoproteasomes [147].
Disease targets and ligands
The three different proteolytically active sites of the 20S proteasome use the same catalytic mechanism in which the N-terminal threonine residue is the active nucleophile. Therefore, many proteasome inhibitors that modify this threonine residue, for example aldehydes [148], epoxyketones [149], vinyl sulfones [150], lactacystin [151] and boronates [152], affect all three peptide bond cleaving activities, though to differing extents due to different binding affinities within the substrate binding pockets and mostly with a preference for chymotrypsin-like activity [153]. By using more ‘site-specific’ inhibitors, Kisselev et al.[154] found that the relative importance of each active site for the degradative process depends on the individual substrate. Thus, site-specific inhibitors, as long as they are membrane permeable, could be helpful in treating malignant neoplastic diseases other than multiple myeloma, since proteasome-dependent processes such as neovascularization, cell adhesion and intravasation might be more sensitive to this type of inhibitor. Site-specific proteasome inhibitors could also be especially useful as immunosuppressive agents in autoimmune diseases by modulating antigen processing and MHC class I-restricted antigen presentation, and in retroviral infections [155, 156]. For similar purposes, compounds that specifically inhibit immunoproteasomes are conceivable [157].
In addition to the ability to permeate biological membranes, proteasome-inhibiting compounds should also exhibit low toxicity. A lead compound for the development of non-toxic compounds could be the naturally existing, commercially available proteasome inhibitor PR39, a 39 amino acid peptide rich in proline and arginine residues with antimicrobial activity. This peptide was shown to allosterically inhibit the chymotrypsin- and caspase-like activities of 20S proteasomes. In addition, the N-terminal 11mer fragment of PR39, known as PR11, was also shown to display inhibitory activity [158]. PR39 attenuates post-ischemic microvascular injuries and inflammatory reactions [159, 160] and appears to bind to the outer α-ring of 20S proteasomes [161].
Originally, proteasome inhibitors such as bortezomib (at the time still designated MG341) were also tested in animal models for treatment of acute and chronic inflammatory reactions, for example delayed type hypersensitivity and arthritis. Oral doses of the inhibitor effectively cured or even suppressed allergic and inflammatory reactions [162]. As proteasomes are involved in key signalling pathways regulating inflammation and sepsis [163, 164], the design of new proteasome inhibitors aimed at these important targets would be useful in the treatment of related diseases.
In contrast to the use of proteasome inhibitors, there are many pathological situations characterized by a depressed proteasomal proteolytic activity that could be ameliorated by proteasome-activating compounds, an area of the field that has received little attention to-date [165]. Although the exact mechanism of the age-related decrease in proteasome activity is unknown, proteasome-activating hydrophobic peptides have been found that most probably bind as modifiers at non-catalytic sites, thus mimicking the effect of the proteasome activator PA28 by opening the gated α-ring pore [166]. These peptides and other compounds that were shown to activate 20S proteasomes such as fatty acids, phosphatidyl-glycerol and di-phosphatidyl-glycerol [167], could be used as starting points for generating proteasome activating compounds.
Next Frontiers
Although a great deal has been published about changes in proteasome activity and their possible involvement in the development of diseases, there are clear gaps in our knowledge of the definite molecular mechanisms that underlie these alterations. This holds especially true with regard to the fact that proteasomes in various cells and tissues are not a multitude of identical 20S proteasomes but a mixture of several proteasome subpopulations (standard- and immuno-proteasomes) and intermediates of each subpopulation [16, 168], which have different substrate specificities [16]. The distribution within the different cell compartments of these subpopulations and the proportions of the various proteasome complexes, for example 26S proteasomes, hybrid proteasomes and proteasome–PA28 complexes are only scarcely known. Additionally, knowledge of the molecular differences between the various proteasome subtypes, which appear to have different susceptibilities to inhibitors [A. Kloß and B. Dahlmann, unpublished observation] is only superficial [169]. These aspects, along with post-translational modifications such as phosphorylation, O-GlcNAc addition and others [170, 171] that affect proteasome activity, need to be investigated intensively with regard to disease-related alterations to the proteasome system. Since the proteasome is considered to be a therapeutic target, this knowledge will also help to design more specific and probably more effective compounds that modulate its activity.
It is clear that modulation of proteasome activity is primarily aimed at inhibiting the complex's proteolytic functions. However, the finding that the association between 19S REG and 20S proteasomes is regulated by a proteasomal ATPase-associated factor provides another promising aspect to explore in the search for compounds that modulate proteasome activity [172]. To mimic or inhibit the activity of this factor might help to tune the activity of 20S/26S proteasomes without risking a complete shut off of this essential system. Similarly, compounds modulating the ATPase activity of the 19S regulator, as well as its polyubiquitin-binding ability, could be considered for therapeutic use.
A completely different aspect for future research with regard to the benefit of proteasome inhibitors is based on the finding that inhibition of proteasomal activity induces de novo synthesis of proteasomes [173]. This knowledge may be applicable to situations where disease processes appear to be based on reduced amounts of, or defects in, proteasomes, for example as occur during aging. Similarly, treatment of endothelial cells with low, non-toxic doses of the proteasome inhibitor MG132 was found to activate an antioxidant defence programme that included up-regulation of endothelial nitric oxide synthase (eNOS), glutathione peroxidase-3, glutathione S-transferase and others, resulting in an improvement in endothelial functions [174, 175]. Recognition of this effect is not only promising in the search for treatments for patients suffering from atherosclerosis and coronary heart diseases, but also to provide a possible measure for preventing these diseases and others such as neurodegeneration [176]. Based on the same biochemical background, vascular endothelial dysfunction and neuronal death after embolic stroke were found to be restricted and terminable in animal models after proteasome inhibitors such as bortezomib [177, 178] or MLN519 [179] were administered within a certain therapeutic window of a few hours post-stroke. MLN519 ((1R-[1S,4R,5S])-1-(1hydroxy-2-methylpropyl-6-oxa-2-azabi-cyclo [3.2.1] heptane-3,7-dione) is a synthetic analog of lactacystin developed by Millennium Pharmaceuticals and already tested in phase I clinical trials [180]. An exact knowledge of the susceptibility of the different forms of proteasomes to this and other inhibitors will help to develop optimal treatments and therapies for a wide range of diseases.
Finally, aside from proteasome inhibitors or activators, proteasomes per se could be useful as diagnostic and even prognostic markers, since the level of circulating proteasomes reflects the state of health of patients suffering from cancer and autoimmune diseases [181].
Abbreviations
- Hsp90:
-
heat shock protein 90
- HIV:
-
human immunodeficiency virus
- HTLV:
-
human T-cell leukaemia virus
- PGPH:
-
post-glutamyl peptide hydrolysing
- Ub:
-
ubiquitin
- UPS:
-
ubiquitin proteasome system
- 19S REG:
-
19S regulator.
References
Hershko A, Ciechanover A: The ubiquitin system. Annu Rev Biochem. 1998, 67: 425-479. 10.1146/annurev.biochem.67.1.425.
Hough R, Pratt G, Rechsteiner M: Ubiquitin-lysozyme conjugates. Identification and characterization of an ATP-dependent protease from rabbit reticulocytes lysates. J Biol Chem. 1986, 261: 2400-2408.
Groll M, Ditzel L, Löwe J, Stock D, Bochtler M, Bartunik HD, Huber R: Structure of 20S proteasome from yeast at 2.4 Å resolution. Nature. 1997, 386: 463-471. 10.1038/386463a0.
Peters J-M, Harris JR, Kleinschmidt JA: Ultrastructure of the ~26S complex containing the ~20S cylinder particle (multicatalytic proteinase/proteasome). Eur J Cell Biol. 1991, 56: 422-432.
Walz J, Erdmann A, Kania M, Typke D, Koster AJ, Baumeister W: 26S proteasome structure revealed by three-dimensional electron microscopy. J Struct Biol. 1998, 121: 19-29. 10.1006/jsbi.1998.3958.
Glickman MH, Rubin DM, Coux O, Wefes I, Pfeifer G, Cjeka Z, Baumeister W, Fried VA, Finley D: A subcomplex of the regulatory particle required for ubiquitin-conjugate degradation and related to the COP9-signalosome and eIF3. Cell. 1998, 94: 615-623. 10.1016/S0092-8674(00)81603-7.
Verma R, Oania R, Graumann J, Deshaies RJ: Multiubiquitin chain receptors define a layer of substrate selectivity in the ubiquitin-proteasome-system. Cell. 2004, 118: 99-110. 10.1016/j.cell.2004.06.014.
Yao T, Cohen RE: A cryptic protease couples deubiquitination and degradation by the proteasome. Nature. 2002, 419: 403-407. 10.1038/nature01071.
Guterman A, Glickman MH: Complementary roles for RPN11 and Ubp6 in deubiquitination and proteolysis by the proteasomes. J Biol Chem. 2004, 279: 1729-1738. 10.1074/jbc.M307050200.
Navon A, Goldberg AL: Proteins are unfolded on the surface of the ATPase ring before transport into the proteasomes. Mol Cell. 2001, 8: 1339-1349. 10.1016/S1097-2765(01)00407-5.
Smith DM, Kafri G, Cheng Y, Ng D, Walz T, Goldberg AL: ATP binding to PAN or the 26S ATPases causes association with the 20S proteasomes, gate opening, and translocation of unfolded proteins. Mol Cell. 2005, 20: 687-698. 10.1016/j.molcel.2005.10.019.
Dick TP, Nussbaum AK, Deeg M, Heinemeyer W, Groll M, Schirle M, Keilholz W, Stevanovic S, Wolf DH, Huber R, Rammensee HG, Schild H: Contribution of proteasomal β-subunits to the cleavage of peptide substrates analysed with yeast mutants. J Biol Chem. 1998, 273: 25637-26646. 10.1074/jbc.273.40.25637.
Groll M, Clausen T: Molecular shredders: how proteasomes fulfil their role. Curr Opin Stuct Biol. 2003, 13: 665-673. 10.1016/j.sbi.2003.10.005.
Heink S, Ludwig D, Kloetzel P-M, Krüger E: IFN-γ-induced immune adaptation of the proteasomes system is an accelerated and transient response. Proc Natl Acad Sci USA. 2005, 102: 9241-9246. 10.1073/pnas.0501711102.
Kloetzel P-M, Ossendorp F: Proteasome and peptidase function in MHC-class-I-mediated antigen presentation. Curr Opin Immunol. 2004, 16: 76-81. 10.1016/j.coi.2003.11.004.
Dahlmann B, Ruppert T, Kuehn L, Merforth S, Kloetzel P-M: Different proteasome subtypes in a single tissue exhibit different enzymatic properties. J Mol Biol. 2000, 303: 643-53. 10.1006/jmbi.2000.4185.
Rechsteiner M, Hill CP: Mobilizing the proteolytic machine: cell biological roles of proteasome activators and inhibitors. Trends Cell Biol. 2005, 15: 27-33. 10.1016/j.tcb.2004.11.003.
Wigley WC, Fabunmi RP, Lee MG, Marino CR, Muallem S, DeMartino GN, Thomas PJ: Dynamic association of proteasomal machinery with the centrosome. J Cell Biol. 1999, 145: 481-490. 10.1083/jcb.145.3.481.
Brooks P, Murray RZ, Mason GGF, Hendil KB, Rivett AJ: Association of immunoproteasomes with the endoplasmic reticulum. Biochem J. 2000, 352: 611-615. 10.1042/0264-6021:3520611.
Tanaka K, Yoshimura T, Tamura T, Fujiwara T, Kumatori A, Ichihara A: Possible mechanism of nuclear translocation of proteasomes. FEBS-Lett. 1990, 271: 41-46. 10.1016/0014-5793(90)80367-R.
Reits EAJ, Benham AM, Plougastel B, Neefjes J, Trowsdale J: Dynamics of proteasome distribution in living cells. EMBO J. 1997, 16: 6087-6094. 10.1093/emboj/16.20.6087.
Ogiso Y, Tomida A, Tsuruo T: Nuclear localization of proteasomes participates in stress-inducible resistance of solid tumor cells to topoisomerase II-directed drugs. Cancer Res. 2002, 62: 5008-5012.
von Mikecz A: The nuclear ubiquitin-proteasome system (nUPS). J Cell Sci. 2006, 119: 1977-1984. 10.1242/jcs.03008.
Wada M, Kosoka M, Saito S, Sano T, Tanaka K, Ichihara A: Serum concentration and localization in tumor cells of proteasomes in patients with hematologic malignancy and their pathophysiologic significance. J Lab Clin Med. 1993, 121: 215-223.
Lavabre-Bertrand T, Henry L, Carillo S, Guiraud I, Ouali A, Dutaud D, Aubry L, Rossi J-F, Bureau JP: Plasma proteasome level is a potential marker in patients with solid tumors and hemopoietic malignancies. Cancer. 2001, 92: 2493-2500. 10.1002/1097-0142(20011115)92:10<2493::AID-CNCR1599>3.0.CO;2-F.
Egerer K, Kuckelkorn U, Rudolph PE, RÜckert JC, Dörner T, Burmester G-R, Kloetzel P-M, Feist E: Circulating proteasomes are markers of cell damage and immunologic activity in autoimmune diseases. J Rheumatol. 2002, 29: 2045-2052.
Russell SJ, Steger KA, Johnston SA: Subcellular localization, stoichiomety, and protein levels of 26S proteasomes subunits in yeast. J Biol Chem. 1999, 274: 21943-21952. 10.1074/jbc.274.31.21943.
Aguilar RC, Wendland B: Ubiquitin: not just for proteasomes any more. Curr Opin Cell Biol. 2003, 15: 184-190. 10.1016/S0955-0674(03)00010-3.
Schnell JD, Hicke L: Non-traditional functions of ubiquitin and ubiquitin-binding proteins. J Biol Chem. 2003, 278: 35857-35860. 10.1074/jbc.R300018200.
Bowerman B, Kurz T: Degrade to create: developmental requirements for ubiquitin-mediated proteolysis during early C.elegans embryogenesis. Development. 2006, 133: 773-784. 10.1242/dev.02276.
Naujokat C, Hoffmann S: Role and function of the 26S proteasome in proliferation and apoptosis. Lab Invest. 2002, 82: 965-980.
Collins GA, Tansey WP: The proteasome: a utility tool for transcription?. Curr Opin Genet Dev. 2006, 16: 197-202. 10.1016/j.gde.2006.02.009.
Taylor C, Jobin C: Ubiquitin protein modification and signal transduction: Implications for inflammatory bowel diseases. Inflamm Bowel Dis. 2005, 11: 1097-1107. 10.1097/01.MIB.0000187577.26043.e5.
Asher G, Bercovich Z, Tsvetkov P, Shaul Y, Kahana C: 20S proteasomal degradation of ornithine decarboxylase is regulated by NQO1. Mol Cell. 2005, 17: 645-655. 10.1016/j.molcel.2005.01.020.
Strehl B, Seifert U, Krüger E, Heink S, Kuckelkorn U, Kloetzel PM: Interferon-γ, the functional plasticity of the ubiquitin-proteasome system, and MHC class I antigen processing. Immunol Rev. 2005, 207: 19-30. 10.1111/j.0105-2896.2005.00308.x.
Sakai N., Sawada MT, Sawada H: Non-traditional roles of ubiquitin-proteasome system in fertilization and gametogenesis. Int J Biochem Cell Biol. 2004, 36: 776-784. 10.1016/S1357-2725(03)00263-2.
Heinemeyer W, Kleinschmidt JA, Saidowsky J, Escher C, Wolf DH: Proteinase yscE, the yeast proteasome/multicatalytic-multifunctional proteinase: Mutants unravel its function in stress proteolysis and uncover its necessity for cell survival. EMBO J. 1991, 10: 555-562.
Muller S, Schwartz LM: Ubiquitin in homeostasis, development and disease. BioEssays. 1995, 17: 677-684. 10.1002/bies.950170804.
Schwartz AL, Ciechanover A: The ubiquitin-proteasome pathway and pathogenesis of human diseases. Annu Rev.Med. 1999, 50: 57-74. 10.1146/annurev.med.50.1.57.
Ciechanover A, Schwarz AL: Ubiquitin-mediated degradation of cellular proteins in health and disease. Hepatology. 2002, 35: 3-6. 10.1053/jhep.2002.30316.
Sakamoto KL: Ubiquitin-dependent proteolysis: its role in human diseases and the design of therapeutic strategies. Mol Genet Metab. 2002, 77: 44-56. 10.1016/S1096-7192(02)00146-4.
Wong BR, Parlati F, Qu K, Demo S, Pray T, Huang J, Payan DG, Bennett MK: Drug discovery in the ubiquitin regulatory pathway. Drug Discovery Today. 2003, 8: 746-754. 10.1016/S1359-6446(03)02780-6.
Ciechanover A: The ubiquitin proteolytic system. From an idea to the patient bed. Proc Am Thorac Soc. 2006, 3: 21-31. 10.1513/pats.200510-106JH.
Kuehn L, Dahlmann B, Reinauer H: Tissue distribution of the multicatalytic proteinase in rat: An immunological and enzymic study. Cienc Biol (Portugal). 1986, 11: 101-107.
Kessler B, Hong X, Petrovic J, Borodovsky A, Dantuma NP, Bogyo M, Overkleeft HS, Ploegh H, Glas R: Pathways accessory to proteasomal proteolysis are less efficient in major histocompatibility complex class I antigen production. J Biol Chem. 2003, 278: 10013-10021. 10.1074/jbc.M211221200.
Celec P: Nuclear factor kappa B-molecular biomedicine: the next generation. Biomed Pharmacother. 2004, 58: 365-371. 10.1016/j.biopha.2003.12.015.
Wagner BJ, Margolis JW: Age-dependent association of isolated bovine lens multicatalytic proteinase complex (proteasome) with heat shock protein 90, an endogenous inhibitor. Arch Biochem Biophys. 1995, 323: 455-462. 10.1006/abbi.1995.0067.
Shibatani T, Nazir M, Ward WF: Alteration of rat liver 20S proteasome activities by age and food restriction. J Gerontol A Biol Sci Med Sci. 1996, 51: B316-B322.
Conconi M, Szweda LI, Levine RL, Stadtman ER, Friguet B: Age-related decline of rat liver multicatalytic proteinase activity and protection from oxidative inactivation by heat-shock protein 90. Arch Biochem Biophys. 1996, 331: 232-240. 10.1006/abbi.1996.0303.
Petropoulos I, Conconi M, Friguet B: Increase of oxidatively modified protein is associated with a decrease of proteasome activity and content in aging epidermal cells. J Gerontol A Biol Sci Med Sci. 2000, 55: B220-B227.
Bulteau AL, Petropoulos I, Friguet B: Age-related alterations of proteasome structure and function in aging epidermis. Exp Gerontol. 2000, 35: 767-777. 10.1016/S0531-5565(00)00136-4.
Bulteau AL, Szweda LI, Friguet B: Age-dependent declines in proteasome activity in the heart. Arch Biochem Biophys. 2002, 397: 298-304. 10.1006/abbi.2001.2663.
Carrard G, Dieu M, Raes M, Toussaint O, Friguet B: Impact of ageing on human proteasome structure and function in human lymphocytes. Int J Biochem Cell Biol. 2003, 35: 728-739. 10.1016/S1357-2725(02)00356-4.
Chondrogianni N, Stratford FLL, Trougakos IP, Friguet B, Rivett AJ, Gonos ES: Central role of the proteasome in senescence and survival of human fibroblasts. J Biol Chem. 2003, 278: 28026-28037. 10.1074/jbc.M301048200.
Keller JN, Hanni KB, Markesbery WR: Possible involvement of proteasome inhibition in aging: implications for oxidative stress. Mech Ageing Dev. 2000, 113: 61-70. 10.1016/S0047-6374(99)00101-3.
Bardag-Gorce F, Farout L, Veyrat-Durebex C, Briand Y, Briand M: Changes in 20S proteasome activity during aging of the LOU rat. Mol Biol Rep. 1999, 26: 89-93. 10.1023/A:1006968208077.
Husom AD, Peters EA, Kolling EA, Fugere NA, Thompson LV, Ferrington DA: Altered proteasome function and subunit composition in aged muscle. Arch Biochem Biophys. 2004, 421: 67-76. 10.1016/j.abb.2003.10.010.
Davies KJA: Degradation of oxidized proteins by the 20S proteasome. Biochimie. 2001, 83: 301-310. 10.1016/S0300-9084(01)01250-0.
Chondrogianni N, Gonos E: Proteasome inhibition induces senescence-like phenotype in primary human fibroblasts cultures. Biogerontoloy. 2004, 5: 55-61. 10.1023/B:BGEN.0000017687.55667.42.
Elofsson M, Splittgerber U, Myung J, Mohan R, Crews CM: Towards subunit-specific proteasome inhibitors: synthesis and evaluation of peptid α', β'-epoxyketones. Chem Biol. 1999, 6: 811-822. 10.1016/S1074-5521(99)80128-8.
Myung J, Kim KB, Crews CM: The ubiquitin-proteasome pathway and proteasome inhibitors. Med Research Rev. 2001, 21: 245-273. 10.1002/med.1009.
Ding Q, Dimayuga E, Markesbery WR, Keller JN: Proteasome inhibition induces reversible impairments in protein synthesis. FASEB J. 2006, 20: 1055-1063. 10.1096/fj.05-5495com.
Kodadek T, Sikder D, Nalley K: Keeping transcriptional activators under control. Cell. 2006, 127: 261-264. 10.1016/j.cell.2006.10.002.
Vabulas RM: Proteasome function and protein biosynthesis. Curr Opin Clin Nutr Metab Care. 2007, 10: 24-31.
Stavreva DA, Kawasaki M, Dundr M, Koberna K, Mülller WG, Tsujimura-Takahashi T, Komatsu W, Hayano T, Isobe T, Raska I, et al.: Potential roles for ubiquitin and the proteasome during ribosome biogenesis. Mol Cell Biol. 2006, 26: 5131-5145. 10.1128/MCB.02227-05.
Zetterberg M, Petersen A, Sjostrand J, Karlsson J: Proteasome activity in human lens nuclei and correlation with age, gender and severity of cataract. Curr Eye Res. 2003, 27: 45-53. 10.1076/ceyr.27.2.45.15457.
Divald A, Powell SR: Proteasome mediates removal of proteins oxidized during myocardial ischemia. Free Radic Biol Med. 2006, 40: 156-164. 10.1016/j.freeradbiomed.2005.09.022.
Tsukamoto O, Minamino T, Okada K, Shintani Y, Takashima S, Kato H, Liao Y, Okazaki H, Asai M, Hirata A, et al.: Depression of proteasomes activities during the progression of cardiac dysfunction in pressure-overloaded heart of mice. Biochem Biophys Res Commun. 2006, 340: 125-1133. 10.1016/j.bbrc.2005.12.120.
Keller JN, Huang FF, Markesbery WR: Decreased levels of proteasome activity and proteasome expression in aging spinal cord. Neurosci. 2000, 98: 149-156. 10.1016/S0306-4522(00)00067-1.
Zeng BY, Medhurst AD, Jackson M, Rose S, Jenner P: Proteasomal activity in brain differs between species and brain regions and changes with age. Mech Ageing Dev. 2005, 126: 760-766. 10.1016/j.mad.2005.01.008.
Abd El Mohsen MM, Iravani MM, Spencer JPE, Rose S, Fahim AT, Motawi TMK, Ismail NAF, Jenner P: Age-associated changes in protein oxidation and proteasome activities in rat brain: Modulation by antioxidants. Biochem Biophys Res Commun. 2005, 336: 386-391. 10.1016/j.bbrc.2005.07.201.
Keller JN, Hanni KB, Markesbery WR: Impaired proteasome function in Alzheimer's disease. J Neurochem. 2000, 75: 436-439. 10.1046/j.1471-4159.2000.0750436.x.
McNaught KSP, Jenner P: Proteasomal function is impaired in substantia nigra in Parkinson's disease. Neurosci Lett. 2001, 297: 191-194. 10.1016/S0304-3940(00)01701-8.
Rideout HJ, Larsen KE, Sulzer D, Stefanis L: Proteasomal inhibition leads to formation of ubiquitin/α-synuclein-immunoreactive inclusions in PC12 cells. J Neurochem. 2001, 78: 899-908. 10.1046/j.1471-4159.2001.00474.x.
Kabashi E, Agar JN, Taylor DM, Minotti S, Durham HD: Focal dysfunction of the proteasome: a pathogenic factor in a mouse model of amyotrophic lateral sclerosis. J Neurochem. 2004, 89: 1325-1335. 10.1111/j.1471-4159.2004.02453.x.
Zhou H, Cao F, Wang Z, Yu ZX, Nguyen HP, Evans J, Li SH, Li XJ: Huntingtin forms toxic NH2-terminal fragment complexes that are promoted by the age-dependent decrease in proteasome activity. J Cell Biol. 2003, 163: 109-118. 10.1083/jcb.200306038.
Oh S, Hong HS, Hwang E, Sim HJ, Lee W, Shin SJ, Mook-Jung I: Amyloid peptide attenuates the proteasome activity in neuronal cells. Mech Aging Dev. 2005, 126: 1292-1299. 10.1016/j.mad.2005.07.006.
Keck S, Nitsch R, Grune T, Ullrich O: Proteasome inhibition by paired helical filament-tau in brains of patients with Alzheimer's disease. J Neurochem. 2003, 85: 115-122.
Lindersson E, Beedholm R, Højrup P, Moos T, Gai W, Hendil KB, Jensen PH: Proteasomal inhibition by α-synuclein filaments and oligomers. J Biol Chem. 2004, 279: 12924-12934. 10.1074/jbc.M306390200.
Bence NF, Sampat RM, Kopito RR: Impairment of the ubiquitin-proteasome system by protein aggregation. Science. 2001, 292: 1552-1555. 10.1126/science.292.5521.1552.
Reinheckel T, Sitte N, Ullrich O, Kuckelkorn U, Davies KJ, Grune T: Comparative resistance of the 20S and 26S proteasome to oxidative stress. Biochem J. 1998, 335: 637-642.
Ding Q, Reinacker K, Dimayuga E, Nukula V, Drake J, Butterfield DA, Dunn JC, Martin S, Bruce-Keller AJ, Keller JN: Role of the proteasome in protein oxidation and neural viability following low-level oxidative stress. FEBS-Lett. 2003, 546: 228-232. 10.1016/S0014-5793(03)00582-9.
Grune T, Jung T, Merker K, Davies KJA: Decreased proteolysis caused by protein aggregates, inclusion bodies, plaques, lipofuscin, ceroid, and ‘aggresomes’ during oxidative stress, aging, and disease. Int J Biochem Cell Biol. 2004, 36: 2519-2530. 10.1016/j.biocel.2004.04.020.
El-Khodor BF, Kholodilov NG, Yarygina O, Burke RE: The expression of mRNAs for the proteasome complex is developmentally regulated in rat mesencephalon. Develop Brain Res. 2001, 129: 47-56. 10.1016/S0165-3806(01)00181-X.
Pryor WA, Porter NA: Suggested mechanisms for the production of 4-hydroxy-2-nonenal from the autoxidation of polyunsaturated fatty acids. Free Radical Biol Med. 1990, 8: 541-543. 10.1016/0891-5849(90)90153-A.
Okada K, Wangpoengtrakul C, Osawa T, Toyokuni S, Tanaka K, Uchida K: 4-Hydroxy-2-nonenal-mediated impairement of intracellular proteolysis during oxidative stress. J Biol Chem. 1999, 274: 23787-23793. 10.1074/jbc.274.34.23787.
Hyun D-H, Lee M-H, Halliwell B, Jenner P: Proteasomal dysfunction induced by 4-hydroxy-2,3-trans-nonenal, an end-product of lipid peroxidation: a mechanism contributing to neurodegeneration?. J Neurochem. 2002, 83: 360-370. 10.1046/j.1471-4159.2002.01125.x.
Ferrington DA, Kapphahn RJ: Catalytic site-specific inhibition of the 20S proteasome by 4-hydroxynonenal. FEBS-Lett. 2004, 578: 217-223. 10.1016/j.febslet.2004.11.003.
Keller JN, Huang FF, Zhu H, Yu J, Ho J-S, Kindy MS: Oxidative stress- associated impairment of proteasome activity during ischemia-reperfusion injury. J Cereb Blood Flow Metab. 2000, 20: 1467-1473. 10.1097/00004647-200010000-00008.
Bulteau AL, Lundberg KC, Humphries KM, Sadek HA, Szweda PA, Friguet B, Szweda LI: Oxidative modification and inactivation of the proteasome during coronary occlusion/reperfusion. J Biol Chem. 2001, 276: 30057-30069. 10.1074/jbc.M100142200.
Conconi M, Petropoulos I, Emod I, Turlin E, Biville F, Friguet B: Protection from oxidative inactivation of the 20S proteasome by heat-shock protein 90. Biochem J. 1998, 333: 407-415.
Kapphahn RJ, Bigelow EJ, Ferrington DA: Age-dependent inhibition of proteasome chymotrypsin-like activity in the retina. Exp Eye Res. 2007,
Dudek EJ, Shang F, Liu Q, Valverde P, Hobbs M, Taylor A: Selectivity of the ubiquitin pathway for oxidatively modified proteins. Relevance to protein precipitation diseases. FASEB J. 2005, 19: 1707-1709.
Husom AD, Peters EA, Kolling EA, Fugere NA, Thompson LV, Ferrington DA: Altered proteasome function and subunit composition in aged muscle. Arch Biochem Biophys. 2004, 421: 67-76. 10.1016/j.abb.2003.10.010.
Ferrington DA, Husom AD, Thompson LV: Altered proteasome structure, function, and oxidation in aged muscle. FASEB J. 2005, 19: 644-646.
Mishto M, Bellavista E, Santoro A, Stolzing A, Ligorio C, Nacmias B, Spazzafumo L, Chiapelli M, Licastro F, Sorbi S, et al.: Immunoproteasome and LMP2 polymorphism in aged and Alzheimer's disease brains. Neurobiol Aging. 2006, 27: 54-66. 10.1016/j.neurobiolaging.2004.12.004.
Mitch WE, Medina R, Grieber S, May C, England BK, Price SR, Bailey JL, Goldberg AL: Metabolic acidosis stimulates muscle protein degradation by activating the adenosin triphosphate-dependent pathway involving ubiquitin and proteasomes. J Clin Invest. 1994, 93: 2127-2133.
Temparis S, Asensi M, Taillandier D, Aurousseau E, Larbaud D, Obled A, Bechet D, Ferrara M, Estrela JM, Attaix D: Increased ATP-ubiquitin-dependent proteolysis in skeletal muscles of tumor-bearing rats. Cancer Res. 1994, 54: 5568-5573.
Medina R, Wing SS, Goldberg AL: Increase in levels of polyubiquitin and proteasome mRNA in skeletal muscle during starvation and denervation atrophy. Biochem J. 1995, 307: 631-637.
Hobler SC, Williams A, Fischer D, Wang JJ, Sun X, Fischer JE, Monaco JJ, Hasselgren P-O: Activity and expression of the 20S proteasome are increased in skeletal muscle during sepsis. Am J Physiol. 1999, 277: R434-R440.
Price SR: Increased transcription of ubiquitin-proteasome system components: molecular responses associated with muscle atrophy. Int J Biochem Cell Biol. 2003, 35: 617-628. 10.1016/S1357-2725(02)00385-0.
Kee AJ, Combaret L, Tilignac T, Souweine B, Aurousseau E, Dalle M, Taillandier D, Attaix D: Ubiquitin-proteasome-dependent muscle proteolysis responds slowly to insulin release and refeeding in starved rats. J Physiol. 2003, 546: 765-776. 10.1113/jphysiol.2002.032367.
Fang C-H, Wang JJ, Hobler S, Li BG, Fischer JE, Hasselgren P-O: Proteasome blockers inhibit protein breakdown in skeletal muscle after burn injury in rats. Clin Sci. 1998, 95: 225-233. 10.1042/CS19980092.
Vinitsky A, Michaud C, Powers JC, Orlowski M: Inhibition of the chymotrypsin-like activity of the pituitary multicatalytic proteinase complex. Biochemistry USA. 1992, 31: 9421-9428.
Hobler SC, Tiao G, Fischer JE, Monaco J, Hasselgren P-O: Sepsis-induced increase in muscle proteolysis is blocked by specific proteasome inhibitors. Am J Physiol. 1998, 274: R30-R37.
Figueiredo-Pereira ME, Berg KA, Wilk S: A new inhibitor of the chymotrypsin-like activity of the multicatalytic proteinase complex (20S proteasome) induces accumulation of ubiquitin-protein conjugates in a neuronal cell. J Neurochem. 1994, 63: 1578-1581.
Fischer D, Gang G, Pritts T, Hasselgren PO: Sepsis-induced muscle proteolysis is prevented by a proteasome inhibitor in vivo. Biochem Biophys Res Commun. 2000, 270: 215-221. 10.1006/bbrc.2000.2398.
Du J, Hu Z, Mitch WE: Molecular mechanisms activating muscle protein degradation in chronic kidney disease and other catabolic conditions. Eur J Clin Invest. 2005, 35: 157-163. 10.1111/j.1365-2362.2005.01473.x.
Attaix D, Ventadour S, Codran A, Béchet D, Taillandier D, Combaret L: The ubiquitin-proteasome system and skeletal muscle wasting. Essays Biochem. 2005, 41: 173-186.
Solomon V, Goldberg AL: Importance of the ATP-ubiquitin-proteasome pathway in degradation of soluble and myofibrillar proteins in rabbit muscle extracts. J Biol Chem. 1996, 271: 26690-26697. 10.1074/jbc.271.41.25240.
Du J, Wang X, Miereles C, Bailey JL, Debigare R, Zheng B, Price SR, Mitch WE: Activation of caspase-3 is an initial step triggering accelerated muscle proteolysis in catabolic conditions. J Clin Invest. 2004, 113: 115-123. 10.1172/JCI200418330.
Lee SW, Dai G, Hu Z, Wang X, Du J, Mitch WE: Regulation of muscle protein degradation: Coordinated control of apoptotic and ubiquitin-proteasome systems by phosphatidylinositol 3 kinase. J Am Soc Nephrol. 2004, 15: 1537-1545. 10.1097/01.ASN.0000127211.86206.E1.
Cao PR, Kim HJ, Lecker SH: Ubiquitin-protein ligases in muscle wasting. Int J Biochem Cell Biol. 2005, 37: 2088-2097. 10.1016/j.biocel.2004.11.010.
Lecker S, Jagoe RT, Gilbert A, Gomes M, Baracos V, Bailey J, Price SR, Mitch WE, Goldberg AL: Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J. 2004, 18: 39-51. 10.1096/fj.03-0610com.
Hunter RB, Kandarian SC: Disruption of either the Nfkb1 or the Bcl3 gene inhibits skeletal muscle atrophy. J Clin Invest. 2004, 114: 1504-1511. 10.1172/JCI200421696.
Cai D, Frantz JD, Tawa NE, Melendez PA, Oh B-C, Lidov HGW, Hasselgren P-O, Frontera WR, Lee J, Glass DJ, Shoelson SE: IKKβ/NF-κB activation causes severe muscle wasting in mice. Cell. 2004, 119: 285-298. 10.1016/j.cell.2004.09.027.
Catz SD, Johnson IL: Transkriptional regulation of bcl-2 by nuclear factor kappa B and its significance in prostate cancer. Oncogene. 2001, 20: 7342-7351. 10.1038/sj.onc.1204926.
Li B, Dou P: Bax degradation by the ubiquitin/proteasome-dependent pathway: involvement in tumor survival and progression. Proc Natl Acad Sci USA. 2000, 97: 3850-3855. 10.1073/pnas.070047997.
Breitschopf K, Zeiher AM, Dimmler S: Ubiquitin-mediated degradation of the proapoptotic active form of bid. A functional consequence on apoptosis induction. J Biol Chem. 2000, 275: 21648-21652. 10.1074/jbc.M001083200.
Maki CG, Huibregtse JM, Howley PM: In vivo ubiquitination and proteasome-mediated degradation of p53. Cancer Res. 1996, 56: 2649-2654.
Reed SI: Ratchets and clocks: The cell cycle, ubiquitylation and protein turnover. Nat Rev Mol Cell Biol. 2003, 4: 855-864. 10.1038/nrm1246.
Cavo M: Proteasome inhibitor bortezomib for the treatment of multiple myeloma. Leukemia. 2006, 20: 1341-1352. 10.1038/sj.leu.2404278.
Chauhan D, Catley L, Li G, Podar K, Hideshima T, Velankar M, Mitsiades C, Mitsiades N, Yasui H, Letai A, Ovaa H, et al.: A novel orally active proteasome inhibitor induces apoptosis in multiple myeloma cells with mechanisms distinct from Bortezomib. Cancer Cell. 2005, 8: 407-419. 10.1016/j.ccr.2005.10.013.
Adams J, Palombella VJ, Sausville EA, Johnson J, Destree A, Lazarus DD, Maas J, Pien CS, Prakash S, Elliott PJ: Proteasome inhibitors: a novel class of potent and effective antitumor agent. Cancer Res. 1999, 59: 2615-2622.
Chauhan D, Hideshima T, Anderson KC: A novel proteasome inhibitor NPI-0052 as an anticancer therapy. Brit J Cancer. 2006, 95: 961-965. 10.1038/sj.bjc.6603406.
Breitschopf K, Haendeler J, Malchow P, Zeiher AM, Dimmler S: Posttranslational modification of Bcl-2 facilitates its proteasome-dependent degradation: Molecular characterization of the involved signalling pathway. Mol Cell Biol. 2000, 20: 1886-1896. 10.1128/MCB.20.5.1886-1896.2000.
Yang Y, Fang S, Jensen JP, Weissman A, Aswell JD: Ubiquitin protein ligase of IAPs and their degradation in proteasomes in response to apoptotic stimuli. Science. 2000, 288: 874-877. 10.1126/science.288.5467.874.
Sadoul R, Fernandez PA, Quiquerez AL, Martinou I, Maki M, Schroeter M, Becherer JD, Irmler M, Tschopp J, Martinou JC: Involvement of the proteasomes in the programmed cell death of NGF-deprived sympathetic neurons. EMBO J. 1996, 15: 3845-3852.
Saez ME, Ramirez-Lorca R, Moron FJ, Ruiz A: The therapeutic potential of the calapin family: new aspects. Drug Discovery Today. 2006, 11: 917-923. 10.1016/j.drudis.2006.08.009.
Low P, Bussel K, Dawson SP, Billett MA, Mayer RJ, Reynolds SE: Expression of a 26S proteasome ATPase subunit, MS73, in muscles that undergo developmentally programmed cell death and its control by ecdysteroid hormones in the insect Manduca sexta. FEBS Lett. 1997, 400: 345-349. 10.1016/S0014-5793(96)01413-5.
Dallaporta B, de Pablo M, Maisse C, Daugas E, Loeffler M, Zamzami N, Kroemer G: Proteasome activation as a critical event of thymocyte apoptosis. Cell Death Diff. 2000, 7: 368-373. 10.1038/sj.cdd.4400661.
Singh S, Khar A: Activation of the NFκB and UB-proteasome pathway during apoptosis induced by a serum factor is mediated through the upregulation of the 26S proteasome subunits. Apoptosis. 2006, 11: 843-859. 10.1007/s10495-006-5878-4.
Chatterjee-Kishore M, Wright KL, Ting JPY, Stark GR: How Stat1 mediates constitutive gene expression: a complex of unphosphorylated Stat1 and IRF1 supports transcription of the LMP2 gene. EMBO J. 2000, 19: 4111-4122. 10.1093/emboj/19.15.4111.
Ferrer I, Martín B, Castaño JG, Lucas JJ, Moreno D, Olivé M: Proteasomal expression, induction of immunoproteasome subunits, and local MHC class I presentation in myofibrillar myopathy and inclusion body myositis. J Neuropathol Exp Neurol. 2004, 63: 484-498.
Diaz-Hernandez M, Martin-Aparicio E, Avila J, Hernandez F, Lucas JJ: Enhanced induction of the immunoproteasome by interferon gamma in neurons expressing mutant Huntingtin. Neurotox Res. 2004, 6: 463-468.
Kotamraju S, Matalon S, Matsunaga T, Shang T, Hickman-Davis JM, Kalyanaraman B: Upregulation of immunoproteasomes by nitric oxide: Potential antioxidative mechanism in endothelial cells. Free Radical Biol Med. 2006, 40: 1034-1044. 10.1016/j.freeradbiomed.2005.10.052.
Klahr S: The role of nitric oxide in hypertension and renal disease progression. Nephrol Dial Transplant. 2001, 16 (Suppl 1): 60-62.
Moreno-Lopez B, Gonzalez-Forero D: Nitric oxide and synaptic dynamics in the adult brain: physiopathological aspects. Rev Neurosci. 2006, 17: 309-357.
Niedbala W, Cai B, Liew FY: Role of nitric oxide in the reguation of T cell functions. Ann Rheum Dis. 2006, 65 (Suppl 3): 37-40.
Thomas DD, Espey MG, Ridnour LA, Hofseth LJ, Mancardi D, Harris CC, Wink DA: Hypoxic inducible factor 1α, extracellular signal-regulated kinase, and p53 are regulated by distinct treshold concentrations of nitric oxide. Proc Natl Acad Sci USA. 2004, 101: 8894-8899. 10.1073/pnas.0400453101.
Stratford FLL, Chondrioganni N, Trougakos IP, Gonos ES, Rivett AJ: Proteasome response to interferon-γ is altered in senescent human fibroblasts. FEBS-Lett. 2006, 580: 3989-3994. 10.1016/j.febslet.2006.06.029.
Johnson A, France J, Sy MS, Harding CV: Down-regulation of the transporter for antigen presentation, proteasome subunits, and class I major histocompatibility complex in tumor cell lines. Cancer Res. 1998, 58: 3660-3667.
Kageshita T, Hirai S, Ono T, Hicklin DJ, Ferrone S: Down-regulation of HLA class I antigen-processing molecules in malignant melanoma: association with disease progression. Am J Pathol. 1999, 154: 745-754.
Murakami Y, Kanda K, Yokota K, Kanayama H, Kagawa S: Prognostic significance of immuno-proteasome subunit expression in patients with renal cell carcinoma: a preliminary study. Mol Urol. 2001, 5: 113-119. 10.1089/10915360152559594.
Meidenbauer N, Zippelius A, Pittet MJ, Laumer M, Vogl S, Heymann J, Rehli M, Seliger B, Schwarz S, Le-Gal FA, et al.: High frequency of functionally active Melan-A-specific T cells in a patient with progressive immunoproteasome-deficient melanoma. Cancer Res. 2004, 64: 6319-6326. 10.1158/0008-5472.CAN-04-1341.
Heink S, Fricke B, Ludwig D, Kloetzel PM, KrÜger E: Tumor cell lines expressing the proteasome subunit isoform LMP7E1 exhibit immunoproteasome deficiency. Cancer Res. 2006, 66: 649-652. 10.1158/0008-5472.CAN-05-2872.
Remoli AL, Marsili G, Perrotti E, Gallerani E, Ilari R, Nappi F, Cafaro A, Ensoli B, Gavioli R, Battistini A: Intracellular HIV-1 tat protein represses constitutive LMP2 transcription increasing proteasome activity by interfering with the binding of IRF1 to STAT1. Biochem J. 2006, 396: 371-380. 10.1042/BJ20051570.
Vinitsky A, Michaud C, Powers JC, Orlowski M: Inhibition of the chymotrypsin-like activity of the pituitary multicatalytic proteinase complex. Biochemistry. 1992, 31: 9421-9428. 10.1021/bi00154a014.
Kim KB, Fonseca FB, Crews CM: Development and characterization of proteasome inhibitors. Meth Enzymol. 2005, 399: 585-609.
Bogyo M, McMaster JS, Gaczynska M, Tortorella D, Goldberg AL, Ploegh H: Covalent modification of the active site threonine of proteasomal β subunits and the Escherichia coli homolog HsIV by a new class of inhibitors. Proc Natl Acad Sci USA. 1997, 94: 6629-6634. 10.1073/pnas.94.13.6629.
Dick LR, Cruikshank AA, Grenier L, Melandr FD, Nunes SL, Stein RL: Mechanistic studies on the inactivation of the proteasome by lactacystin. J Biol Chem. 1996, 271: 7273-7276. 10.1074/jbc.271.13.7273.
Iqbal M, Chatterjee S, Kauer JC, Mallamo JP, Messina PA, Reiboldt A, Siman R: Potent α-ketocarbonyl and boronic ester derived inhibitors of proteasome. Bioorg Med Chem Lett. 1996, 6: 287-290. 10.1016/0960-894X(96)00014-5.
Kisselev AF, Goldberg AL: Proteasome inhibitors: from research tools to drug candidates. Chemistry & Biology. 2001, 8: 739-758. 10.1016/S1074-5521(01)00056-4.
Kisselev AF, Callard A, Goldberg AL: Importance of the different proteolytic sites of the proteasome and the efficacy of inhibitors varies with the protein substrate. J Biol Chem. 2006, 281: 8582-8590. 10.1074/jbc.M509043200.
Groettrup M, Schmidtke G: Selective proteasome inhibitors: modulators of antigen presentation. DDT. 1999, 4: 63-71.
Ott DE, Coren LV, Sowder RC, Adams J, Schubert U: Retroviruses have differing requirements for proteasome function in budding process. J Virol. 2003, 77: 3384-3393. 10.1128/JVI.77.6.3384-3393.2003.
Ho A, Cyrus K, Kim K.B: Towards immunoproteasome-specific inhibitors: An improved synthesis of dihydroeponemycin. Eur J Org Chem. 2005, 22: 4829-4834. 10.1002/ejoc.200500437.
Gaczynska M, Osmulski PA, Gao PA, Post MJ, Simons M: Proline-and arginine-rich peptides constitute a novel class of allosteric inhibitors of proteasome activity. Biochemistry. 2003, 42: 8663-8670. 10.1021/bi034784f.
Hoffmeyer MR, Scalia R, Ross CR, Jones SP, Lefer DJ: PR-39, a potent neutrophil inhibitor, attenuates myocardial ischemia-reperfusion injury in mice. Am J Physiol Heart Circ Physiol. 2000, 279: H2824-H2828.
Bao J, Sato K, Li M, Gao Y, Abid R, Aird W, Simons M, Post M: PR-39 and PR-11 peptides inhibit ischemia-reperfusion injury by blocking proteasome-mediated IκBα degradation. Am J Physiol Heart Circ Physiol. 2001, 281: H2612-H2618.
Gao Y, Lecker S, Post MJ, Hietaranta AJ, Li J, Volk R, Li M, Sato K, Saluja AK, Steer ML, Goldberg AL, Simons M: Inhibition of ubiquitin-proteasome pathway-mediated IκBα degradation by naturally occurring antibacterial peptide. J Clin Invest. 2000, 106: 439-448.
Adams J, Stein R: Novel inhibitors of the proteasome and their therapeutic use in inflammation. Ann Rep Med Chem. 1996, 31: 279-288.
Safranek R, Ishibashi N, Oka Y, Ozasa H, Shirouzu K, Holecek M: Modulation of inflammatory response in sepsis by proteasome inhibition. Int J Exp Pathol. 2006, 87: 369-372. 10.1111/j.1365-2613.2006.00490.x.
Shen J, Reis J, Morrison DC, Papasian C, Raghavakaimal S, Kolbert C, Qureshi AA, Vogel SN, Qureshi N: Key inflammatory signalling pathways are regulated by the proteasome. Shock. 2006, 25: 472-484. 10.1097/01.shk.0000209554.46704.64.
Wilk S, Chen WE: Synthetic peptide-based activators of the proteasome. Mol Biol Rep. 1997, 24: 119-124. 10.1023/A:1006851428691.
Kisselev AF, Kaganovich D, Goldberg AL: Binding of hydrophobic peptides to several non-catalytic sites promotes peptide hydrolysis by all active sites of 20S proteasomes. Evidence for peptide induced channel opening in the α-rings. J Biol Chem. 2002, 277: 22260-22270. 10.1074/jbc.M112360200.
Dahlmann B, Becher B, Sobek A, Ehlers C, Kopp F, Kuehn L: In vitro activation of the 20S proteasome. Enzyme Protein. 1993, 47: 274-284.
Zoeger A, Blau M, Egerer K, Feist E, Dahlmann B: Circulating proteasomes are functional and have a subtype pattern distinct from 20S proteasomes in major blood cells. Clin Chem. 2006, 52: 2079-2086. 10.1373/clinchem.2006.072496.
Schmidt F, Dahlmann B, Janek K, Kloß A, Wacker M, Ackermann R, Thiede B, Jungblut PR: Comprehensive quantitative proteome analysis of 20S proteasome subtypes from rat liver by isotope coded affinity tag and 2-D gel-based approaches. Proteomics. 2006, 6: 4622-4632. 10.1002/pmic.200500920.
Bose S, Stratford FL, Broadfoot KI, Mason GG, Rivett AJ: Phosphorylation of 20S proteasome alpha subunit C8 (alpha7) stabilizes the 26S proteasome and plays a role in the regulation of proteasome complexes by γ-interferon. Biochem J. 2004, 378: 177-184. 10.1042/BJ20031122.
Zachara NE, Hart GW: O-GlcNAc modification: a nutritional sensor that modulates proteasome function. Trends Cell Biol. 2004, 14: 218-221. 10.1016/j.tcb.2004.03.005.
Park Y, Hwang YP, Lee JS, Seo SH, Yoon SK, Yoon JB: Proteasomal ATPase-associated factor 1 negatively regulates proteasome activity by interacting with proteasomal ATPases. Mol Cell Biol. 2005, 25: 3842-3852. 10.1128/MCB.25.9.3842-3853.2005.
Meiners S, Heyken D, Weller A, Ludwig A, Stangl K, Kloetzel P-M, Krüger E: Inhibition of proteasome activity induces concerted expression of proteasome genes and de novo formation of mammalian proteasomes. J Biol Chem. 2003, 278: 21517-21525. 10.1074/jbc.M301032200.
Stangl V, Lorenz M, Meiners S, Ludwig A, Bartsch C, Moobed M, Vietzke A, Kinkel H-T, Baumann G, Stangl K: Long-term up-regulation of eNOS and improvement of endothelial function by inhibition of the ubiquitin-proteasome pathway. FASEB J. 2004, 18: 272-279. 10.1096/fj.03-0054com.
Meiners S, Ludwig A, Lorenz M, Dreger H, Baumann G, Stangl V, Stangl K: Nontoxic proteasome inhibition activates a protective antioxidant defense in endothelial cells. Free Radical Biol Med. 2006, 40: 2232-2241. 10.1016/j.freeradbiomed.2006.03.003.
Yew EHJ, Cheung NS, Choy MS, Qi RZ, Lee AY-W, Peng ZF, Melendez AJ, Manikandan J, Koay ES-C, Chiu L-L, Ng WL, Whiteman M, Kandiah J, Halliwell B: Proteasome inhibition by lactacystin in primary neuronal cells induces both potentially neuroprotective and pro-apoptotic transcriptional responses: a microarray analysis. J Neurochem. 2005, 94: 943-956. 10.1111/j.1471-4159.2005.03220.x.
Henninger N, Sicard KM, Bouley J, Fisher M, Stagliano NE: The proteasome inhibitor VELCADE reduces infarction in rat models of focal cerebral ischemia. Neurosci Lett. 2006, 398: 300-305. 10.1016/j.neulet.2006.01.015.
Zhang L, Zhang ZG, Liu X, Hozeska A, Stagliano N, Riordan W, Lu M, Chopp M: Treatment of embolic stroke in rats with bortezomib and recombinant human tissue plasminogen activator. Thromb Haemost. 2006, 95: 166-173.
Williams AJ, Dave JR, Tortella FC: Neuroprotection with the proteasome inhibitor MLN519 in focal ischemic brain injury: Relation to nuclear factor κB (NFκB), inflammatory gene expression, and leukocyte infiltration. Neurochem Int. 2006, 49: 106-112. 10.1016/j.neuint.2006.03.018.
Shah IM, Lees KR, Pien CP, Elliott PJ: Early clinical experience with the novel proteasome inhibitor PS-519. Br J Clin Pharmocol. 2002, 54: 269-276. 10.1046/j.1365-2125.2002.01638.x.
Jakob C, Egerer K, Liebisch P, Turkmen S, Zavrski I, Kuckelkorn U, Heider U, Kaiser M, Fleissner C, Sterz J, et al.: Circulating proteasome levels are an independent prognostic factor for survival in multiple myeloma. Blood. 2006, 109: 2100-2105. 10.1182/blood-2006-04-016360.
Seeger M, Ferrel K, Frank R, Dubiel W: HIV-1 tat inhibits the 20S proteasome and its 11S regulator-mediated activation. J Biol Chem. 1997, 272: 8145-8148. 10.1074/jbc.272.13.8145.
Hu Z, Zhang Z, Doo E, Coux O, Goldberg AL, Liang TJ: Hepatitis B virus X protein is both a substrate and a potential inhibitor of the proteasome complex. J Virol. 1999, 73: 7231-7240.
Hemelaar J, Bex F, Booth B, Cerundolo V, McMichael A, Daenke S: Human T-cell leukaemia virus type 1 tax protein binds to assembled nuclear proteasomes and ehances their proteolytic activity. J Virol. 2001, 75: 11106-11115. 10.1128/JVI.75.22.11106-11115.2001.
Krause S, Kuckelkorn U, Dörner T, Burmester G-R, Feist E, Kloetzel P-M: Immunoproteasome subunit LMP2 expression is deregulated in Sögren's syndrom but not in other autoimmune disorders. Ann Rheum Dis. 2006, 65: 1021-1027. 10.1136/ard.2005.045930.
Publication history
Republished from Current BioData's Targeted Proteins database (TPdb; http://www.targetedproteinsdb.com).
Acknowledgements
This article has been published as part of BMC Biochemistry Volume 8 Supplement 1, 2007: Ubiquitin-Proteasome System in Disease Part 1. The full contents of the supplement are available online at http://www.biomedcentral.com/1471-2091/8?issue=S1.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an open access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Dahlmann, B. Role of proteasomes in disease. BMC Biochem 8 (Suppl 1), S3 (2007). https://doi.org/10.1186/1471-2091-8-S1-S3
Published:
DOI: https://doi.org/10.1186/1471-2091-8-S1-S3