
Abstract
Parkinson's disease (PD) is the most common neurodegenerative movement disorder. Although a subject of intense research, the etiology of PD remains poorly understood. Recently, several lines of evidence have implicated an intimate link between aberrations in the ubiquitin proteasome system (UPS) and PD pathogenesis. Derangements of the UPS, which normally functions as a type of protein degradation machinery, lead to alterations in protein homeostasis that could conceivably promote the toxic accumulation of proteins detrimental to neuronal survival. Not surprisingly, various cellular and animal models of PD that are based on direct disruption of UPS function reproduce the most prominent features of PD. Although persuasive, new developments in the past few years have in fact raised serious questions about the link between the UPS and PD. Here I review current thoughts and controversies about their relationship and discuss whether strategies aimed at mitigating UPS dysfunction could represent rational ways to intervene in the disease.
Publication history: Republished from Current BioData's Targeted Proteins database (TPdb; http://www.targetedproteinsdb.com).
Similar content being viewed by others
Parkinson's disease
Parkinson's disease (PD) is the most common neurodegenerative movement disorder, affecting millions of elderly individuals worldwide [1, 2]. Clinically, the disease is characterized by major motoric difficulties that include bradykinesia, postural instability, rigidity and tremor. Although neurodegeneration in PD involves multiple areas of the brain such as the dorsal motor nucleus of the vagus, locus ceruleus (LC) and olfactory nuclei [3], the principal neuropathology that gives rise to the array of motoric deficits seen in PD patients is the loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc), which results in a severe depletion of striatal dopamine (DA) [4]. Often, the neuronal degeneration is accompanied by eosinophilic intracytoplasmic inclusions known as Lewy bodies (LBs) that are present in surviving neurons of the SN as well as in other affected brain areas [5]. However, the LB is not an obligate marker of the disease as some familial PD cases are apparently devoid of its presence [6–9].
Although a subject of intense research, the etiology of PD remains poorly understood and no treatment exists to prevent, cure or retard the progression of this disabling disease. Current favored hypotheses for sporadic PD include combinations of the aging process, genetic propensity and environmental exposures leading to oxidative stress, mitochondrial dysfunction, microglial activation and excitotoxicity [2, 10]. More recently, evidence from several lines of study strongly implicate a key role for the ubiquitin proteasome system (UPS) in PD pathogenesis [11–14].
UPS
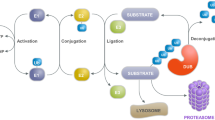
The UPS is an intracellular protein degradation system that is responsible for the majority of protein turnover within the cell [15] (Figure 1). In this system, proteins destined for degradation are covalently tagged with ubiquitin, a 76 amino acid residue protein, through the formation of an iso-peptide bond between the ε-amino group of a lysine residue of the substrate and the C-terminal carboxylate (G76) of ubiquitin. This ligation reaction is elaborate and requires the sequential actions of ubiquitin activating (E1), conjugating (E2) and ligating (E3) enzymes [16] (Figure 1). Usually, the multienzyme-mediated ubiquitylation process is repeated many times to allow the formation of a polyubiquitin chain on the substrate. Ubiquitin molecules in the chain are linked together via iso-peptidic bonds between the terminal G76 residue of each ubiquitin unit and a specific lysine (K) residue (most commonly K48) of the previous ubiquitin [17]. The G76–K48 polyubiquitylated substrate is then targeted for degradation by the 26S proteasome, a large protease complex consisting of a barrel-shaped 20S proteolytic core in association with two 19S (PA700) regulatory caps, one on each side of the barrel's openings (Figure 1). The 20S catalytic core is characterized by three distinct proteolytic activities: chymotrypsin-like, trypsin-like and peptidyl glutamylpeptide hydrolytic, which can be measured in vitro by means of a fluorimeter using Suc-Leu-Leu-Val-Try-AMC, Boc-Leu-Arg-Arg-AMC and Z-Leu-Leu-Glu-AMC, respectively, as fluorogenic peptide substrates [18]. The components of the 19S cap play vital roles in the initial steps of substrate proteolysis, including the recognition, unfolding and translocation of substrate proteins into the lumen of the proteolytic core [19–21]. Individual ubiquitin monomers are regenerated in the process by the actions of deubiquitylating enzymes (DUBs) (Figure 1). It is important to note that the ubiquitin molecule contains a total of seven lysine residues (at positions 6, 11, 27, 29, 33, 48 and 63) and that polyubiquitin chain assembly via alternative lysine linkages has also been described [22]. Notably, K63-linked polyubiquitylation of proteins is not typically associated with proteasomal degradation [23–27]. It is also noteworthy that an enormous number of cellular proteins (>1000), comprising various E1s, E2s, E3s and other related members, are involved in the UPS [28]. Indeed, protein ubiquitylation rivals protein phosphorylation as a major post-translational event in the cell. Conceivably, therefore, derangements of UPS function could lead to dire cellular consequences, particularly in post-mitotic cells.

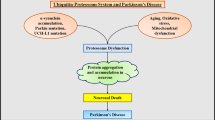
The ubiquitin proteasome system (UPS) and Parkinson's disease (PD). Under normal conditions, proteins destined for proteasomal degradation are tagged with a chain of ubiquitin (Ub) proteins via multiple rounds of a linear reaction catalyzed by ubiquitin activating (E1), conjugating (E2) and ligating enzymes (E3). An example of an E3 is parkin. Ubiquitylation reactions are reversed by the action of deubiquitylating enzymes (DUBs), of which UCHL1 is a member. Energy in the form of ATP is required to drive the UPS machinery. Age-related changes, exogenous stress, mitochondrial alterations and PD-linked genetic mutations in parkin, UCHL1 and α-synuclein could promote disruption of the UPS and conceivably result in the accumulation of protein aggregates or abnormal protein intermediates that could be directly detrimental to neuronal survival. Lewy bodies are thought to form as a result of an attempt by the cell to sequester these abnormal proteins. The enhancement of protein re-folding by chaperones such as Hsp70 and the clearance of protein aggregates via the stimulation of autophagy could help mitigate UPS dysfunction. Such strategies may offer innovative approaches in the treatment of PD.
UPS and PD
Evidence implicating a direct role for the UPS in PD came from the association of genetic mutations in the parkin (PRKN2) gene with familial parkinsonism about a decade ago [13], and the subsequent demonstration by three independent groups that parkin functions as a ubiquitin ligase associated with proteasomal degradation [29–31]. Importantly, using in vitro assays to measure the ubiquitylation activity of parkin immunoprecipitated from transfected cells, these pioneer investigators found that disease-causing mutations in parkin compromise its normal role as an E3 enzyme [29–31]. This property of parkin thus directly links UPS aberrations to dopaminergic neuronal survival, the association of which is further supported by the discovery of a missense mutation (I93M) in UCHL1, a DUB, in a pair of German siblings with inherited PD [14]. Notably, the I93M UCHL1 mutant has markedly reduced ubiquitin hydrolase activity in vitro[14], suggesting that impaired polyubiquitin hydrolysis could also contribute to dopaminergic neuronal death. Another PD-linked gene product, α-synuclein, is a major component of LBs [32]. Although α-synuclein is not a member of the UPS, overexpression of wild-type or mutant α-synuclein in cultured cells and in the brains of transgenic mice is known to inhibit proteasome function (as determined by in vitro 20S proteasome assay), with disease-relevant mutants eliciting stronger effects [33–35]. Notably, triplication of the α-synuclein gene is also causative of PD [36].
Consistent with an implicated role for the UPS in familial parkinsonism, McNaught and colleagues (Mount Sinai School of Medicine) observed a significant reduction in the levels of PA700 expression in the SNpc of post-mortem sporadic PD brains relative to control brains [11] (PA700 levels are otherwise elevated in other regions of the PD brain such as the frontal cortex and striatum [11, 37]). Selective decrease in the level of proteasomal core subunits within the SN was also noted [11]. Further, in vitro assay of SN extracts from PD brains revealed a marked decrease in the activity of the 20S proteasome [11]. Corroborating evidence came from a parallel report by Spillantini and co-workers (Cambridge Centre for Brain Repair) who demonstrated a 55% reduction in the proteasomal chymotrypsin-like activity in the SN, but not in the cingulate, frontal and occipital cortices, of PD brains [38]. Together, these results suggest structural and functional impairments of the UPS in sporadic PD. Although the precise basis of UPS dysfunction in sporadic PD awaits further clarifications, it likely involves age-related changes in the brain resulting in increased oxidative stress and energy depletion, as well as exposures to environmental toxins [39, 40]. Supporting this, the herbicide maneb and a range of pesticides linked to sporadic PD, including rotenone, have been shown to produce proteasome inhibition in vitro[41, 42]. Similarly, we recorded a marked decrease in proteasome activity in extracts derived from rotenone- and paraquat-treated cells relative to untreated controls [43]. Further, Betarbet et al. (Emory University) found that proteasome function is selectively decreased in the ventral midbrain region in rotenone-induced rats [42], while a similar study by another group (University of Pisa) showed that continuous infusion of the parkinsonian neurotoxin, MPTP, in mice induces proteasome impairments in their brains [44]. Taken together, these results suggest a co-operative relationship between environmental exposures and proteasome dysfunction in promoting neurodegeneration in sporadic PD.
How does failure of the UPS lead to dopaminergic neurodegeneration? Conceivably, aberrant protein homeostasis could result in the toxic accumulation of intracellular proteins detrimental to neuronal survival. Supporting this, the accumulation of misfolded and aggregated α-synuclein is thought to be the primary pathogenic event in familial PD linked to mutations or multiplication of the α-synuclein gene [45]. Notably, aggregated α-synuclein selectively interacts with the 19S cap and concomitantly inhibits the function of the 26S proteasome [46], thereby perpetuating a vicious cycle. Similarly, in parkin-linked parkinsonism, the accumulation of certain parkin substrates could interfere with cellular functions and induce neurotoxicity [45]. Indeed, numerous reported substrates of parkin, including CDCrel (cell division control-related protein) 1 and 2a [47], Pael-R (parkin-associated endothelin-like receptor) [48], cyclin E [49], p38 (JTV-1/AIMP2) [50] and FBP1 (far upstream sequence element-binding protein 1) [51], accumulate in the brains of PD patients carrying parkin mutations. Further, both p38 and FBP1 also accumulate in the brains of parkin-deficient mice [50, 51], and overexpression of p38 in cell culture or SN of mice leads to marked neurotoxicity [50]. Taken together, these results suggest that parkin-mediated ubiquitylation of its substrates promotes their turnover via the UPS and offer a simple hypothesis that toxic substrate accumulation in the absence of functional parkin triggers neurodegeneration. However, because none of these parkin substrates are exclusively expressed in dopaminergic neurons, it remains puzzling as to why dopaminergic neurons are selectively vulnerable to parkin deficiency. Nonetheless, the importance of parkin function to neuronal survival is currently widely accepted. This is exemplified by its broad neuroprotective capacity, which includes conferring protection against manganese-induced cell death [52], α-synuclein toxicity [53], Pael-R [54] and p38/JTv-1 accumulation [50], kainate-induced excitotoxicity [49], DA-mediated toxicity [55], and importantly, proteasome dysfunction [43, 56]. Accordingly, the loss of parkin function following its mutation is expected to compromise neuronal survival. Interestingly, a recent ubiquitylation assay conducted by Tanaka (Tokyo Metropolitan Institute of Medical Science) and Corti (INSERM) laboratories using purified recombinant parkin demonstrated that the majority of disease-associated parkin missense mutations do not compromise its enzymatic activity in vitro[57, 58]. Conversely, several other groups, including ours, found that parkin mutations often alter the protein's solubility and concomitantly promote its aggregation within the cell [59–62]. Further, we and others also found that normal brain aging, as well as a wide variety of PD-linked stressors, including DA, induce similar alterations in parkin [43, 63, 64] or otherwise inactivate the enzyme [65, 66], thereby suggesting a mechanism for parkin dysfunction in the pathogenesis of idiopathic PD. Loss of soluble, functional parkin resulting from normal aging or stress-induced modifications could therefore be as relevant to sporadic PD as parkin dysfunction triggered by disease-associated mutations is to parkin-related parkinsonism.
Disease models, knockouts and assays
Using fetal rat ventral mesencephalic cultures as a model, several groups have demonstrated that lactacystin-induced proteasome dysfunction results in a dose-dependent and preferential degeneration of dopaminergic neurons, accompanied by the formation of inclusion bodies that stain positively for ubiquitin and α-synuclein [67, 68]. Interestingly, inactivation of ubiquitin hydrolases with ubiquitin aldehyde produces similar effects in these primary cultures [67]. However, these studies were performed using acute doses (5–10 µM for 24–48 hours) of pharmacological inhibitors. To better reflect the events occurring in susceptible neurons during PD pathogenesis, Keller and co-workers (University of Kentucky) subjected SH-SY5Y neuroblastoma cells to low levels of proteasome inhibition (100 nM MG115) for an extended period lasting several weeks. They observed in these cells elevated levels of protein oxidation and protein aggregates, as well as altered mitochondrial homeostasis [69, 70]. Neural cells exposed to chronic proteasome inhibition thus reproduce the key features of sporadic PD faithfully [69, 70], thereby supporting the involvement of UPS aberrations in PD pathogenesis. Consistent with this, two groups observed retrograde dopaminergic neurodegeneration in rodent brains following intrastriatal administration of lactacystin [71, 72]. In further pursuit of this hypothesis, McNaught and colleagues subjected adult Sprague-Dawley rats to six regularly-spaced subcutaneous injections of either the naturally occurring proteasome inhibitor epoxomicin (1.5 mg/kg) or the synthetic proteasome inhibitor PSI (3 or 6 mg/kg) over a period of two weeks [73]. Remarkably, after a latency of two weeks, both inhibitors induced progressive motor dysfunction characterized by bradykinesia, rigidity and tremor in treated rats that could be alleviated by L-DOPA administration [73]. Consistent with the loss of dopaminergic nerve terminals in the striatum, 11C-CFT-based positron emission tomography imaging revealed a progressive reduction of the DA transporter ligand in these rats [73]. The nigral pathology, as well as extra-nigral degeneration in brain regions such as the LC, dorsal motor nucleus of the vagus and nucleus basalis of Meynert, was confirmed at post mortem [73]. Whereas a reduction in proteasome activity, as measured in vitro, appears to accompany the degeneration, elevated proteasome activity was observed in areas that remained intact, suggesting a compensatory mechanism [73]. Notably, inclusions that stained positively for ubiquitin and α-synuclein formed within neurons that survived the degeneration process in affected areas [73]. This rat model of PD, based on systemic administration of proteasome inhibitors, thus recapitulates the key clinical and pathological hallmarks of PD faithfully and provides solid support for the role of UPS disruption in PD pathogenesis. Unfortunately, the model is currently controversial as several laboratories have recently reported failures in their attempts to replicate the results despite diligently adhering to McNaught's original protocol [74–76]. These conflicting findings include the absence of nigral pathology, motor impairment and neuronal inclusions [74–76]. However, some other groups did manage to reproduce the nigral pathology [77, 78]. Though the reason for this variability is currently unclear, it could be due to variations in the source and hence quality of the inhibitors, differences in methodologies or exogenous factors present in feeds or in the environment [79, 80]. Hopefully, these issues will soon be resolved as the novel model of PD generated by McNaught and colleagues is arguably one that reflects the disease most accurately and as such has important implications for the development of PD therapeutics.
Concurrent to the proteasome inhibition model, several investigators have also generated genetic models of PD to probe the link between the disruption of key UPS players and PD pathogenesis. Two groups, led by Shen (Harvard Medical School) and Rooney (Aventis Pharma) respectively, first reported mouse models that are ablated of orthologous parkin[81, 82]. However, none of these models exhibit parkinsonian phenotypes. Indeed, with the exception of cell loss in the LC in a subsequent model generated by the Dawson laboratory (Johns Hopkins School of Medicine) [83], parkin null mice have generally failed to exhibit apparent signs of parkinsonism [81, 82, 84]. Further, the majority of previously identified parkin substrates (except p38 and FBP) do not show any detectable accumulation in these mice [50, 51, 81, 82, 84]. Similarly, inactivation of UCHL1 in mice leads not to dopaminergic neuronal death but to gracile axonal dystrophy syndrome, a degenerative disease associated with sensory and motor ataxia [85]. However, Drosophila parkin null mutants, originally developed by Greene et al. (University of Washington) [86], exhibit selective dopaminergic neurodegeneration in the adult brains and concomitant locomotion defects [87, 88], a phenotype that appears to mirror the human condition. Although the anatomy of the Drosophila brain differs from that of the vertebrate brain, many aspects of nervous system development and function are conserved between flies and humans. The parkin fly model could thus offer opportunities for the discovery and testing of experimental PD therapeutics, especially those aimed at restoring parkin function or compensating for parkin dysfunction.
Disease targets and ligands
Since disruption of the UPS appears to play a key role in PD pathogenesis, therapies aimed directly or indirectly at mitigating UPS dysfunction could potentially offer respite for individuals afflicted with PD. Some strategies aimed at alleviating UPS dysfunction are discussed below.
(i) Managing protein misfolding and aggregation – The accumulation and subsequent aggregation of proteins as a result of proteasome inhibition is a likely contributor to pathogenicity. Conceivably, strategies aimed at reducing proteasomal load or stimulating proteolysis could promote beneficial outcomes for the PD patient. This could involve decreasing the burden of pathogenic proteins through RNA silencing [89]. Alternatively, gene-based approaches that enhance the expression levels of chaperones to facilitate protein re-folding could also be effective [90–92]. For example, co-expression of hsp70/Hsp70 with α-synuclein reduces α-synuclein aggregation and concomitantly attenuates α-synuclein-mediated dopaminergic neuronal death in both flies and mice [90, 92]. Further, Hsp70 gene transfer by adeno-associated virus vector inhibits MPTP-induced nigrostriatal degeneration in mice [91], presumably by reducing the levels of misfolded proteins induced by the neurotoxin. Not surprisingly, pharmacological promotion of Hsp70 expression by Geldanamycin, a commercially available drug whose analogs are currently entering/undergoing Phase II clinical trials, also mitigates neurotoxicity in several models of PD [93, 94]. Aside from these, compounds that directly stimulate UPS activity are also expected to reduce protein aggregation and pathology. Dexamethasone, an anti-inflammatory drug commercially marketed under brand names such as Decadron and Solurex, is known to increase the expression levels of various UPS components including subunits of the proteasomal core and PA700 activator [95]. These combined effects could explain dexamethasone's protection against dopaminergic neurodegeneration in MPTP-treated mice [96]. Potentially, several other UPS modulators currently being tested for varied disease conditions could also be relevant to PD (see UPS Drugs and Biologicals table).
(ii) Autophagy induction - Autophagy is a major cellular process by which cytoplasmic components, including organelles, are catabolized [97, 98]. Although autophagy represents a distinct degradation system from the UPS, recent evidence suggests a complementary relationship between these two systems in the maintenance of protein homeostasis [99]. Further, two recent independent reports provide compelling evidence regarding the importance of autophagy in neurodegeneration [100, 101]. Of particular relevance to PD, both the UPS and autophagy are apparently involved in α-synuclein clearance [102]. Harnessing this pathway could thus offer innovative approaches in the treatment of PD. The mammalian target of rapamycin (mTOR) of the phosphatidylinositol 3-kinase pathway, which normally functions to shut down autophagy, represents a good target to modulate the lysosome-mediated degradation system [97, 98]. Rapamycin-mediated inhibition of mTOR activates autophagy and increases the clearance of aggregate-prone proteins, including α-synuclein, and concomitantly reduces their neurotoxicity [103]. Similarly, activation of autophagy via mTOR-independent pathways through the actions of lithium (which inhibit inositol monophosphatase thereby leading to the activation of autophagy) or trehalose (mechanism of activation unknown), promotes beneficial outcomes in cell culture models of protein aggregation [104, 105].
(iii) Parkin gene therapy – Given that parkin apparently functions as a broad-spectrum neuroprotectant, it is conceivable that parkin gene delivery could offer a novel avenue of PD therapy. Indeed, virus-mediated delivery of parkin prevents dopaminergic neurodegeneration in rats either overexpressing α-synuclein [106, 107] or treated with 6-Hyroxydopamine (6-OHDA, a catecholaminergic neurotoxin that triggers cell death through the generation of oxidative stress [108]), as well as in mice treated with MPTP [109]. Concurring with these, a recent study further demonstrated the protective function of parkin delivery in a non-human primate model of α-synuclein overexpression [110]. Similarly, enhanced expression of parkin in Drosophila counteracts PD-like symptoms promoted by the overexpression of α-synuclein [111] or Pael-R [54]. Interestingly, a recent screen for modifiers of parkin dysfunction in flies identified glutathione S-transferase S1 (GstS1) [88]. Overexpression of GstS1 prevents dopaminergic neurodegeneration in parkin mutant flies [88], suggesting that the induction of GstS1 expression could be useful in the treatment of PD.
Although the above strategies aimed at mitigating UPS function could represent promising therapeutic approaches for the PD patient, it is important to note that sporadic PD is likely multifactorial and that the UPS could just be one of several players responsible for its pathogenesis. Given this, PD could perhaps be cured not through the administration of a singular therapy, but through a combination of therapies targeted at major problem centers in the pathogenic cascade, with the UPS possibly being one of the focal points.
Next frontiers
Despite what appears to be compelling evidence implicating an association between the UPS and PD, certain gaps and controversies surrounding their relationship need to be clarified before we can move forward with UPS-based therapeutics. One of these relates to the validity of parkin and UCHL1 mutations in supporting a role for the UPS in PD. Although the association of parkin and UCHL1 mutations with familial parkinsonism supports a link between the UPS and PD, it is apparent that the involvement of the UCHL1 I93M mutation in PD pathogenesis has become contentious in recent years as its occurrence to date is restricted to the pair of German siblings [112]. Furthermore, novel roles for parkin-mediated ubiquitylation that are uncoupled from the proteasome are progressively being elucidated. Emerging evidence suggests that parkin is a multifaceted ubiquitin ligase capable of mediating alternative modes of ubiquitylation that are not typically associated with proteolysis [57, 58, 113, 114]. These non-proteolytic forms of parkin-mediated ubiquitylation, which include K63-linked polyubiquitylation and monoubiquitylation, have been linked to diverse cellular processes such as DNA repair, IkBa kinase activation, endocytosis and translational regulation [23–27], thereby suggesting that parkin-mediated ubiquitylation could play regulatory roles. Supporting this, Fallon et al. (Montreal Neurological Institute) recently identified Eps15, an adaptor protein that is involved in epidermal growth factor receptor endocytosis and trafficking, as a substrate of parkin-mediated monoubiquitylation [115]. Similarly, Hampe et al. (INSERM) demonstrated that parkin mediates multi-monoubiquitylation of p38 in vitro[57], an observation that apparently contradicts the finding by the Dawson laboratory that p38 accumulates in the brains of parkin-deficient mice and parkin-related PD patients [50]. The reason for this discrepancy remains unclear. Conversely, we have shown that parkin normally mediates K63-linked polyubiquitylation of the α-synuclein-interacting protein, synphilin-1, which promotes the stability of the protein and concomitantly its sequestration into LB-like inclusions [113]. Our result suggests the interesting possibility that parkin-mediated K63-linked ubiquitylation could contribute to LB biogenesis and at the same time provide an explanation to the general absence of LBs in parkin-related parkinsonism [6–8]. Depending on the mode of ubiquitylation, parkin thus appears to have the capacity to promote either degradation or stability of its substrates. This is seemingly paradoxical, but is consistent with parkin's multidimensional roles in the cell [116] and recent observations by several groups that previously identified parkin substrates (except p38 and FBP1) did not accumulate in the brains of parkin-deficient mice [50, 81, 82, 84]. Importantly, it cautions against fixation on the traditional view that substrates of parkin must exhibit an accelerated, proteasome-dependent turnover in the presence of the enzyme. At present, it would appear that disruption of proteasome-independent events following parkin mutations could also account for the susceptibility of neurons to degeneration in patients with parkin-related parkinsonism. Clearly, considerably more basic work needs to be done to clarify the multifunctional roles of parkin, and its interactions with the UPS, be it direct or indirect. The concurrent elucidation of the determinants governing the choice of parkin-mediated ubiquitylation would be illuminating. Because antibodies that specifically recognize different ubiquitin chain topologies are currently unavailable, I envisage that mass spectrometry-based proteomics techniques would be useful here.
Another urgent endeavour is to resolve the conflicting findings between the proteasome inhibition models generated by McNaught and by others, since reproduction of the original model could provide unequivocal support for a direct role of UPS disruption in PD pathogenesis. Interestingly, Landau et al. (McGill University) recently repeated McNaught's experiment in mice and found that the ethanol vehicle produces marked dopaminergic neurotoxicity and significantly eclipses the effects of the synthetic proteasome inhibitor PSI in the dopaminergic system [117]. Notably, ethanol is known to elicit proteasome inhibition and oxidative stress in the liver [118]. The ethanol vehicle for PSI thus appears to be a confounding problem for this model. Meanwhile, two prominent PD researchers have suggested the generation of additional series of animals by the original laboratory and sending them to other laboratories for blinded assessment [79]. This will certainly help to validate the model.
Finally, the relationship between LB inclusions and UPS impairment remains controversial. While the presence of α-synuclein-enriched LBs is compatible with proteasome dysfunction, a recent study from Spillantini's laboratory found no generalized impairment of the proteasome in brain regions with LB pathology [38]. Obviously, a better understanding of how misfolded proteins are targeted for degradation or inclusion formation would be useful. It is possible, as we and others have recently speculated, that inclusion formation could represent attempts by the cell to divert misfolded proteins away from an overloaded proteasome (thus prolonging its survival), especially in times of chronic stress [119, 120]. I anticipate that live cell imaging techniques coupled with a reliable in vivo proteasome reporter assay would help to address some of the questions regarding the life cycle of an inclusion body and its relationship with the UPS.
Abbreviations
- DA:
-
dopamine
- GstS1:
-
glutathione S-transferase S1
- PD:
-
Parkinson's disease
- LC:
-
locus ceruleus
- SNpc:
-
substantia nigra pars compacta
- UPS:
-
ubiquitin proteasome system.
References
Dorsey E.R., Constantinescu R., Thompson J.P., Biglan K.M., Holloway R.G., Kieburtz K., Marshall F.J., Ravina B.M., Schifitto G., Siderowf A., Tanner C.M.: Projected number of people with Parkinson disease in the most populous nations, 2005 through 2030. Neurology. 2007, 68: 384-386. 10.1212/01.wnl.0000247740.47667.03.
Savitt J.M., Dawson V.L., Dawson T.M.: Diagnosis and treatment of Parkinson disease: molecules to medicine. J Clin Invest. 2006, 116: 1744-1754. 10.1172/JCI29178.
Braak H., Del Tredici K., Rub U., de Vos R.A., Jansen Steur E.N., Braak E.: Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging. 2003, 24: 197-211. 10.1016/S0197-4580(02)00065-9.
Birkmayer W., Hornykiewicz O.: [The L-3,4-dioxyphenylalanine (DOPA)-effect in Parkinson-akinesia.]. Wien Klin Wochenschr. 1961, 73: 787-788.
Forno L.S.: Neuropathology of Parkinson's disease. J Neuropathol Exp Neurol. 1996, 55: 259-272.
Hayashi S., Wakabayashi K., Ishikawa A., Nagai H., Saito M., Maruyama M., Takahashi T., Ozawa T., Tsuji S., Takahashi H.: An autopsy case of autosomal-recessive juvenile parkinsonism with a homozygous exon 4 deletion in the parkin gene. Mov Disord. 2000, 15: 884-888. 10.1002/1531-8257(200009)15:5<884::AID-MDS1019>3.0.CO;2-8.
Mori H., Kondo T., Yokochi M., Matsumine H., Nakagawa-Hattori Y., Miyake T., Suda K., Mizuno Y.: Pathologic and biochemical studies of juvenile parkinsonism linked to chromosome 6q. Neurology. 1998, 51: 890-892.
Takahashi H., Ohama E., Suzuki S., Horikawa Y., Ishikawa A., Morita T., Tsuji S., Ikuta F.: Familial juvenile parkinsonism: clinical and pathologic study in a family. Neurology. 1994, 44: 437-441.
Zimprich A., Biskup S., Leitner P., Lichtner P., Farrer M., Lincoln S., Kachergus J., Hulihan M., Uitti R.J., Calne D.B., Stoessl A.J., Pfeiffer R.F., Patenge N., Carbajal I.C., Vieregge P., Asmus F., Muller-Myhsok B., Dickson D.W., Meitinger T., Strom T.M., Wszolek Z.K., Gasser T.: Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004, 44: 601-607. 10.1016/j.neuron.2004.11.005.
Gandhi S., Wood N.W.: Molecular pathogenesis of Parkinson's disease. Hum Mol Genet. 2005, 14 (Spec No. 2): 2749-2755. 10.1093/hmg/ddi308.
McNaught K.S., Belizaire R., Isacson O., Jenner P., Olanow C.W.: Altered proteasomal function in sporadic Parkinson's disease. Exp Neurol. 2003, 179: 38-46. 10.1006/exnr.2002.8050.
McNaught K.S., Jenner P.: Proteasomal function is impaired in substantia nigra in Parkinson's disease. Neurosci Lett. 2001, 297: 191-194. 10.1016/S0304-3940(00)01701-8.
Kitada T., Asakawa S., Hattori N., Matsumine H., Yamamura Y., Minoshima S., Yokochi M., Mizuno Y., Shimizu N.: Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998, 392: 605-608. 10.1038/33416.
Leroy E., Boyer R., Auburger G., Leube B., Ulm G., Mezey E., Harta G., Brownstein M.J., Jonnalagada S., Chernova T., Dehejia A., Lavedan C., Gasser T., Steinbach P.J., Wilkinson K.D., Polymeropoulos M.H.: The ubiquitin pathway in Parkinson's disease. Nature. 1998, 395: 451-452. 10.1038/26652.
Hershko A., Ciechanover A.: The ubiquitin system. Annu Rev Biochem. 1998, 67: 425-479. 10.1146/annurev.biochem.67.1.425.
Hershko A., Heller H., Elias S., Ciechanover A.: Components of ubiquitin-protein ligase system Resolution, affinity purification, and role in protein breakdown. J Biol Chem. 1983, 258: 8206-8214.
Chau V., Tobias J.W., Bachmair A., Marriott D., Ecker D.J., Gonda D.K., Varshavsky A.: A multiubiquitin chain is confined to specific lysine in a targeted short-lived protein. Science. 1989, 243: 1576-1583. 10.1126/science.2538923.
Stein R.L., Melandri F., Dick L.: Kinetic characterization of the chymotryptic activity of the 20S proteasome. Biochemistry. 1996, 35: 3899-3908. 10.1021/bi952262x.
Braun B.C., Glickman M., Kraft R., Dahlmann B., Kloetzel P.M., Finley D., Schmidt M.: The base of the proteasome regulatory particle exhibits chaperone-like activity. Nat Cell Biol. 1999, 1: 221-226. 10.1038/12043.
Glickman M.H., Rubin D.M., Coux O., Wefes I., Pfeifer G., Cjeka Z., Baumeister W., Fried V.A., Finley D.: A subcomplex of the proteasome regulatory particle required for ubiquitin-conjugate degradation and related to the COP9-signalosome and eIF3. Cell. 1998, 94: 615-623. 10.1016/S0092-8674(00)81603-7.
Navon A., Goldberg A.L.: Proteins are unfolded on the surface of the ATPase ring before transport into the proteasome. Mol Cell. 2001, 8: 1339-1349. 10.1016/S1097-2765(01)00407-5.
Peng J., Schwartz D., Elias J.E., Thoreen C.C., Cheng D., Marsischky G., Roelofs J., Finley D., Gygi S.P.: A proteomics approach to understanding protein ubiquitination. Nat Biotechnol. 2003, 21: 921-926. 10.1038/nbt849.
Arnason T., Ellison M.J.: Stress resistance in Saccharomyces cerevisiae is strongly correlated with assembly of a novel type of multiubiquitin chain. Mol Cell Biol. 1994, 14: 7876-7883.
Fisk H.A., Yaffe M.P.: A role for ubiquitination in mitochondrial inheritance in Saccharomyces cerevisiae. J Cell Biol. 1999, 145: 1199-1208. 10.1083/jcb.145.6.1199.
Galan J.M., Haguenauer-Tsapis R.: Ubiquitin lys63 is involved in ubiquitination of a yeast plasma membrane protein. Embo J. 1997, 16: 5847-5854. 10.1093/emboj/16.19.5847.
Spence J., Sadis S., Haas A.L., Finley D.: A ubiquitin mutant with specific defects in DNA repair and multiubiquitination. Mol Cell Biol. 1995, 15: 1265-1273.
Wang C., Deng L., Hong M., Akkaraju G.R., Inoue J., Chen Z.J.: TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature. 2001, 412: 346-351. 10.1038/35085597.
Hicke L., Schubert H.L., Hill C.P.: Ubiquitin-binding domains. Nat Rev Mol Cell Biol. 2005, 6: 610-621. 10.1038/nrm1701.
Imai Y., Soda M., Takahashi R.: Parkin suppresses unfolded protein stress-induced cell death through its E3 ubiquitin-protein ligase activity. J Biol Chem. 2000, 275: 35661-35664. 10.1074/jbc.C000447200.
Shimura H., Hattori N., Kubo S., Mizuno Y., Asakawa S., Minoshima S., Shimizu N., Iwai K., Chiba T., Tanaka K., Suzuki T.: Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet. 2000, 25: 302-305. 10.1038/77060.
Zhang Y., Gao J., Chung K.K., Huang H., Dawson V.L., Dawson T.M.: Parkin functions as an E2-dependent ubiquitin- protein ligase and promotes the degradation of the synaptic vesicle-associated protein, CDCrel-1. Proc Natl Acad Sci U S A. 2000, 97: 13354-13359. 10.1073/pnas.240347797.
Spillantini M.G., Schmidt M.L., Lee V.M., Trojanowski J.Q., Jakes R., Goedert M.: Alpha-synuclein in Lewy bodies. Nature. 1997, 388: 839-840. 10.1038/42166.
Tanaka Y., Engelender S., Igarashi S., Rao R.K., Wanner T., Tanzi R.E., Sawa A., V L.D., Dawson T.M., Ross C.A.: Inducible expression of mutant alpha-synuclein decreases proteasome activity and increases sensitivity to mitochondria-dependent apoptosis. Hum Mol Genet. 2001, 10: 919-926. 10.1093/hmg/10.9.919.
Stefanis L., Larsen K.E., Rideout H.J., Sulzer D., Greene L.A.: Expression of A53T mutant but not wild-type alpha-synuclein in PC12 cells induces alterations of the ubiquitin-dependent degradation system, loss of dopamine release, and autophagic cell death. J Neurosci. 2001, 21: 9549-9560.
Chen L., Thiruchelvam M.J., Madura K., Richfield E.K.: Proteasome dysfunction in aged human alpha-synuclein transgenic mice. Neurobiol Dis. 2006, 23: 120-126. 10.1016/j.nbd.2006.02.004.
Singleton A.B., Farrer M., Johnson J., Singleton A., Hague S., Kachergus J., Hulihan M., Peuralinna T., Dutra A., Nussbaum R., Lincoln S., Crawley A., Hanson M., Maraganore D., Adler C., Cookson M.R., Muenter M., Baptista M., Miller D., Blancato J., Hardy J., Gwinn-Hardy K.: alpha-Synuclein locus triplication causes Parkinson's disease. Science. 2003, 302: 841-10.1126/science.1090278.
Furukawa Y., Vigouroux S., Wong H., Guttman M., Rajput A.H., Ang L., Briand M., Kish S.J., Briand Y.: Brain proteasomal function in sporadic Parkinson's disease and related disorders. Ann Neurol. 2002, 51: 779-782. 10.1002/ana.10207.
Tofaris G.K., Razzaq A., Ghetti B., Lilley K.S., Spillantini M.G.: Ubiquitination of alpha-synuclein in Lewy bodies is a pathological event not associated with impairment of proteasome function. J Biol Chem. 2003, 278: 44405-44411. 10.1074/jbc.M308041200.
Zeng B.Y., Medhurst A.D., Jackson M., Rose S., Jenner P.: Proteasomal activity in brain differs between species and brain regions and changes with age. Mech Ageing Dev. 2005, 126: 760-766. 10.1016/j.mad.2005.01.008.
Keller J.N., Dimayuga E., Chen Q., Thorpe J., Gee J., Ding Q.: Autophagy, proteasomes, lipofuscin, and oxidative stress in the aging brain. Int J Biochem Cell Biol. 2004, 36: 2376-2391. 10.1016/j.biocel.2004.05.003.
Wang X.F., Li S., Chou A.P., Bronstein J.M.: Inhibitory effects of pesticides on proteasome activity: implication in Parkinson's disease. Neurobiol Dis. 2006, 23: 198-205. 10.1016/j.nbd.2006.02.012.
Betarbet R., Canet-Aviles R.M., Sherer T.B., Mastroberardino P.G., McLendon C., Kim J.H., Lund S., Na H.M., Taylor G., Bence N.F., Kopito R., Seo B.B., Yagi T., Yagi A., Klinefelter G., Cookson M.R., Greenamyre J.T.: Intersecting pathways to neurodegeneration in Parkinson's disease: effects of the pesticide rotenone on DJ-1, alpha-synuclein, and the ubiquitin-proteasome system. Neurobiol Dis. 2006, 22: 404-420. 10.1016/j.nbd.2005.12.003.
Wang C., Ko H.S., Thomas B., Tsang F., Chew K.C., Tay S.P., Ho M.W., Lim T.M., Soong T.W., Pletnikova O., Troncoso J., Dawson V.L., Dawson T.M., Lim K.L.: Stress-induced alterations in parkin solubility promote parkin aggregation and compromise parkin's protective function. Hum Mol Genet. 2005, 14: 3885-3897. 10.1093/hmg/ddi413.
Fornai F., Schluter O.M., Lenzi P., Gesi M., Ruffoli R., Ferrucci M., Lazzeri G., Busceti C.L., Pontarelli F., Battaglia G., Pellegrini A., Nicoletti F., Ruggieri S., Paparelli A., Sudhof T.C.: Parkinson-like syndrome induced by continuous MPTP infusion: convergent roles of the ubiquitin-proteasome system and alpha-synuclein. Proc Natl Acad Sci U S A. 2005, 102: 3413-3418. 10.1073/pnas.0409713102.
Lim K.L., Lim T.M.: Molecular mechanisms of neurodegeneration in Parkinson's disease: clues from Mendelian syndromes. IUBMB Life. 2003, 55: 315-322.
Snyder H., Mensah K., Theisler C., Lee J., Matouschek A., Wolozin B.: Aggregated and monomeric alpha-synuclein bind to the S6' proteasomal protein and inhibit proteasomal function. J Biol Chem. 2003, 278: 11753-11759. 10.1074/jbc.M208641200.
Choi P., Snyder H., Petrucelli L., Theisler C., Chong M., Zhang Y., Lim K., Chung K.K., Kehoe K., D'Adamio L., Lee J.M., Cochran E., Bowser R., Dawson T.M., Wolozin B.: SEPT5_v2 is a parkin-binding protein. Brain Res Mol Brain Res. 2003, 117: 179-189. 10.1016/S0169-328X(03)00318-8.
Imai Y., Soda M., Inoue H., Hattori N., Mizuno Y., Takahashi R.: An unfolded putative transmembrane polypeptide, which can lead to endoplasmic reticulum stress, is a substrate of Parkin. Cell. 2001, 105: 891-902. 10.1016/S0092-8674(01)00407-X.
Staropoli J.F., McDermott C., Martinat C., Schulman B., Demireva E., Abeliovich A.: Parkin Is a Component of an SCF-like Ubiquitin Ligase Complex and Protects Postmitotic Neurons from Kainate Excitotoxicity. Neuron. 2003, 37: 735-749. 10.1016/S0896-6273(03)00084-9.
Ko H.S., von Coelln R., Sriram S.R., Kim S.W., Chung K.K., Pletnikova O., Troncoso J., Johnson B., Saffary R., Goh E.L., Song H., Park B.J., Kim M.J., Kim S., Dawson V.L., Dawson T.M.: Accumulation of the authentic parkin substrate aminoacyl-tRNA synthetase cofactor, p38/JTV-1, leads to catecholaminergic cell death. J Neurosci. 2005, 25: 7968-7978. 10.1523/JNEUROSCI.2172-05.2005.
Ko H.S., Kim S.W., Sriram S.R., Dawson V.L., Dawson T.M.: Identification of far upstream element-binding protein-1 as an authentic Parkin substrate. J Biol Chem. 2006, 281: 16193-16196. 10.1074/jbc.C600041200.
Higashi Y., Asanuma M., Miyazaki I., Hattori N., Mizuno Y., Ogawa N.: Parkin attenuates manganese-induced dopaminergic cell death. J Neurochem. 2004, 89: 1490-1497. 10.1111/j.1471-4159.2004.02445.x.
Petrucelli L., O'Farrell C., Lockhart P.J., Baptista M., Kehoe K., Vink L., Choi P., Wolozin B., Farrer M., Hardy J., Cookson M.R.: Parkin protects against the toxicity associated with mutant alpha-synuclein: proteasome dysfunction selectively affects catecholaminergic neurons. Neuron. 2002, 36: 1007-1019. 10.1016/S0896-6273(02)01125-X.
Yang Y., Nishimura I., Imai Y., Takahashi R., Lu B.: Parkin suppresses dopaminergic neuron-selective neurotoxicity induced by Pael-R in Drosophila. Neuron. 2003, 37: 911-924. 10.1016/S0896-6273(03)00143-0.
Jiang H., Ren Y., Zhao J., Feng J.: Parkin protects human dopaminergic neuroblastoma cells against dopamine-induced apoptosis. Hum Mol Genet. 2004, 13: 1745-1754. 10.1093/hmg/ddh180.
Tsai Y.C., Fishman P.S., Thakor N.V., Oyler G.A.: Parkin facilitates the elimination of expanded polyglutamine proteins and leads to preservation of proteasome function. J Biol Chem. 2003, 278: 22044-22055. 10.1074/jbc.M212235200.
Hampe C., Ardila-Osorio H., Fournier M., Brice A., Corti O.: Biochemical analysis of Parkinson's disease-causing variants of Parkin, an E3 ubiquitin-protein ligase with monoubiquitylation capacity. Hum Mol Genet. 2006, 15: 2059-2075. 10.1093/hmg/ddl131.
Matsuda N., Kitami T., Suzuki T., Mizuno Y., Hattori N., Tanaka K.: Diverse effects of pathogenic mutations of Parkin that catalyze multiple monoubiquitylation in vitro. J Biol Chem. 2006, 281: 3204-3209. 10.1074/jbc.M510393200.
Cookson M.R., Lockhart P.J., McLendon C., O'Farrell C., Schlossmacher M., Farrer M.J.: RING finger 1 mutations in Parkin produce altered localization of the protein. Hum Mol Genet. 2003, 12: 2957-2965. 10.1093/hmg/ddg328.
Gu W.J., Corti O., Araujo F., Hampe C., Jacquier S., Lucking C.B., Abbas N., Duyckaerts C., Rooney T., Pradier L., Ruberg M., Brice A.: The C289G and C418R missense mutations cause rapid sequestration of human Parkin into insoluble aggregates. Neurobiol Dis. 2003, 14: 357-364. 10.1016/j.nbd.2003.08.011.
Wang C., Tan J.M., Ho M.W., Zaiden N., Wong S.H., Chew C.L., Eng P.W., Lim T.M., Dawson T.M., Lim K.L.: Alterations in the solubility and intracellular localization of parkin by several familial Parkinson's disease-linked point mutations. J Neurochem. 2005, 93: 422-431. 10.1111/j.1471-4159.2005.03023.x.
Winklhofer K.F., Henn I.H., Kay-Jackson P.C., Heller U., Tatzelt J.: Inactivation of parkin by oxidative stress and C-terminal truncations: a protective role of molecular chaperones. J Biol Chem. 2003, 278: 47199-47208. 10.1074/jbc.M306769200.
LaVoie M.J., Ostaszewski B.L., Weihofen A., Schlossmacher M.G., Selkoe D.J.: Dopamine covalently modifies and functionally inactivates parkin. Nat Med. 2005, 11: 1214-1221. 10.1038/nm1314.
Pawlyk A.C., Giasson B.I., Sampathu D.M., Perez F.A., Lim K.L., Dawson V.L., Dawson T.M., Palmiter R.D., Trojanowski J.Q., Lee V.M.: Novel monoclonal antibodies demonstrate biochemical variation of brain parkin with age. J Biol Chem. 2003, 278: 48120-48128. 10.1074/jbc.M306889200.
Chung K.K., Thomas B., Li X., Pletnikova O., Troncoso J.C., Marsh L., Dawson V.L., Dawson T.M.: S-nitrosylation of parkin regulates ubiquitination and compromises parkin's protective function. Science. 2004, 304: 1328-1331. 10.1126/science.1093891.
Yao D., Gu Z., Nakamura T., Shi Z.Q., Ma Y., Gaston B., Palmer L.A., Rockenstein E.M., Zhang Z., Masliah E., Uehara T., Lipton S.A.: Nitrosative stress linked to sporadic Parkinson's disease: S-nitrosylation of parkin regulates its E3 ubiquitin ligase activity. Proc Natl Acad Sci U S A. 2004, 101: 10810-10814. 10.1073/pnas.0404161101.
McNaught K.S., Mytilineou C., Jnobaptiste R., Yabut J., Shashidharan P., Jennert P., Olanow C.W.: Impairment of the ubiquitin-proteasome system causes dopaminergic cell death and inclusion body formation in ventral mesencephalic cultures. J Neurochem. 2002, 81: 301-306. 10.1046/j.1471-4159.2002.00821.x.
Rideout H.J., Lang-Rollin I.C., Savalle M., Stefanis L.: Dopaminergic neurons in rat ventral midbrain cultures undergo selective apoptosis and form inclusions, but do not up-regulate iHSP70, following proteasomal inhibition. J Neurochem. 2005, 93: 1304-1313. 10.1111/j.1471-4159.2005.03124.x.
Ding Q., Dimayuga E., Martin S., Bruce-Keller A.J., Nukala V., Cuervo A.M., Keller J.N.: Characterization of chronic low-level proteasome inhibition on neural homeostasis. J Neurochem. 2003, 86: 489-497. 10.1046/j.1471-4159.2003.01885.x.
Sullivan P.G., Dragicevic N.B., Deng J.H., Bai Y., Dimayuga E., Ding Q., Chen Q., Bruce-Keller A.J., Keller J.N.: Proteasome inhibition alters neural mitochondrial homeostasis and mitochondria turnover. J Biol Chem. 2004, 279: 20699-20707. 10.1074/jbc.M313579200.
Miwa H., Kubo T., Suzuki A., Nishi K., Kondo T.: Retrograde dopaminergic neuron degeneration following intrastriatal proteasome inhibition. Neurosci Lett. 2005, 380: 93-98. 10.1016/j.neulet.2005.01.024.
Fornai F., Lenzi P., Gesi M., Ferrucci M., Lazzeri G., Busceti C.L., Ruffoli R., Soldani P., Ruggieri S., Alessandri M.G., Paparelli A.: Fine structure and biochemical mechanisms underlying nigrostriatal inclusions and cell death after proteasome inhibition. J Neurosci. 2003, 23: 8955-8966.
McNaught K.S., Perl D.P., Brownell A.L., Olanow C.W.: Systemic exposure to proteasome inhibitors causes a progressive model of Parkinson's disease. Ann Neurol. 2004, 56: 149-162. 10.1002/ana.20186.
Bove J., Zhou C., Jackson-Lewis V., Taylor J., Chu Y., Rideout H.J., Wu D.C., Kordower J.H., Petrucelli L., Przedborski S.: Proteasome inhibition and Parkinson's disease modeling. Ann Neurol. 2006, 60: 260-264. 10.1002/ana.20937.
Kordower J.H., Kanaan N.M., Chu Y., Suresh Babu R., Stansell J., Terpstra B.T., Sortwell C.E., Steece-Collier K., Collier T.J.: Failure of proteasome inhibitor administration to provide a model of Parkinson's disease in rats and monkeys. Ann Neurol. 2006, 60: 264-268. 10.1002/ana.20935.
Manning-Bog A.B., Reaney S.H., Chou V.P., Johnston L.C., McCormack A.L., Johnston J., Langston J.W., Di Monte D.A.: Lack of nigrostriatal pathology in a rat model of proteasome inhibition. Ann Neurol. 2006, 60: 256-260. 10.1002/ana.20938.
Zeng B.Y., Bukhatwa S., Hikima A., Rose S., Jenner P.: Reproducible nigral cell loss after systemic proteasomal inhibitor administration to rats. Ann Neurol. 2006, 60: 248-252. 10.1002/ana.20932.
Schapira A.H., Cleeter M.W., Muddle J.R., Workman J.M., Cooper J.M., King R.H.: Proteasomal inhibition causes loss of nigral tyrosine hydroxylase neurons. Ann Neurol. 2006, 60: 253-255. 10.1002/ana.20934.
Beal F., Lang A.: The proteasomal inhibition model of Parkinson's disease: “Boon or bust”?. Ann Neurol. 2006, 60: 158-161. 10.1002/ana.20939.
McNaught K.S., Olanow C.W.: Proteasome inhibitor-induced model of Parkinson's disease. Ann Neurol. 2006, 60: 243-247. 10.1002/ana.20936.
Goldberg M.S., Fleming S.M., Palacino J.J., Cepeda C., Lam H.A., Bhatnagar A., Meloni E.G., Wu N., Ackerson L.C., Klapstein G.J., Gajendiran M., Roth B.L., Chesselet M.F., Maidment N.T., Levine M.S., Shen J.: Parkin-deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. J Biol Chem. 2003, 278: 43628-43635. 10.1074/jbc.M308947200.
Itier J.M., Ibanez P., Mena M.A., Abbas N., Cohen-Salmon C., Bohme G.A., Laville M., Pratt J., Corti O., Pradier L., Ret G., Joubert C., Periquet M., Araujo F., Negroni J., Casarejos M.J., Canals S., Solano R., Serrano A., Gallego E., Sanchez M., Denefle P., Benavides J., Tremp G., Rooney T.A., Brice A., Garcia de Yebenes J.: Parkin gene inactivation alters behaviour and dopamine neurotransmission in the mouse. Hum Mol Genet. 2003, 12: 2277-2291. 10.1093/hmg/ddg239.
Von Coelln R., Thomas B., Savitt J.M., Lim K.L., Sasaki M., Hess E.J., Dawson V.L., Dawson T.M.: Loss of locus coeruleus neurons and reduced startle in parkin null mice. Proc Natl Acad Sci U S A. 2004, 101: 10744-10749. 10.1073/pnas.0401297101.
Perez F.A., Palmiter R.D.: Parkin-deficient mice are not a robust model of parkinsonism. Proc Natl Acad Sci U S A. 2005, 102: 2174-2179. 10.1073/pnas.0409598102.
Saigoh K., Wang Y.L., Suh J.G., Yamanishi T., Sakai Y., Kiyosawa H., Harada T., Ichihara N., Wakana S., Kikuchi T., Wada K.: Intragenic deletion in the gene encoding ubiquitin carboxy-terminal hydrolase in gad mice. Nat Genet. 1999, 23: 47-51.
Greene J.C., Whitworth A.J., Kuo I., Andrews L.A., Feany M.B., Pallanck L.J.: Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci U S A. 2003, 100: 4078-4083. 10.1073/pnas.0737556100.
Cha G.H., Kim S., Park J., Lee E., Kim M., Lee S.B., Kim J.M., Chung J., Cho K.S.: Parkin negatively regulates JNK pathway in the dopaminergic neurons of Drosophila. Proc Natl Acad Sci U S A. 2005, 102: 10345-10350. 10.1073/pnas.0500346102.
Whitworth A.J., Theodore D.A., Greene J.C., Benes H., Wes P.D., Pallanck L.J.: Increased glutathione S-transferase activity rescues dopaminergic neuron loss in a Drosophila model of Parkinson's disease. Proc Natl Acad Sci U S A. 2005, 102: 8024-8029. 10.1073/pnas.0501078102.
Abbas-Terki T., Blanco-Bose W., Deglon N., Pralong W., Aebischer P.: Lentiviral-mediated RNA interference. Hum Gene Ther. 2002, 13: 2197-2201. 10.1089/104303402320987888.
Auluck P.K., Chan H.Y., Trojanowski J.Q., Lee V.M., Bonini N.M.: Chaperone suppression of alpha-synuclein toxicity in a Drosophila model for Parkinson's disease. Science. 2002, 295: 865-868. 10.1126/science.1067389.
Dong Z., Wolfer D.P., Lipp H.P., Bueler H.: Hsp70 gene transfer by adeno-associated virus inhibits MPTP-induced nigrostriatal degeneration in the mouse model of Parkinson disease. Mol Ther. 2005, 11: 80-88. 10.1016/j.ymthe.2004.09.007.
Klucken J., Shin Y., Masliah E., Hyman B.T., McLean P.J.: Hsp70 Reduces alpha-Synuclein Aggregation and Toxicity. J Biol Chem. 2004, 279: 25497-25502. 10.1074/jbc.M400255200.
Auluck P.K., Meulener M.C., Bonini N.M.: Mechanisms of Suppression of {alpha}-Synuclein Neurotoxicity by Geldanamycin in Drosophila. J Biol Chem. 2005, 280: 2873-2878. 10.1074/jbc.M412106200.
Shen H.Y., He J.C., Wang Y., Huang Q.Y., Chen J.F.: Geldanamycin induces heat shock protein 70 and protects against MPTP-induced dopaminergic neurotoxicity in mice. J Biol Chem. 2005, 280: 39962-39969. 10.1074/jbc.M505524200.
Combaret L., Taillandier D., Dardevet D., Bechet D., Ralliere C., Claustre A., Grizard J., Attaix D.: Glucocorticoids regulate mRNA levels for subunits of the 19 S regulatory complex of the 26 S proteasome in fast-twitch skeletal muscles. Biochem J. 2004, 378: 239-246. 10.1042/BJ20031660.
Kurkowska-Jastrzebska I., Litwin T., Joniec I., Ciesielska A., Przybylkowski A., Czlonkowski A., Czlonkowska A.: Dexamethasone protects against dopaminergic neurons damage in a mouse model of Parkinson's disease. Int Immunopharmacol. 2004, 4: 1307-1318. 10.1016/j.intimp.2004.05.006.
Cuervo A.M.: Autophagy in neurons: it is not all about food. Trends Mol Med. 2006, 12: 461-464. 10.1016/j.molmed.2006.08.003.
Levine B., Yuan J.: Autophagy in cell death: an innocent convict?. J Clin Invest. 2005, 115: 2679-2688. 10.1172/JCI26390.
Kruse K.B., Brodsky J.L., McCracken A.A.: Characterization of an ERAD gene as VPS30/ATG6 reveals two alternative and functionally distinct protein quality control pathways: one for soluble Z variant of human alpha-1 proteinase inhibitor (A1PiZ) and another for aggregates of A1PiZ. Mol Biol Cell. 2006, 17: 203-212. 10.1091/mbc.E04-09-0779.
Hara T., Nakamura K., Matsui M., Yamamoto A., Nakahara Y., Suzuki-Migishima R., Yokoyama M., Mishima K., Saito I., Okano H., Mizushima N.: Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006, 441: 885-889. 10.1038/nature04724.
Komatsu M., Waguri S., Chiba T., Murata S., Iwata J., Tanida I., Ueno T., Koike M., Uchiyama Y., Kominami E., Tanaka K.: Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006, 441: 880-884. 10.1038/nature04723.
Webb J.L., Ravikumar B., Atkins J., Skepper J.N., Rubinsztein D.C.: Alpha-Synuclein is degraded by both autophagy and the proteasome. J Biol Chem. 2003, 278: 25009-25013. 10.1074/jbc.M300227200.
Williams A., Jahreiss L., Sarkar S., Saiki S., Menzies F.M., Ravikumar B., Rubinsztein D.C.: Aggregate-prone proteins are cleared from the cytosol by autophagy: therapeutic implications. Curr Top Dev Biol. 2006, 76: 89-101.
Sarkar S., Davies J.E., Huang Z., Tunnacliffe A., Rubinsztein D.C.: Trehalose, a Novel mTOR-independent Autophagy Enhancer, Accelerates the Clearance of Mutant Huntingtin and {alpha}-Synuclein. J Biol Chem. 2007, 282: 5641-5652. 10.1074/jbc.M609532200.
Sarkar S., Floto R.A., Berger Z., Imarisio S., Cordenier A., Pasco M., Cook L.J., Rubinsztein D.C.: Lithium induces autophagy by inhibiting inositol monophosphatase. J Cell Biol. 2005, 170: 1101-1111. 10.1083/jcb.200504035.
Lo Bianco C., Schneider B.L., Bauer M., Sajadi A., Brice A., Iwatsubo T., Aebischer P.: Lentiviral vector delivery of parkin prevents dopaminergic degeneration in an alpha-synuclein rat model of Parkinson's disease. Proc Natl Acad Sci U S A. 2004, 101: 17510-17515. 10.1073/pnas.0405313101.
Yamada M., Mizuno Y., Mochizuki H.: Parkin gene therapy for alpha-synucleinopathy: a rat model of Parkinson's disease. Hum Gene Ther. 2005, 16: 262-270. 10.1089/hum.2005.16.262.
Vercammen L., Van der Perren A., Vaudano E., Gijsbers R., Debyser Z., Van den Haute C., Baekelandt V.: Parkin protects against neurotoxicity in the 6-hydroxydopamine rat model for Parkinson's disease. Mol Ther. 2006, 14: 716-723. 10.1016/j.ymthe.2006.06.009.
Paterna J.C., Leng A., Weber E., Feldon J., Bueler H.: DJ-1 and Parkin modulate dopamine-dependent behavior and inhibit MPTP-induced nigral dopamine neuron loss in mice. Mol Ther. 2007, 15: 698-704. 10.1038/sj.mt.6300154.
Yasuda T., Miyachi S., Kitagawa R., Wada K., Nihira T., Ren Y.R., Hirai Y., Ageyama N., Terao K., Shimada T., Takada M., Mizuno Y., Mochizuki H.: Neuronal specificity of alpha-synuclein toxicity and effect of Parkin co-expression in primates. Neuroscience. 2007, 144: 743-753. 10.1016/j.neuroscience.2006.09.052.
Haywood A.F., Staveley B.E.: Parkin counteracts symptoms in a Drosophila model of Parkinson's disease. BMC Neurosci. 2004, 5: 14-10.1186/1471-2202-5-14.
Healy D.G., Abou-Sleiman P.M., Wood N.W.: Genetic causes of Parkinson's disease: UCHL-1. Cell Tissue Res. 2004, 318: 189-194. 10.1007/s00441-004-0917-3.
Lim K.L., Chew K.C., Tan J.M., Wang C., Chung K.K., Zhang Y., Tanaka Y., Smith W., Engelender S., Ross C.A., Dawson V.L., Dawson T.M.: Parkin mediates nonclassical, proteasomal-independent ubiquitination of synphilin-1: implications for Lewy body formation. J Neurosci. 2005, 25: 2002-2009. 10.1523/JNEUROSCI.4474-04.2005.
Doss-Pepe E.W., Chen L., Madura K.: Alpha-synuclein and parkin contribute to the assembly of ubiquitin lysine 63-linked multiubiquitin chains. J Biol Chem. 2005, 280: 16619-16624. 10.1074/jbc.M413591200.
Fallon L., Belanger C.M., Corera A.T., Kontogiannea M., Regan-Klapisz E., Moreau F., Voortman J., Haber M., Rouleau G., Thorarinsdottir T., Brice A., van Bergen En Henegouwen P.M., Fon E.A.: A regulated interaction with the UIM protein Eps15 implicates parkin in EGF receptor trafficking and PI(3)K-Akt signalling. Nat Cell Biol. 2006, 8: 834-842. 10.1038/ncb1441.
Feany M.B., Pallanck L.J.: Parkin: a multipurpose neuroprotective agent?. Neuron. 2003, 38: 13-16. 10.1016/S0896-6273(03)00201-0.
Landau A.M., Kouassi E., Siegrist-Johnstone R., Desbarats J.: Proteasome inhibitor model of Parkinson's disease in mice is confounded by neurotoxicity of the ethanol vehicle. Mov Disord. 2007, 22: 403-407. 10.1002/mds.21306.
Donohue T.M., Kharbanda K.K., Casey C.A., Nanji A.A.: Decreased proteasome activity is associated with increased severity of liver pathology and oxidative stress in experimental alcoholic liver disease. Alcohol Clin Exp Res. 2004, 28: 1257-1263. 10.1097/01.ALC.0000134233.89896.19.
Lim K.L., Dawson V.L., Dawson T.M.: Parkin-mediated lysine 63-linked polyubiquitination: a link to protein inclusions formation in Parkinson's and other conformational diseases?. Neurobiol Aging. 2006, 27: 524-529. 10.1016/j.neurobiolaging.2005.07.023.
Mukhopadhyay D., Riezman H.: Proteasome-independent functions of ubiquitin in endocytosis and signaling. Science. 2007, 315: 201-205. 10.1126/science.1127085.
Publication history
Republished from Current BioData's Targeted Proteins database (TPdb; http://www.targetedproteinsdb.com).
Acknowledgements
The authors would like to acknowledge the following agencies for their support: Singapore Millennium Foundation (JT), Singhealth Foundation, National Medical Research Council and A*STAR Biomedical Research Council (LKL).
This article has been published as part of BMC Biochemistry Volume 8 Supplement 1, 2007: Ubiquitin-Proteasome System in Disease Part 1. The full contents of the supplement are available online at http://www.biomedcentral.com/1471-2091/8?issue=S1.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an open access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Lim, KL., Tan, J.M. Role of the ubiquitin proteasome system in Parkinson's disease. BMC Biochem 8 (Suppl 1), S13 (2007). https://doi.org/10.1186/1471-2091-8-S1-S13
Published:
DOI: https://doi.org/10.1186/1471-2091-8-S1-S13