
Abstract
Background
The rapid desensitization of the human prostacyclin (IP) in response to agonist binding has been shown in cell culture. Phosphorylation of the IP receptor by protein kinase C (PKC) has been suggested to be involved in this process.
Methods and results
In this study we investigated the vasodilatory effects of iloprost, a stable prostacyclin analogue, in perfused rabbit lungs. Continuous infusion of the thromboxane mimetic U46619 was employed to establish stable pulmonary hypertension. A complete loss of the vasodilatory response to iloprost was observed in experiments with continuous iloprost perfusion, maintaining the intravascular concentration of this prostanoid over a 180 min period. When lungs under chronic iloprost infusion were acutely challenged with inhaled iloprost, a corresponding complete loss of vasoreactivity was observed. This desensitization was not dependent on upregulation of cAMP-specific phosphodiesterases or changes in adenylate cyclase activity, as suggested by unaltered dose-response curves to agents directly affecting these enzymes. Application of a prostaglandin E1 receptor antagonist 6-isopropoxy-9-oxoxanthene-2-carboxylic acid (AH 6809) or the PKC inhibitor bisindolylmaleimide I (BIM) enhanced the vasodilatory response to infused iloprost and partially prevented tachyphylaxis.
Conclusion
A three-hour infusion of iloprost in pulmonary hypertensive rabbit lungs results in complete loss of the lung vasodilatory response to this prostanoid. This rapid desensitization is apparently not linked to changes in adenylate cyclase and phosphodiesterase activation, but may involve PKC function and co-stimulation of the EP1 receptor in addition to the IP receptor by this prostacyclin analogue.
Similar content being viewed by others


Background
Prostacyclin (PGI2) is the major product of cyclooxygenases (COX) in the vascular endothelium and mediates potent anti-platelet, vasodilator, and anti-inflammatory actions by a prostacyclin receptor (IP) [1]. This receptor is a member of the G protein-coupled receptor (GPCR) superfamily and is coupled to adenylate cyclase (AC) and phospholipase C (PLC) [2–4]. The prostanoid receptors are classified into DP, IP, EP (EP 1–4), FP and TP receptors [5–7] with different affinities for agonists and different roles in signal transduction. The IP, EP2, EP4 and DP receptors are coupled to stimulation of adenylate cyclase, while the TP, EP1 and FP receptors are coupled to Ca2+ mobilization. The EP3 receptor is an alternative spliced gene, with at least 8 isoforms identified so far. Depending on the subtype, this receptor can be negatively or positively coupled to Gs [8, 9]. Agonist (PGI2 or PGI2 mimetics such as cicaprost, iloprost, carbacyclin, and prostaglandin E1)-binding to the IP receptor leads to activation of protein kinase A (PKA) by cyclic adenosine monophosphate (cAMP) [10]. Although, the above mentioned agonist specificities for IP receptors vary and none of them were highly selective for IP receptors alone. The affinity of cicaprost for human IP receptors is merely 3-fold higher than for the prostaglandin E2 EP4 subtype of receptor, which also couples to Gs [11], and only 17-fold higher than for the EP3 receptor in the mouse [12]. Iloprost is equipotent at both human and mouse IP and EP1 receptors, and carbacyclin and prostaglandin E1 show even greater affinity for EP3 than for IP receptors [11, 12].
Disturbances to PGI2 synthesis [13, 14], as well as polymorphisms of the PGI2-synthase (PGIS) [15], have been linked to severe pulmonary hypertension. Substitution of PGI2, either by overexpression of the PGIS [16] in an experimental model or by application of PGI2 [17, 18] or its analogues iloprost [19, 20] or beraprost [21, 22] in patients decreases pulmonary artery pressure. However, tolerance of the lung vasodilatory response to continuously infused PGI2 rapidly develops in patients with severe pulmonary hypertension, and dose adjustments have to be made [17, 23]. In COPD patients with pulmonary hypertension, the pulmonary vasodilatory response to continuously infused PGI2 was found to dissipate within 24 h [24]. Marked tolerance to the anti-mitogenic actions of PGI2 developed within 24 h in coronary artery smooth muscle cells [25]. Basic studies demonstrated that desensitization of the IP receptor occurs within minutes after exposure to agonists and is due to agonist-induced receptor phosphorylation, mainly mediated by PKC [26], with subsequent sequestration of the intact receptor and removal from the cell surface [26, 27]. In addition, there is evidence for changes in adenylate cyclase and phosphodiesterase (PDE) activation occurring in response to IP receptor stimulation, which may contribute to loss of the vasodilatory response to PGI2 and its analogues [28–30]. In the present study, we employed the model of pulmonary hypertensive perfused rabbit lungs to investigate the dose-response relationship and features of tolerance development upon continuous iloprost infusion in the intact lung vasculature.
Materials and methods
Materials
Sterile Krebs-Henseleit buffer (KHB) was obtained from Serag-Wiessner (Naila, Germany). The thromboxane-A2 mimetic U46619 was supplied by Paesel-Lorei (Frankfurt, Germany) and iloprost by Schering (Berlin, Germany). All other chemicals were purchased from Merck (Darmstadt, Germany).
Isolated lung model
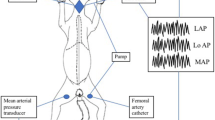
The perfused rabbit lung model has previously been described in detail [31]. Briefly, rabbits of either sex weighing 2.6 to 2.9 kg were anticoagulated with heparin (1000 U/kg) and anaesthetized with intravenous ketamine/xylazine. Tracheostomy was performed and the animals were ventilated with room air. After mid-sternal thoracotomy, catheters were placed into the pulmonary artery and the left atrium, and perfusion with KHB was started. For washout of blood, perfusate was initially not recirculated and the lungs were removed without interruption of ventilation and perfusion. The lungs were placed in a temperature-equilibrated chamber at 37.5°C, freely suspended from a force transducer for monitoring of organ weight. In a recirculating system the flow was slowly increased to 120 ml/min (total volume 350 ml). Left atrial pressure was set at 2 mmHg in all experiments. In parallel with onset of artificial perfusion, an air mixture of 80.5% N2, 15% O2 and 4.5% CO2 was used for ventilation. Tidal volume (11 ml/kg) and frequency (10 – 13 breaths/min) were adapted to maintain the pH of the recirculating buffer in the range between 7.35 and 7.37. A positive end-expiratory pressure of 1 mmHg was used throughout. The PO2 and PCO2 values in the post-lung buffer fluid ranged between 100 and 120 mmHg and 38 and 43 mmHg, respectively. Pressures in the pulmonary artery, the left atrium and the trachea were registered by means of small diameter tubing threaded into the perfusion catheters and the trachea and connected to pressure transducers (zero referenced at the hilum). The whole system was heated to 37.5°C.
Lungs included in the study had 1) a homogeneous white appearance with no signs of hemostasis, edema or atelectasis; 2) pulmonary artery and ventilation pressures in the normal range; and were 3) isogravimetric (lung weight gain < 0.2 g/h) during an initial steady state period of at least 45–60 min.
Experimental protocols
Isolated lung experiments
As described previously [32], a sustained increase of PPA from ≈6 to ≈24 mmHg was achieved by continuous infusion of the thromboxane-mimetic U46619 with a dose range of 70–160 pmol·kg-1·min-1. Individual titration was performed. This level of pulmonary hypertension was then maintained for at least 320 min with variations in PPA of less than 2 mmHg. The following experimental groups were employed:
Control lungs (n = 4): No interventions were undertaken.
U46619-infused lungs (n = 4): After termination of the steady state period, U46619 was continuously infused for 320 min to provoke an increase in PPA to ~24 mmHg.
Iloprost infusion (n = 4): U46619 was administered as described. 20 min after onset of U46619 infusion, iloprost was applied by a bolus injection of 200 ng, followed by a continuous infusion of 33 ng/kg/h to achieve stable buffer concentrations in the range of 350 pg/ml.
Intravenous PGI 2 application (n = 4): U-46619 was administered as described. 20 min after onset of U-46619 infusion, PGI2 was continuously applied at a dose of 50 ng· kg-1·min-1 until termination of the experiments.
Rolipram i.v. (n = 4): After adjusting stable pulmonary hypertension, the PDE4 inhibitor rolipram was applied in increasing doses (dose range 0.001–10 μM).
Iloprost infusion and rolipram i.v. (n = 4): Iloprost was applied by a bolus injection of 200 ng, followed by a continuous infusion of 33 ng/kg/h. 240 min after starting the iloprost infusion, rolipram was applied in increasing doses (dose range 0.001–10 μM).
Motapizone i.v. (n = 4): After adjusting stable pulmonary hypertension, the PDE3 inhibitor motapizone was applied in increasing doses (dose range 0.01–100 μM).
Iloprost infusion and motapizone i.v. (n = 4): Iloprost was applied by a bolus injection of 200 ng, followed by a continuous infusion of 33 ng/kg/h. 240 min after starting the iloprost infusion, motapizone was applied in increasing doses (dose range 0.01–100 μM).
Iloprost inhalation (n = 4): When stable pulmonary hypertension was achieved, iloprost was nebulized (deposited dose 75 ng) by means of an ultrasonic nebulizer as described previously [33, 34].
Iloprost infusion and iloprost inhalation (n = 4): U46619 was continuously infused and iloprost was applied by a bolus injection of 200 ng, followed by a continuous infusion of 33 ng/kg/h. 240 min after starting the iloprost infusion, iloprost was nebulized (deposited dose 75 ng).
Forskolin i.v. (n = 4): After adjusting stable pulmonary hypertension, the adenylate cyclase stimulator forskolin was applied in increasing doses (dose range 10–100 μM).
Iloprost infusion and forskolin i.v. (n = 4): Iloprost was applied by a bolus injection of 200 ng, followed by a continuous infusion of 33 ng/kg/h. 240 min after starting the iloprost infusion, forskolin was applied in increasing doses (dose range 10–100 μM).
Bisindolylmaleimide I (BIM) infusion (n = 4): BIM (1 μM) was applied by a bolus injection after stable adjustment of pulmonary hypertension with U46619. As described previously, stable buffer levels of BIM are achieved by a single application in the setup of isolated perfused rabbit lungs [35].
Iloprost infusion in presence of BIM (n = 4): U46619 was administered as described. After stable adjustment of pulmonary hypertension, BIM (1 μM) was applied by a bolus injection. Iloprost was bolus injected and infused as described above.
6-isopropoxy-9-oxoxanthene-2-carboxylic acid (AH 6809) infusion (n = 4): AH 6809 (3 μM) was applied by a bolus injection after stable adjustment of pulmonary hypertension with U46619. As described previously, AH6809 demonstrated long acting EP1 blocking activity in fluid perfused small intestine segments after a single application [36].
Iloprost infusion in presence of AH 6809 (n = 4): U46619 was administered as described. 45 min after onset of U46619 infusion, AH 6809 (3 μM) was applied as a bolus injection and the bolus injection plus infusion of iloprost was started as in the preceding groups.
Cell culture
Rabbit pulmonary smooth muscle cells (PSMC) were prepared as described. Primary SMC were isolated from rabbit pulmonary artery by carefully preparing <1 mm3 pieces of media, devoid of adventitial tissue as assessed by microscopic control. The pieces of media were placed into 12-well cell culture plates with 500 μl culture medium. The isolated pulmonary smooth muscle cells (PSMC) identity was verified by characteristic appearance in phase-contrast microscopy, indirect immunofluorescent antibody staining for smooth muscle-specific isoforms of α-actin and myosin (at least 95% of cells stained positive), and lack of staining for von Willebrand factor and vimentin, indicating that the cultures did not contain significant numbers of endothelial cells or adventitial fibroblasts [37]. Smooth muscle cells were grown in Dulbecco's modified Eagle's high glucose medium supplemented with 10% fetal bovine serum, 100 μg/ml streptomycin and 100 units/ml penicillin.
Desensitization assays and cAMP measurement
Smooth muscle cells were grown to 90% confluence in 24-well plates, as previously described. For desensitization assays, cells were treated with iloprost for the indicated times. Then, cells were washed three times with HBSS and preincubated in HBSS containing 1 mg/ml BSA, 10 mM HEPES (pH 7.3) and 1 mM IBMX for 10 min at 37°C. Afterwards cells were again stimulated with iloprost (100 nM) or forskolin (10 μM) for 10 min and cAMP measurements were performed as described below. Reactions were stopped by aspiration and addition of ice-cold 96% ethanol. Dried samples were overlaid with 300 μl RIA-buffer (150 mM NaCl, 8 mM Na2HPO4, 2 mM NaH2PO4, pH 7.4) and frozen overnight at -80°C. cAMP in the supernatant was determined by radioimmunoassay [Steiner et al., 1972]. Protein determination was performed according to the method of Bradford. Cells treated with vehicle alone (no ligand) or forskolin (50 μM) were served as negative and positive controls respectively for the cAMP measurement.
Data analysis
Values are means ± SEM. Student's t-test was performed for comparison of two values. For multiple comparisons, ANOVA was followed by post hoc test (Bonferroni/Dunn method). Statistical significance was considered at p < 0.05.
Results
Baseline conditions
After termination of the steady state period, all lungs displayed PPA values in the range between 5 and 7 mmHg.
U46619-elicited pulmonary hypertension
Continuous infusion of U46619 provoked an increase in PPA to 23.4 ± 0.9 mmHg within 25 min, with subsequent plateau of the pulmonary hypertension. This level of pulmonary hypertension was then maintained for at least 300 min with variations in PPA of less than 2 mmHg. Total weight gain at the end of the experiments was 6.5 ± 2.7 g.
U46619-elicited pulmonary hypertension and infusion of iloprost or PGI2
As in the preceding group, U46619 infusion for establishing stable pulmonary hypertension was performed. Bolus injection of 200 ng iloprost was followed by an infusion of 33 ng iloprost per hour. This procedure resulted in a significant vasodilatory response with PPA values decreasing by a mean of 3.3 mmHg. Similarly, the continuous infusion of a more selective IP receptor ligand, PGI2 also resulted in significant vasodilatory responses (Fig 1). Stable iloprost buffer concentrations in the range of 350–380 pg/ml were documented over the entire perfusion period. A slow increase in PPA toward pre-infusion values was noted to commence after 100–120 min, and 200 min after beginning of the iloprost infusion, the vasodilatory effect was virtually fully lost. The total weight gain of the lungs was 5.3 ± 3.2 g, and thus did not differ from the groups with mono U46619 infusion.

Prostacyclin receptor (IP) desensitization in U46619-induced pulmonary hypertension in intact rabbit lungs in response to PGI 2 or iloprost infusion. Pulmonary hypertension was adjusted by continuous infusion of the thromboxane mimetic U46619. After a bolus injection of 200 ng iloprost followed by an infusion of 33 ng iloprost per hour (stable buffer levels of ~350 pg/ml) or continuous infusion of PGI2 (50 ng/min) is administered and measured their influence on U46619-elicited pulmonary hypertension.
Dose-effect curves of PDE-inhibitors with or without infusion of iloprost
As shown in Fig. 2A, the intravascular administered PDE3 inhibitor motapizone (dose range 0.01–100 μM) effected a dose-dependent reduction of the elevated PPA values in lungs with U46619-elicited pulmonary hypertension. There was no shift in the dose response curve when applying identical doses of motapizone 240 min after onset of iloprost infusion. Similar experiments were performed with the PDE4 inhibitor rolipram, which dose-dependently reduced pulmonary artery pressure in U46619 preconstricted lungs, with identical dose-effect curves in the absence and presence of iloprost infusion (Fig. 2B). Total weight gain was 5.7 ± 2.9 g and 5.3 ± 1.9 g (iloprost infusion) in the motapizone treated lungs and 4.6 ± 2.2 g and 5.0 ± 2.6 g (iloprost infusion) in the rolipram treated lungs, respectively.

Dose effect curve of the PDE3 inhibitor motapizone or PDE4 inhibitor rolipram on U46619-elevated pulmonary artery pressure in untreated and desensitized (post iloprost infusion) lungs. Motapizone and rolipram dose-dependently reduced the elevated pulmonary artery pressure. There was no change in the dose response curve when applying identical doses of motapizone 240 min after iloprost infusion. (A) The decrease in pulmonary artery pressure (ΔPAP, as decrease of U46619-induced pulmonary hypertension) in response to motapizone or (B) rolipram (concentrations related to the recirculating perfusate) is given. (mean ± SEM of four independent experiments each; SEM bars are not included when smaller than symbol). *: p < 0.05 as compared to U46619-elevated pulmonary artery pressure.
Nebulization of iloprost
Inhalation of aerosolized iloprost (total dose 75 ng inhaled over 10 min) resulted in a significant reduction of the U46619-induced pulmonary hypertension, with PAP values decreasing by a mean of 6.3 mmHg (Fig. 3). In separate experiments, iloprost was infused in advance for 240 min, and then nebulization was started. There was virtually no response to inhaled iloprost after this preceding iloprost infusion period. The total weight gain of the lungs was 6.7 ± 2.9 g at the end of experiments.

Effect of inhaled iloprost on U46619-elevated pulmonary artery pressure in untreated and desensitized (post iloprost infusion) lungs. Inhalation of aerosolized iloprost for 10 min (deposited dose 75 ng) resulted in a significant reduction of the U46619-induced pulmonary hypertension, with PAP values decreasing by a mean of 6.3 mmHg. The vasodilatory potency of inhaled iloprost was completely lost in the desensitization group (iloprost infusion for 240 min). *: p < 0.05 as compared to U46619-elevated pulmonary artery pressure.
Dose-effect curves of forskolin
As shown in Fig. 4, the adenylate cyclase activator forskolin (dose range 10–300 μM) reduced the U46619-elicited pulmonary hypertension in a dose dependant manner. After iloprost infusion for 240 min, an identical dose response curve was observed. Total weight gain was 5.6 ± 2.1 g at the end of the experiments.

Dose effect curve of the adenylate cyclase activator forskolin on U46619-elevated pulmonary artery pressure in untreated and desensitized (post iloprost infusion) lungs. Forskolin dose-dependently reduced the elevated pulmonary artery pressure. There was no change in the dose response curve when applying identical doses of forskolin 240 min after iloprost infusion. The decrease in pulmonary artery pressure (ΔPAP, as decrease of U46619-induced pulmonary hypertension) in response to forskolin (concentrations related to the recirculating perfusate) is given (mean ± SEM of 4 independent experiments each; SEM bars are not included when smaller than symbol). *: p < 0.05 as compared to U46619-elevated pulmonary artery pressure.
Infusion of iloprost: effects of BIM
As in the preceding groups, U46619 infusion for establishing stable pulmonary hypertension was performed. Subsequently, the PKC inhibitor BIM at a dose of 1 μM was applied alone or in combination with iloprost. In combination group, after 5 min of BIM application, bolus injection of 200 ng iloprost was followed by an infusion of 33 ng iloprost per hour. This procedure resulted in a significant vasodilatory response with PPA values decreasing by a mean of 4.3 mmHg (Fig. 5A). However, the sole BIM application demonstrated no effects on U46619-elicited pulmonary hypertension (Fig. 5A). The calculated area under the curve (AUC) of the pressure curve was significantly increased as compared to sole iloprost infusion (Fig. 6). The loss of the vasodilatory response to iloprost was somewhat retarded, but not prevented, as compared to single iloprost application. The total weight gain of the lungs was 5.8 ± 2.5 g.

Influence of iloprost infusion and its combination with AH 6809 and BIM on U46619-elicited pulmonary hypertension. Pulmonary artery pressure (PAP) is given (mean ± SEM of 4 independent experiments each, SEM bars are missing when enclosed by the symbol). (A) The prostaglandin E1 receptor antagonist AH 6809 alone or in combination with iloprost (pre-applied followed by an iloprost infusion). (B) The PKC inhibitor BIM alone or in combination with iloprost (pre-applied followed by an iloprost infusion).

Influence of iloprost infusion and its combination with AH 6809 and BIM on the area under the curve of the U46619-elicited pulmonary hypertension. Area under the curve (AUC) is given (mean ± SEM of 4 independent experiments each). The AUC was calculated by standard techniques and is given as mmHg × min. **: p < 0.01; ***: p < 0.001 as compared to AUC of sole iloprost infusion.
Infusion of iloprost: effects of AH 6809
After adjusting stable pulmonary hypertension, the EP1 receptor antagonist AH 6809 at a dose of 3 μM was applied alone or in combination with iloprost, without effecting pulmonary artery pressure. In combination group, after 5 min of AH 6809 application, a bolus injection of 200 ng iloprost was followed by an infusion of 33 ng iloprost per hour. The maximum vasodilatory response to iloprost was 11.4 mmHg (Fig. 5B) and the area under the curve (AUC) of the pressure curve was 305 mmHg × min (p < 0.05 versus sole iloprost infusion) (Fig. 6). The total weight gain of the lungs was 4.8 ± 2.0 g. In contrary, the AH 6809 application alone had no effect on U46619-induced tension (Fig. 5B).
Measurement of cyclic AMP
We assayed the desensitization kinetics of iloprost and forskolin in rabbit pulmonary smooth muscle cells (Fig. 7). Incubation of cells with iloprost led to a time-dependent reduction of iloprost induced cAMP formation to 88% (1 h), 71% (2 h), 76% (3 h), 66% (5 h), 54% (7 h) and 26% (24 h) of control. Incubation of cells with iloprost and stimulation of cells with forskolin led to a reduction of cAMP formation to 83% (1 h), 84% (2 h), 91% (3 h), 83% (5 h), 79% (7 h) and 75% (24 h) of control.

Prostacyclin receptor (IP) desensitization in rabbit pulmonary smooth muscle cells. Cells were continuously stimulated with 100 nM iloprost for the indicated preincubation times. cAMP response to iloprost (100 nM) or forskolin (10 μM) was assayed in these cells and is given as a percent of the non-desensitized control. Values are expressed as mean ± SEM of three independent experiments performed in triplicate. *: p < 0.05; ***: p < 0.001 as compared to non-desensitized control.
Discussion
The desensitization of the IP receptor after incubation of platelets [38, 39] or IP receptor overexpressing cells [26, 27, 40, 41] with PGI2 or its analogues in vitro is a well-known phenomenon. Tolerance development of the lung vasodilatory response to PGI2 has also been observed in patients with severe pulmonary hypertension being chronically treated by intravenous infusion of the prostanoid [23, 24]. In the present study we show that such desensitization occurs within 180 min in perfused rabbit lungs.
Desensitization is not dependant on increased PDE activity
Phosphodiesterases (PDE) are enzymes that hydrolyze cyclic AMP and cyclic GMP, the second messengers of PGI2, and NO [42, 43]. The characterization of the various PDEs currently known has profited from the employment of selective PDE inhibitors. Concerning the lung vasculature, the presence of the PDE isoenzymes 1, 3, 4 and 5 in the cytosolic and particulate phases (homogenized human pulmonary artery tissue) has been demonstrated [44]. There is strong evidence of PDE activation and upregulation in response to intracellular elevation of cAMP. It has been shown recently that cyclic AMP upregulates isoforms of PDE4 in airway smooth muscle cells [45], monocytes [46], endothelial cells [47] and Jurkat T-cells [48]. In addition, short term activation (phosphorylation) of PDE4 via PKA was demonstrated by treatment of cells with forskolin, a stimulator of adenylate cyclase [49]. Furthermore, treatment of rats with 7-oxo-prostacyclin resulted in a significant increase in PDE4 activity, which attenuated the elevation of isoprenaline-induced cAMP levels as well as the contractile force development in a Langedorff-heart preparation [50]. Similar results were obtained for PDE3 which has been shown to be regulated on the level of activity [48] and expression [32] by increased cAMP levels. In the current study, we employed motapizone and rolipram, selective inhibitors of PDE3 and PDE4, respectively and performed dose response studies on vasodilation in normal and desensitized (iloprost-treated) isolated rabbit lungs. As reported earlier, these PDE inhibitors are potent pulmonary vasodilators [51] and we found no shift in the dose response curve in desensitized lungs. Thus, there is no evidence of increased PDE activity in iloprost-treated isolated rabbit lungs.
Desensitization is not dependant on adenylate cyclase activity
We employed forskolin as direct activator of adenylate cyclase and performed dose response studies on pulmonary vasodilation in isolated rabbit lungs. The response to forskolin in the desensitized (iloprost-treated) organs remained intact (and seemed to be slightly increased), indicating no regulation of adenylate cyclase. Similar results were obtained in rats chronically treated with albuterol in which acetylcholine (ACh)-induced bronchoconstriction was reversible by forskolin, but not by albuterol itself, suggesting that the intracellular site of desensitization is upstream of adenylate cyclase [27].
Moreover, the cAMP production of iloprost-pretreated rabbit pulmonary smooth muscle cells in response to forskolin was also not significantly reduced, except in 24 hours of iloprost-pretreated group (to 75% of control). This is quite contrary to iloprost responses, where a significantly reduced cAMP levels (to 25% of control) was observed, thereby excluding the down-regulation of adenylate cyclase as a possible reason for reduced cAMP formation after continuous stimulation of cells with iloprost. In this context, desensitization of IP receptors by iloprost may possibly explained by its direct influence on the receptor, a mechanism upstream of adenylate cyclases. This involves rapid receptor phosphorylation, which results in uncoupling of receptor-G protein interactions and subsequent receptor internalization. These events result in a diminished response to agonist [26]. This may also be due to preferential reduction of domain-bound pool of the cognate Gproteins GsαL and GsαS [52].
On the other hand, iloprost induced vasorelaxation responses may also be via cAMP-independent manner. Consistent with the observation, PGI2 analogues iloprost were shown to activate MaxiK and KIR channel and subsequent vasorelaxation through a cAMP-independent, G(s)-protein mediated mechanism in vascular smooth muscle cells [53, 54]. Concomitantly in guinea-pig aorta, cyclic AMP-dependent and -independent pathways were recently proposed to underlie the relaxation induced by the PGI2 analogues, iloprost and Beraprost [55–57]. In contrast, in rat tail artery, it is thought that channel phosphorylation by cyclic AMP-dependent PKA is responsible for the MaxiK channel-mediated vascular relaxation due to IP receptor stimulation by iloprost [58, 59]. This is well in line with investigations in human fibroblasts, where cells were challenged at any time point of iloprost pre-treatment with forskolin and fully maintained their cAMP response [30]. In contrast, Sobolewski et al., demonstrated in rat pulmonary smooth muscle cells (PSMCs) which were exposed to cicaprost, another PGI2 analogue, a significant reduction in forskolin-induced cAMP production [60] as well as a decrease in adenylate cyclase isoform 2 and 5/6 expression after one hour of exposure to this prostanoid. However, both isoforms were expressed at control levels when PSMCs were incubated for longer time periods with cicaprost. It is possible that the agonists employed, as well as species differences are responsible for these conflicting data.
Iloprost influences the EP1 receptor in addition to the IP receptor
It is well known that iloprost, the clinically approved compound for treatment of thromboangiitis obliterans [20] and pulmonary arterial hypertension [5], is not highly selective for the IP receptor but also activates the EP1 receptor which is coupled to Gq and signals via IP3 generation and increased intracellular calcium (Ca2+) [61]. To investigate the effects of possible involvement of the EP1 receptor we employed the EP1 receptor antagonist AH 6809 and found a dramatic increase in the efficacy of intravenous iloprost. In contrary, the AH 6809 application alone had no effect on U46619-induced tension. Thus, the agonistic effects on EP1 receptor explain the differences between iloprost and selective IP agonists in inhibition of smooth muscle cell response [26, 40].
PKC inhibition and IP receptor desensitization
There is evidence that the initial desensitization process is dependent on phosphorylation of the IP receptor by PKC [41, 62]. After phosphorylation, the IP receptor is sequestered to clathrin-coated vesicles and by a dynamin-dependant process internalized into endosomes [41]. This receptor sequestration is, unlike desensitization, PKC-independent [27] and a time of approximately 18 h is needed for a resensitization in human fibroblasts [27, 30]. However, several studies including our work employed inhibitors of PKC (GF-109203X, BIM) and could not or could only partially prevent receptor desensitization, suggesting a minor role of PKC in the desensitization process.
In conclusion, this study demonstrates for the first time in an intact lung model that the PGI2 analogue iloprost caused desensitization of the IP receptor within 2 to 3 hours, resulting in complete loss of responsiveness after this time period. Desensitization was not due to increased PDE activity or loss of adenylate cyclase activity, but may involve PKC function and co-stimulation of the EP1 receptor in addition to the IP receptor by this PGI2 analogue.
References
Vane JR, Botting RM: Pharmacodynamic profile of prostacyclin. Am J Cardiol 1995, 75:3A-10A.
Boie Y, Rushmore TH, Darmon-Goodwin A, Grygorczyk R, Slipetz DM, Metters KM, Abramovitz M: Cloning and expression of a cDNA for the human prostanoid IP receptor. J Biol Chem 1994, 269:12173–12178.
Coleman RA, Smith WL, Narumiya S: International Union of Pharmacology classification of prostanoid receptors: properties, distribution, and structure of the receptors and their subtypes. Pharmacol Rev 1994, 46:205–229.
Namba T, Oida H, Sugimoto Y, Kakizuka A, Negishi M, Ichikawa A, Narumiya S: cDNA cloning of a mouse prostacyclin receptor. Multiple signaling pathways and expression in thymic medulla. J Biol Chem 1994, 269:9986–9992.
Breyer RM, Bagdassarian CK, Myers SA, Breyer MD: Prostanoid receptors: subtypes and signaling. Annu Rev Pharmacol Toxicol 2001, 41:661–690.
Negishi M, Sugimoto Y, Ichikawa A: Molecular mechanisms of diverse actions of prostanoid receptors. Biochim Biophys Acta 1995, 1259:109–119.
Narumiya S, FitzGerald GA: Genetic and pharmacological analysis of prostanoid receptor function. J Clin Invest 2001, 108:25–30.
Narumiya S, Sugimoto Y, Ushikubi F: Prostanoid receptors: structures, properties, and functions. Physiol Rev 1999, 79:1193–1226.
Hata AN, Breyer RM: Pharmacology and signaling of prostaglandin receptors: multiple roles in inflammation and immune modulation. Pharmacol Ther 2004, 103:147–166.
Wise H, Jones RL: Focus on prostacyclin and its novel mimetics. Trends Pharmacol Sci 1996, 17:17–21.
Abramovitz M, Adam M, Boie Y, Carriere M, Denis D, Godbout C, Lamontagne S, Rochette C, Sawyer N, Tremblay NM, Belley M, Gallant M, Dufresne C, Gareau Y, Ruel R, Juteau H, Labelle M, Ouimet N, Metters KM: The utilization of recombinant prostanoid receptors to determine the affinities and selectivities of prostaglandins and related analogs. Biochim Biophys Acta 2000, 1483:285–293.
Kiriyama M, Ushikubi F, Kobayashi T, Hirata M, Sugimoto Y, Narumiya S: Ligand binding specificities of the eight types and subtypes of the mouse prostanoid receptors expressed in Chinese hamster ovary cells. Br J Pharmacol 1997, 122:217–224.
Christman BW, McPherson CD, Newman JH, King GA, Bernard GR, Groves BM, Loyd JE: An imbalance between the excretion of thromboxane and prostacyclin metabolites in pulmonary hypertension. N Engl J Med 1992, 327:70–75.
Tuder RM, Cool CD, Geraci MW, Wang J, Abman SH, Wright L, Badesch D, Voelkel NF: Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertension. Am J Respir Crit Care Med 1999, 159:1925–1932.
Iwai N, Katsuya T, Ishikawa K, Mannami T, Ogata J, Higaki J, Ogihara T, Tanabe T, Baba S: Human prostacyclin synthase gene and hypertension : the Suita Study. Circulation 1999, 100:2231–2236.
Geraci MW, Gao B, Shepherd DC, Moore MD, Westcott JY, Fagan KA, Alger LA, Tuder RM, Voelkel NF: Pulmonary prostacyclin synthase overexpression in transgenic mice protects against development of hypoxic pulmonary hypertension. J Clin Invest 1999, 103:1509–1515.
Barst RJ, Rubin LJ, Long WA, McGoon MD, Rich S, Badesch DB, Groves BM, Tapson VF, Bourge RC, Brundage BH: A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. The Primary Pulmonary Hypertension Study Group [see comments]. N Engl J Med 1996, 334:296–302.
Rubin LJ, Mendoza J, Hood M, McGoon M, Barst R, Williams WB, Diehl JH, Crow J, Long W: Treatment of primary pulmonary hypertension with continuous intravenous prostacyclin (epoprostenol). Results of a randomized trial. Ann Intern Med 1990, 112:485–491.
Olschewski H, Walmrath D, Schermuly R, Ghofrani A, Grimminger F, Seeger W: Aerosolized prostacyclin and iloprost in severe pulmonary hypertension. Ann Intern Med 1996, 124:820–824.
Olschewski H, Simonneau G, Galie N, Higenbottam T, Naeije R, Rubin LJ, Nikkho S, Speich R, Hoeper MM, Behr J, Winkler J, Sitbon O, Popov W, Ghofrani HA, Manes A, Kiely DG, Ewert R, Meyer A, Corris PA, Delcroix M, Gomez-Sanchez M, Siedentop H, Seeger W: Inhaled iloprost for severe pulmonary hypertension. N Engl J Med 2002, 347:322–329.
Barst RJ, McGoon M, McLaughlin V, Tapson V, Rich S, Rubin L, Wasserman K, Oudiz R, Shapiro S, Robbins IM, Channick R, Badesch D, Rayburn BK, Flinchbaugh R, Sigman J, Arneson C, Jeffs R: Beraprost therapy for pulmonary arterial hypertension. J Am Coll Cardiol 2003, 41:2119–2125.
Galie N, Humbert M, Vachiery JL, Vizza CD, Kneussl M, Manes A, Sitbon O, Torbicki A, Delcroix M, Naeije R, Hoeper M, Chaouat A, Morand S, Besse B, Simonneau G: Effects of beraprost sodium, an oral prostacyclin analogue, in patients with pulmonary arterial hypertension: a randomized, double-blind, placebo-controlled trial. J Am Coll Cardiol 2002, 39:1496–1502.
McLaughlin VV, Genthner DE, Panella MM, Rich S: Reduction in pulmonary vascular resistance with long-term epoprostenol (prostacyclin) therapy in primary pulmonary hypertension [see comments]. N Engl J Med 1998, 338:273–277.
Archer SL, Mike D, Crow J, Long W, Weir EK: A placebo-controlled trial of prostacyclin in acute respiratory failure in COPD. Chest 1996, 109:750–755.
Zucker TP, Bonisch D, Hasse A, Grosser T, Weber AA, Schror K: Tolerance development to antimitogenic actions of prostacyclin but not of prostaglandin E1 in coronary artery smooth muscle cells. Eur J Pharmacol 1998, 345:213–220.
Smyth EM, Nestor PV, FitzGerald GA: Agonist-dependent phosphorylation of an epitope-tagged human prostacyclin receptor. J Biol Chem 1996, 271:33698–33704.
Nilius SM, Hasse A, Kuger P, Schror K, Meyer-Kirchrath J: Agonist-induced long-term desensitization of the human prostacyclin receptor. FEBS Lett 2000, 484:211–216.
Le J I, Shepherd M, Van HG, Houslay MD, Hall IP: Cyclic AMP-dependent transcriptional up-regulation of phosphodiesterase 4D5 in human airway smooth muscle cells. Identification and characterization of a novel PDE4D5 promoter. J Biol Chem 2002, 277:35980–35989.
Mehats C, Tanguy G, Dallot E, Cabrol D, Ferre F, Leroy MJ: Is up-regulation of phosphodiesterase 4 activity by PGE2 involved in the desensitization of beta-mimetics in late pregnancy human myometrium? J Clin Endocrinol Metab 2001, 86:5358–5365.
Sobolewski A, Jourdan KB, Upton PD, Long L, Morrell NW: Mechanism of cicaprost-induced desensitization in rat pulmonary artery smooth muscle cells involves a PKA-mediated inhibition of adenylyl cyclase. Am J Physiol Lung Cell Mol Physiol 2004, 287:L352-L359.
Seeger W, Walmrath D, Grimminger F, Rosseau S, Schutte H, Kramer HJ, Ermert L, Kiss L: Adult respiratory distress syndrome: model systems using isolated perfused rabbit lungs. Methods Enzymol 1994, 233:549–584.
Schermuly RT, Roehl A, Weissmann N, Ghofrani HA, Schudt C, Tenor H, Grimminger F, Seeger W, Walmrath D: Subthreshold doses of specific phosphodiesterase type 3 and 4 inhibitors enhance the pulmonary vasodilatory response to nebulized prostacyclin with improvement in gas exchange. J Pharmacol Exp Ther 2000, 292:512–520.
Schermuly RT, Krupnik E, Tenor H, Schudt C, Weissmann N, Rose F, Grimminger F, Seeger W, Walmrath D, Ghofrani HA: Coaerosolization of phosphodiesterase inhibitors markedly enhances the pulmonary vasodilatory response to inhaled iloprost in experimental pulmonary hypertension. Maintenance of lung selectivity. Am J Respir Crit Care Med 2001, 164:1694–1700.
Schermuly RT, Leuchte H, Ghofrani HA, Weissmann N, Rose F, Kohstall M, Olschewski H, Schudt C, Grimminger F, Seeger W, Walmrath D: Zardaverine and aerosolised iloprost in a model of acute respiratory failure. Eur Respir J 2003, 22:342–347.
Weissmann N, Voswinckel R, Hardebusch T, Rosseau S, Ghofrani HA, Schermuly R, Seeger W, Grimminger F: Evidence for a role of protein kinase C in hypoxic pulmonary vasoconstriction. Am J Physiol 1999, 276:L90-L95.
Shahbazian A, Heinemann A, Peskar BA, Holzer P: Differential peristaltic motor effects of prostanoid (DP, EP, IP, TP) and leukotriene receptor agonists in the guinea-pig isolated small intestine. Br J Pharmacol 2002, 137:1047–1054.
Rose F, Grimminger F, Appel J, Heller M, Pies V, Weissmann N, Fink L, Schmidt S, Krick S, Camenisch G, Gassmann M, Seeger W, Hanze J: Hypoxic pulmonary artery fibroblasts trigger proliferation of vascular smooth muscle cells: role of hypoxia-inducible transcription factors. FASEB J 2002, 16:1660–1661.
Darius H, Binz C, Veit K, Fisch A, Meyer J: Platelet receptor desensitization induced by elevated prostacyclin levels causes platelet-endothelial cell adhesion. J Am Coll Cardiol 1995, 26:800–806.
Fisch A, Tobusch K, Veit K, Meyer J, Darius H: Prostacyclin receptor desensitization is a reversible phenomenon in human platelets. Circulation 1997, 96:756–760.
Smyth EM, Li WH, FitzGerald GA: Phosphorylation of the prostacyclin receptor during homologous desensitization. A critical role for protein kinase c. J Biol Chem 1998, 273:23258–23266.
Smyth EM, Austin SC, Reilly MP, FitzGerald GA: Internalization and sequestration of the human prostacyclin receptor. J Biol Chem 2000, 275:32037–32045.
Manganiello VC, Murata T, Taira M, Belfrage P, Degerman E: Diversity in cyclic nucleotide phosphodiesterase isoenzyme families. Arch Biochem Biophys 1995, 322:1–13.
Torphy TJ: Phosphodiesterase isozymes: molecular targets for novel antiasthma agents. Am J Respir Crit Care Med 1998, 157:351–370.
Rabe KF, Tenor H, Dent G, Schudt C, Nakashima M, Magnussen H: Identification of PDE isozymes in human pulmonary artery and effect of selective PDE inhibitors. Am J Physiol 1994, 266:L536-L543.
Lee R, Wolda S, Moon E, Esselstyn J, Hertel C, Lerner A: PDE7A is expressed in human B-lymphocytes and is up-regulated by elevation of intracellular cAMP. Cell Signal 2002, 14:277–284.
Torphy TJ, Zhou HL, Foley JJ, Sarau HM, Manning CD, Barnette MS: Salbutamol up-regulates PDE4 activity and induces a heterologous desensitization of U937 cells to prostaglandin E2. Implications for the therapeutic use of beta-adrenoceptor agonists. J Biol Chem 1995, 270:23598–23604.
Zhu B, Kelly J, Vemavarapu L, Thompson WJ, Strada SJ: Activation and induction of cyclic AMP phosphodiesterase (PDE4) in rat pulmonary microvascular endothelial cells. Biochem Pharmacol 2004, 68:479–491.
Seybold J, Newton R, Wright L, Finney PA, Suttorp N, Barnes PJ, Adcock IM, Giembycz MA: Induction of phosphodiesterases 3B, 4A4, 4D1, 4D2, and 4D3 in Jurkat T-cells and in human peripheral blood T-lymphocytes by 8-bromo-cAMP and Gs-coupled receptor agonists. Potential role in beta2-adrenoreceptor desensitization. J Biol Chem 1998, 273:20575–20588.
Murthy KS, Zhou H, Makhlouf GM: PKA-dependent activation of PDE3A and PDE4 and inhibition of adenylyl cyclase V/VI in smooth muscle. Am J Physiol Cell Physiol 2002, 282:C508-C517.
Borchert G, Bartel S, Beyerdorfer I, Kuttner I, Szekeres L, Krause EG: Long lasting anti-adrenergic effect of 7-oxo-prostacyclin in the heart: a cycloheximide sensitive increase of phosphodiesterase isoform I and IV activities. Mol Cell Biochem 1994, 132:57–67.
Finney PA, Belvisi MG, Donnelly LE, Chuang TT, Mak JC, Scorer C, Barnes PJ, Adcock IM, Giembycz MA: Albuterol-induced downregulation of Gsalpha accounts for pulmonary beta(2)-adrenoceptor desensitization in vivo. J Clin Invest 2000, 106:125–135.
Moravcova Z, Rudajev V, Stohr J, Novotny J, Cerny J, Parenti M, Milligan G, Svoboda P: Long-term agonist stimulation of IP prostanoid receptor depletes the cognate G(s)alpha protein in membrane domains but does not change the receptor level. Biochim Biophys Acta 2004, 1691:51–65.
Tanaka Y, Yamaki F, Koike K, Toro L: New insights into the intracellular mechanisms by which PGI2 analogues elicit vascular relaxation: cyclic AMP-independent, Gs-protein mediated-activation of MaxiK channel. Curr Med Chem Cardiovasc Hematol Agents 2004, 2:257–265.
Orie NN, Fry CH, Clapp LH: Evidence that inward rectifier K+ channels mediate relaxation by the PGI2 receptor agonist cicaprost via a cyclic AMP-independent mechanism. Cardiovasc Res 2006, 69:107–115.
Yamaki F, Kaga M, Horinouchi T, Tanaka H, Koike K, Shigenobu K, Toro L, Tanaka Y: MaxiK channel-mediated relaxation of guinea-pig aorta following stimulation of IP receptor with beraprost via cyclic AMP-dependent and -independent mechanisms. Naunyn Schmiedebergs Arch Pharmacol 2001, 364:538–550.
Clapp LH, Turcato S, Hall S, Baloch M: Evidence that Ca2+-activated K+ channels play a major role in mediating the vascular effects of iloprost and cicaprost. Eur J Pharmacol 1998, 356:215–224.
Turcato S, Clapp LH: Effects of the adenylyl cyclase inhibitor SQ22536 on iloprost-induced vasorelaxation and cyclic AMP elevation in isolated guinea-pig aorta. Br J Pharmacol 1999, 126:845–847.
Schubert R, Serebryakov VN, Engel H, Hopp HH: Iloprost activates KCa channels of vascular smooth muscle cells: role of cAMP-dependent protein kinase. Am J Physiol 1996, 271:C1203-C1211.
Schubert R, Serebryakov VN, Mewes H, Hopp HH: Iloprost dilates rat small arteries: role of K(ATP)- and K(Ca)-channel activation by cAMP-dependent protein kinase. Am J Physiol 1997, 272:H1147-H1156.
Fiessinger JN, Schafer M: Trial of iloprost versus aspirin treatment for critical limb ischaemia of thromboangiitis obliterans. The TAO Study. Lancet 1990, 335:555–557.
Clapp LH, Finney P, Turcato S, Tran S, Rubin LJ, Tinker A: Differential effects of stable prostacyclin analogs on smooth muscle proliferation and cyclic AMP generation in human pulmonary artery. Am J Respir Cell Mol Biol 2002, 26:194–201.
Smyth EM, Austin SC, FitzGerald GA: Activation-dependent internalization of the human prostacyclin receptor. Adv Exp Med Biol 2002, 507:295–301.
Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft (SFB 547), Project C6.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Schermuly, R.T., Pullamsetti, S.S., Breitenbach, S.C. et al. Iloprost-induced desensitization of the prostacyclin receptor in isolated rabbit lungs. Respir Res 8, 4 (2007). https://doi.org/10.1186/1465-9921-8-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1465-9921-8-4