
Abstract
Background
Hyperoxic exposures are often found in clinical settings of respiratory insufficient patients, although oxygen therapy (>50% O2) can result in the development of acute hyperoxic lung injury within a few days. Upon hyperoxic exposure, the inducible nitric oxide synthase (iNOS) is activated by a variety of proinflammatory cytokines both in vitro and in vivo. In the present study, we used a murine hyperoxic model to evaluate the effects of iNOS deficiency on the inflammatory response.
Methods
Wild-type and iNOS-deficient mice were exposed to normoxia, 60% O2 or >95% O2 for 72 h.
Results
Exposure to >95% O2 resulted in an increased iNOS mRNA and protein expression in the lungs from wild-type mice. No significant effects of iNOS deficiency on cell differential in bronchoalveolar lavage fluid were observed. However, hyperoxia induced a significant increase in total cell count, protein concentration, LDH activity, lipid peroxidation, and TNF-α concentration in the bronchoalveolar lavage fluid compared to iNOS knockout mice. Moreover, binding activity of NF-κB and AP-1 appeared to be higher in wild-type than in iNOS-deficient mice.
Conclusion
Taken together, our results provide evidence to suggest that iNOS plays a proinflammatory role in acute hyperoxic lung injury.
Similar content being viewed by others

Background
Supplemental oxygen therapy is administered for the treatment of tissue hypoxia, most commonly in an intensive care setting of respiratory insufficient patients, though its potent toxicity is well described [1]. The pathophysiology of oxygen injury is characterized by lung inflammation including activation and recruitment of neutrophils and alveolar macrophages, tissue and alveolar edema, surfactant dysfunction, and excess production of free radicals and inflammatory cytokines [2–4]. Although the exact mechanisms of pulmonary oxygen toxicity are still unknown, compelling evidence suggests that reactive oxygen species such as superoxide anion, hydroxyl radical, and hydrogen peroxide are important mediators of lung injury [5–7]. High oxygen concentrations induce cellular damage by several mechanisms such as oxidation of proteins, peroxidation of membrane lipids, and breakage of DNA strands [8–10]. Moreover, hyperoxia also induces the release of a wide spectrum of proinflammatory cytokines such as tumor necrosis factor- [11–13]. However, the precise molecular mechanisms by which hyperoxia produces acute lung injury remain unclear.
[11–13]. However, the precise molecular mechanisms by which hyperoxia produces acute lung injury remain unclear.
Reactive oxygen species can also react with other free radicals such as nitric oxide (NO) to yield more cytotoxic species including peroxynitrite anion [14, 15]. Peroxynitrite is a strong oxidizing agent that can also initiate lipid peroxidation [16, 17]. Since the discovery of NO as a potent vascular smooth muscle relaxant and regulator of blood pressure, NO generated by the inducible nitric oxide synthase (iNOS) has been identified in many cell types such as alveolar macrophages or epithelial cells and implicated in a variety of biological roles [15, 18, 19].
NO is synthesized from L-arginine by two main isoforms of the NO synthase: the constitutive and the inducible isoform [20]. The NOS enzymes are complex homodimeric proteins consisting of a N-terminal oxygenase domain, a central calmodulin binding sequence, and a C-terminal reductase domain [21, 22]. Inducible NOS is expressed following induction by a variety of inflammatory cytokines such as TNF-α [22] or by lipopolysaccharide (LPS) [23–25]. Constitutive iNOS expression has been reported in the lung [26, 27] and several inflammatory processes involving the lung, such as sepsis [23–25], asbestosis-induced lung injury [28, 29] and hyperoxia [30, 31] are associated with an elevated NO production. However, the effect of hyperoxia on endogenous NO production is a matter of controversial discussion, depending on the experimental conditions [30, 32–34].
Inappropriate regulation of nuclear factor-κB (NF-κB)-and activator protein-1 (AP-1)-mediated transcription has also been associated with pathological conditions, including acute inflammation such as hyperoxic exposure [35]. Intracellular signaling pathways leading to an activation of transcriptional regulators such as NF-κB and AP-1 can be affected by reactive oxygen and nitrogen species [36–38]. Both transcription factors are activated in lung cells after short periods of hyperoxic exposure. Binding sites for NF-κB and AP-1 are present in the promotor of the iNOS gene and of proinflammatory cytokines such as TNF-α [35, 39, 40].
The objective of this study was to investigate the effect of iNOS deficiency on acute hyperoxic lung injury. As indicators of lung hyperpermeability, lavageable lung protein and LDH activity were measured; lung lipid oxidation was assessed based on the levels of thiobarbituric acid reactive substances. To characterize the inflammatory process, lavageable cell counts, cell differential, and TNF-
concentration were analyzed. Binding activity of NF-κB and AP-1 was investigated in order to elucidate transcriptional mechanisms for iNOS and TNF-α expression.
Methods
Animals
Inducible NOS-deficient mice (C57BL6/J × 129SvEv) were originally provided by Dr. J. Mudgett (Merck & Co., Rathway, New Jersey, USA), Dr. J. MacMicking, and Dr. C. Nathan (Cornell University Medical College, New York, USA) [41]. As controls, matching wild-type mice were used (C57BL6/J × 129SvEv). Animals were bred in the facilities of the Institute for Surgical Research (Munich, Germany). Protocols used in this study were approved by the appropriate government body.
Hyperoxic exposure
Male mice (12 – 16 weeks old, body weight between 26.1 g and 27.3 g) were kept in groups of seven in a sealed Plexiglas chamber (27 × 27 × 20 cm3). Animals were randomized and exposed to 60% O2 and >95% O2 with a gas flow rate of 6 l/min at atmospheric pressure for 72 h. Mice exposed to room air in the same chamber served as controls. O2 levels were monitored twice daily with an oxygen analyzer (Drägerwerk AG, Lübeck, Germany). The environmental temperature was maintained at 24°C ± 1, relative humidity was 73% ± 13, and air pressure was 947 mbar ± 5. Oxygen was humidified by bubbling through a water chamber. The Plexiglas chamber bottom was lined with soda lime for CO2 absorption (Mallinkrodt Baker B. V., Deventer, Holland). Exposures were continuous for the time indicated except for 5 – 10 min daily when the chamber was opened for housekeeping purposes. Animals were kept on a 12 h light/dark cycle. Standard rodent food and water were available ad libitum.
Bronchoalveolar lavage cell counts and cell differential
Immediately following exposure, mice were anaesthetized by intraperitoneal injection of sodium pentobarbital (10 mg/kg body weight, Narcoren®, Merial, Halbergmoos, Germany). Tracheotomy was performed and a 20 G × 32 mm needle (Abbocath®-T, Venisystems, Sligo, Ireland) was inserted and secured. Bronchoalveolar lavage (BAL) was performed five times with 1 ml of sterile non-pyrogenic phosphate-buffered saline solution (PBS; Serva, Heidelberg, Germany) in each animal. After centrifugation at 300 × g for 10 min, the supernatant was collected and stored at -20°C and -80°C for later protein assays. The BAL cell pellet was resuspended in PBS and washed by centrifugation. Cells were stained with May-Grunwald-Giemsa (Varistain 4, Shandon Labortechnik GmbH, Frankfurt, Germany) to identify cellular populations. Total cell counts were assessed with a hemacytometer (Coulter Ac T 8, Coulter Electronics, Krefeld, Germany).
Lavageable lung protein assay
Cell free BAL fluid was evaluated for total protein content by the bicinchoninic acid assay using bovine serum albumin (PAA Laboratories, Linz, Austria) based on a method of Smith et al. [42].
Lactate dehydrogenase activity assay
To evaluate lactate dehydrogenase (LDH) activity in cell free BAL fluids, a commercially available kit was used (LDH Optimiert, Roche Diagnostics, Mannheim, Germany).
TNF-α assay
Concentration of TNF-α in cell free BAL fluid was measured by an enzyme linked immunosorbent assay using a commercially available kit (EM-TNFA, Endogen, Woburn, Massachusetts, USA). Briefly, 50 ml biotinylated antibody reagent were added to 50 ml-samples in an anti-mouse TNF-α pre-coated strip well plate. After incubation for 2 h at room temperature, the plate was washed, a streptavidin horseradish peroxidase solution and the 3, 3',5, 5'-tetramethylbenzidine substrate solution were added and incubated in the dark. The absorbance was detected at 450 nm in a microplate reader (EAR 400 AT, Salzburger Labortechnik, Salzburg, Austria). A standard curve was used to determine the amount of TNF-α concentration in the samples.
Reverse transcriptase-polymerase chain reaction
Total RNA was isolated from non-lavaged lung homogenate of each mouse (RNeasy Mini Kit, Quiagen, Hilden, Germany), reverse transcribed into cDNA in a volume of 20 ml, containing 2 μg RNA, 1.5 μM Oligo-p(dT)15-primer, 5 × PCR-buffer, 0.1 M DTT, 10 nM dNTP-mix and 200 U/μl of Moloney murine leukemia virus reverse transcriptase. Reverse transcriptase-polymerase chain reaction (RT-PCR) amplifications were performed with aliquots of cDNA (3 μl) in total volume of 50 μl (5 μl 10 × PCR reaction-buffer, 1 μl dNTP-mix, 1 μl each of forward and reverse single strand DNA primers specific for mouse iNOS, 38.8 μl sterile deionized water, 0.2 μl Taq DNA-polymerase 1 U/ml). Oligonucleotide primers for iNOS were 5'-CAC AAG GCC ACA TCG GAT TTC-3' (sense) and 5'-TGC ATA CCA CTT CAA CCC AG-3' (antisense). Co-amplification of the housekeeping gene β-actin served as an internal control, using the following primers, 5'-GGA CTC CTA TGT GGG TGA CGA GG-3' (sense), 5'-GGG AGA GCA TAG CCC TCG TAG AT-3' (antisense). RT-PCR was started with 1 min incubation at 95°C followed by the steps of denaturation at 95°C for 45 sec, annealing at 55 – 64°C for 45 sec, elongation at 72°C for 1 min. The number of cycles (30 – 35 each) was chosen to ensure that the amplification product did not reach the level of saturation. Reactions were electrophoresed in 1% agarose gel and stained with ethidium bromide. The densitometry of each cDNA band was quantified using BIO-1D.V96 software and the ratio of iNOS cDNA to β-actin cDNA was determined.
Electrophoretic mobility shift assay
Nuclear protein extracts were prepared from pooled lung tissue as previously described [43]. Briefly, the oligonucleotides were incubated with a binding buffer (0.04 M Tris, 0.2 M NaCl, 2 mM EDTA, 8% glycerine, 2 μm Ficoll 400, 0.2 mM PMSF, 4 mM DTT). After 5 min incubation, 5 μ l of [γ32P]-dATP end-labeled double-stranded oligonucleotides containing an NF-κB-consensus sequence (5'-AGT TGA GGG GAC TTT CCC AGG C-3') or AP-1-consensus sequence (5'-CGC TTG ATG AGT CAG CCG GAA-3') were added to the reaction followed by an incubation for 1 h at 37°C. The mixture was subjected to electrophoresis on a 6% PAA-Gel (75% H2O, 45 mM Tris, 45 mM bore acid, 1 mM EDTA, pH 8. 6% APS, 60 μl TEMED) for 2 h at 250 V.
Thiobarbituric acid reactive substances assay
Concentration of thiobarbituric acid reactive substances (TBARS) was evaluated with an assay according to Thiery et al. [44]. BAL fluid was prepared to denaturate proteins with 50% trichloroacetic acid. The supernatants were transferred to a clean tube and 75 μl of 1.3% thiobarbituric acid (Sigma Chemie, Deisenhofen, Germany) in 0.3% NaOH were added. After incubation for 1 h at 90°C and subsequent cooling in ice water, samples were centrifuged for 6 min. Finally, 200 μl of sample were transferred to a 96-well plate and the absorbance at 530 nm was read in a microplate reader (Dynex Technologies, Denkendorf, Germany). TBARS were quantified by using a standard curve of malondialdehyde (Sigma Chemie, Germany).
Statistical analysis
Results are presented as the group mean ± standard error of the mean (SEM). Statistical comparison between values of the three oxygen concentrations was performed by using analysis of variance on ranks and Mann-Whitney rank sum test followed by Bonferroni's correction. Statistical comparison between wild-type and iNOS knockout mice was analyzed by using Mann-Whitney rank sum test. Significance was accepted at p < 0.05.
Results
General conditions of the animals and body weight
Wild-type and iNOS knockout mice all survived hyperoxia the entire 72 h. After hyperoxic exposure >95% O2, animals showed signs of reduced general conditions and reactions. Hyperoxic exposure >95% O2 also caused a significant reduction in body weight of wild-type mice compared to normoxic conditions and 60% oxygen exposure within the 72 h experimental period. In contrast, there was no significant change in body weight of iNOS knockout mice before and after normoxia and hyperoxia, respectively (table 1).
Differential and total cell counts
BAL in wild-type and iNOS knockout mice was performed to assess cellular infiltration in the alveolar space upon 72 h hyperoxic exposure (60% and >95% O2). Results presented in table 2 demonstrate no differences in baseline cell differentials between wild-type and iNOS knockout mice. Upon 72 h exposure to >95% O2, there was a significant decrease in the percentage of alveolar macrophages as well as a significant increase in the percentage of neutrophils and lymphocytes in both wild-type and iNOS knockout mice compared to normoxic conditions. No significant differences between wild-type- and iNOS knockout mice were found. However, hyperoxic exposure (>95% O2) resulted in a significant increase in total BAL cell counts after 72 h in wild-type (0.54 ± 0.05 × 106/ml) and in iNOS knockout mice (0.38 ± 0.04 × 106/ml) compared to normoxia (0.20 ± 0.03 × 106/ml and 0.16 ± 0.02 × 106/ml, respectively) and 60% O2 exposure (0.24 ± 0.04 × 106/ml and 0.19 ± 0.04 × 106/ml, respectively). This increase in BAL total cell counts under >95% O2 was significantly higher in wild-type than in iNOS knockout mice (figure 1).

Total cell counts in BAL from wild-type and iNOS knockout mice after 72 h exposure to 21%, 60%, and >95% O2. Data are mean ± SEM of seven mice for each group. #p < 0.05 vs. normoxia; *p < 0.05 vs. 60% O2; $p < 0.05 vs. iNOS knockout mice.
Lavageable lung protein
Total protein concentration in the BAL fluid was determined as an indicator of lung hyperpermeability induced by hyperoxic exposure. Under normoxia and 60% O2, total protein concentration did not differ between wild-type mice (21% O2: 86.4 ± 37.3 μg/ml; 60% O2: 95.5 ± 22.8 μg/ml) and knockout mice (21% O2: 157.1 ± 23.7 μg/ml; 60% O2: 86.0 ± 26.6 μg/ml). Exposure to >95% O2 resulted in a significant increase in protein concentration in wild-type mice (973.8 ± 95.7 μg/ml) and only in a modest increase in iNOS knockout mice (326.8 ± 90.4 μg/ml) that did not reach statistical significance.
Lactate dehydrogenase activity
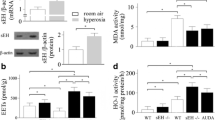
As an indicator of cellular damage, LDH activity was measured in BAL fluid. Under normoxic conditions and 60% O2, LDH activity was comparable between wild-type (21% O2: 3.1 ± 1.0 U/min/ml; 60% O2: 1.6 ± 0.4 U/min/ml) and iNOS knockout mice (21% O2: 2.3 ± 0.5 U/min/ml; 60% O2: 6.5 ± 1.7 U/min/ml). Exposure to >95% O2 resulted in a significant enhancement of LDH activity in wild-type mice (41.8 ± 10.8 U/min/ml) compared to iNOS knockout mice (9.6 ± 3.0 U/min/ml) and to normoxia (figure 3).

Lactate dehydrogenase activity in BAL from wild-type and iNOS knockout mice after 72 h exposure to 21%, 60%, and >95% O2. Data are mean ± SEM of seven mice for each group. #p < 0.05 vs. normoxia; *p < 0.05 vs. 60% O2; $p < 0.05 vs. iNOS knockout mice.
TNF-α concentration
TNF-α concentrations were determined in BAL fluid to investigate inflammatory cytokine release. Under normoxic conditions, TNF-α release did not differ between wild-type (28.5 ± 3.8 pg/ml) and iNOS knockout mice (35.0 ± 5.4 pg/ml), the same as upon 60% O2 exposure (29.2 ± 2.6 pg/ml and 25.4 ± 6.0 pg/ml, respectively). However, there was a significantly enhanced TNF-α release measured upon >95% O2 exposure in wild-type (83.0 ± 9.8 pg/ml) and iNOS knockout mice (54.9 ± 9.0 pg/ml) compared to normoxic conditions. TNF-α concentration was significantly higher in wild-type than in iNOS knockout animals (figure 4).

TNF-α concentration in BAL from wild-type and iNOS knockout mice after 72 h exposure to 21%, 60%, and >95% O2. Data are mean ± SEM of seven mice for each group. #p < 0.05 vs. normoxia; *p < 0.05 vs. 60% O2; $p < 0.05 vs. iNOS knockout mice.
Concentration of thiobarbituric acid reactive substances
Lung lipid peroxidation was assessed based on the levels of thiobarbituric acid reactive substances in BAL (figure 5). Wild-type mice exposed to >95% O2 exhibited a pronounced increase in concentration of TBARS (146.0 ± 62.0 nmol/ml) compared to normoxia (35.0 ± 14.0 nmol/ml) and 60% O2 (31.0 ± 17.0 nmol/ml). In iNOS knockout mice, concentrations of TBARS after >95% O2 (52.0 ± 18.0 nmol/ml) did not differ from those after normoxic conditions (26.0 ± 0.0 nmol/ml) and 60% O2 exposure (35.0 ± 26.0 nmol/ml), respectively.

Concentration of thiobarbituric acid reactive substances in BAL from wild-type and iNOS knockout mice after 72 h exposure to 21%, 60%, and >95% O2. Data are mean ± SEM of seven mice for each group. $p < 0.05 vs. iNOS knockout mice.
Activation of NF-κB and AP-1
In an effort to elucidate transcriptional mechanisms for increased iNOS and TNF-α expression after hyperoxic exposure, electrophoretic mobility shift assays for NF-κB and AP-1 were performed (figure 6). NF-κB and AP-1 were weakly activated under normoxic conditions. Increased activation of both NF-κB and AP-1 was observed after >95% O2 compared to normoxia and 60% O2. This enhancement of binding activity under hyperoxia appeared to be more prominent in the group of wild-type mice in comparison to iNOS knockout animals.

Binding activity of NF-κB and AP-1 in lung tissue from wild-type and iNOS knockout mice after 72 h normoxia or hyperoxia. Figure shown is representative for seven experiments.
iNOS mRNA expression
To investigate the induction of the iNOS gene in lung tissue, expression of iNOS mRNA was analyzed (figure 7). As expected, there was no expression of iNOS mRNA in lung tissues from iNOS knockout mice. In wild-type mice, hyperoxic exposure (60% and >95% O2) induced an increased expression of iNOS mRNA in lung samples compared to normoxic situation. Densitometric analysis was performed by determining the ratio of iNOS cDNA to β-actin cDNA. Results demonstrated a significant increase in iNOS mRNA expression upon >95% O2 (1.2 ± 0.1) compared to 60% O2 (0.8 ± 0.1).

Ethidium bromide stained gels of β-actin and iNOS RT-PCR products in lung tissue from wild-type (A) and iNOS knockout mice (B) after 72 h normoxia or hyperoxia. Data shown are representative for seven experiments.
Discussion
Prolonged exposure to high concentrations of oxygen (>50% O2) during an intensive care setting to maintain arterial pO2 can lead to progressive lung injury. Several cellular systems including alveolar macrophages and leukocytes are involved in this process. Activation of inflammatory cells causes the release of reactive oxygen species and proinflammatory cytokines, resulting in endothelial dysfunction, tissue and alveolar edema formation, and surfactant inactivation. Furthermore, high levels of NO produced by inducible NO synthase may contribute to tissue damage. NO is directly cytotoxic or can combine with superoxide anions to form the more reactive oxidant peroxynitrite. Although a large amount of literature exists concerning the pulmonary response to oxidant exposure, some issues remain unresolved.
Our findings confirm previous results showing that hyperoxia is able to upregulate iNOS expression in lung tissue [30, 34]. As expected, there was no expression of iNOS mRNA in lungs of iNOS knockout mice. In wild-type mice, exposure to 60% and >95% O2 induced a significant increase in iNOS mRNA expression. This enhanced iNOS mRNA expression during hyperoxic exposure seems to contradict findings reported in a study published by Arkovitz and colleagues, in which hyperoxia did not induce iNOS expression in lungs of mice [32]. This may be explained by the fact that the detection of iNOS mRNA by using northern blot technique is not as sensitive as RT-PCR. In accordance with results from others [28–30], we found little iNOS protein immunostaining under normoxic conditions and 60% oxygen exposure, while hyperoxic exposure >95% O2 induced a prominent expression of iNOS protein in the lungs from wild-type mice (data not shown).

Protein concentration in BAL from wild-type and iNOS knockout mice after 72 h exposure to 21%, 60%, and >95% O2. Data are mean ± SEM of seven mice for each group. #p < 0.05 vs. normoxia; *p < 0.05 vs. 60% O2; $p < 0.05 vs. iNOS knockout mice.
The data from the present study demonstrate that in vivo oxygen exposure significantly elevated total BAL cell count after 72 h >95% O2 both in wild-type and in iNOS knockout mice. According to this, oxygen exposure resulted in a significant enhancement in the number of neutrophils and lymphocytes in BAL fluid, combined with a significant reduction in the number of alveolar macrophages both in wild-type and iNOS knockout mice. Dedhia et al. also found elevated numbers of neutrophils and lymphocytes combined with decreased numbers of alveolar macrophages in rat lungs [45]. Recent studies report that, although iNOS deficiency does not affect leukocyte rolling and adhesion following treatment with thrombin [46], iNOS-deficient mice have significantly elevated leukocyte accumulation and enhanced leukocyte-endothelium interactions in endotoxinemia [24]. These results suggest that iNOS expression plays a potent role in regulation of leukocyte recruitment depending on the way of induction. Hyperoxia-induced inflammatory cell influx, particularly of neutrophils, can contribute to oxidant stress through formation of reactive oxygen species. Auten and collaborators demonstrated that DNA damage in hyperoxia-exposed rat lungs may be reduced by blocking neutrophil influx [47]. In our model of oxidant injury, no effect of iNOS deficiency on BAL cell differentials could be made out, whereas total BAL cell counts were significantly elevated in wild-type mice compared to iNOS knockout mice. The increase in the number of neutrophils and lymphocytes in BAL fluid may partially reflect the loss of integrity of the endothelium barrier. This damage is indicated by a significant elevation of total protein concentration and LDH activity after acute hyperoxia in wild-type mice in comparison to iNOS knockout animals. Kleeberger and colleagues previously reported that iNOS expression is involved in ozone-induced lung hyperpermeability showing reduced mean BAL fluid protein and leukocyte accumulation [48].
Recent studies indicate that iNOS also plays a proinflammatory role in the development of asbestosis-related pulmonary disorders, measured as a significantly decreased total protein count, LDH activity, and nitrotyrosine staining in iNOS-deficient mice [27]. In contrast, Kobayashi et al. reported that hyperoxia caused an increased accumulation of leukocytes, elevated LDH activity and albumin concentration, and a higher wet-dry-ratio in lungs from iNOS-deficient mice compared to wild-type animals [31]. Based on their findings, these authors suggest the presence of an iNOS-independent pathway of lung nitration and injury in hyperoxia. In our study, we found that nitrosylation of proteins in the lungs of mice exposed to >95% O2 was attenuated in iNOS-deficient mice (data not shown). Formation of nitrotyrosine was proposed as a relatively specific marker for detecting endogenous generation of peroxynitrite. However, recent evidence indicates that alternate reactions are capable of inducing nitration of tyrosine in proteins, for example the reaction of myeloperoxidase with hydrogen peroxide. Therefore, increased nitrotyrosine staining is considered as an indicator of "increased nitrative stress" rather than a "footprint" for the formation of peroxynitrite [49, 50]. Amplified formation of reactive oxygen and nitrogen species can be proved by determination of thiobarbituric acid reactive substances, a secondary product of lipid peroxidation indicating oxidative and/or nitrative stress [9]. In our study, significantly reduced formation of thiobarbituric acid reactive substances following >95% oxygen exposure was found in iNOS knockout mice, again suggesting a beneficial effect of iNOS deficiency on oxidant lung injury.
Cytokines may also play a role in oxygen toxicity. Several studies point out that TNF-α is produced during hyperoxic exposure [51, 52]. Furthermore, hyperoxia induces sequential formation of pulmonary TNF-α and IL-6, which corresponds to the severity of pathological findings [12]. In our study, iNOS deficiency resulted in a significant decrease in BAL TNF-α concentration during hyperoxic exposure. Findings of Sass et al. also demonstrate that iNOS-derived NO regulates proinflammatory genes in vivo resulting in inflammatory liver injury in mice by stimulation of TNF-α production [53]. To investigate whether hyperoxia-induced TNF-α expression was regulated on the level of protein or mRNA, activation of the redox-sensitive transcription factors NF-κB and AP-1 was analyzed. As recently described, NF-κB was activated following hyperoxia resulting in an increase in TNF-α and IFN-γ gene expression in murine pulmonary lymphocytes [35]. Moreover, we found that the activation of both factors seen in wild-type mice was weaker in iNOS knockout mice suggesting that induction of iNOS upon hyperoxia may in fact activate these transcription factors. These findings contrast the silencing effect of NO on NF-κB demonstrated upon stimulation with LPS or silica [54]. Data from Kupatt et al. [55] also indicate a negative feedback mechanism of eNOS-derived NO on activation of NF-κB following myocardial reoxygenation. In addition to isotype-specific differences in NO forming capacity, the synergistic NF-κB and AP-1 activation upon an reactive oxygen or nitrogen species challenge might diminish the inhibitory effect of NO. Recent studies indicate that exogenously administered NO causes increased c-fos and c-jun gene and protein expression combined with an evident AP-1 binding activity mediated by reactive oxygen and nitrogen species [56].
Conclusions
Taken together, our data show that the absence of the iNOS gene does attenuate, but not fully abolish, oxidation, nitration, and cytotoxicity in response to acute hyperoxic exposure. The degree of transcriptional activation, inflammation, and oxidative lung injury caused by hyperoxia is significantly reduced in iNOS knockout mice compared to wild-type animals. In conclusion, these findings provide evidence to suggest that, upon hyperoxic exposure to >95% O2, proinflammatory effects of iNOS may be predominant, thereby contributing to the extent of acute hyperoxic lung injury.
Abbreviations
- AP-1:
-
protein-1
- BAL:
-
bronchoalveolar lavage
- iNOS:
-
inducible nitric oxide synthase
- LDH:
-
lactate dehydrogenase
- LPS:
-
lipopolysaccharide
- NF-κB:
-
nuclear factor-kappa B
- NO:
-
nitric oxide
- TBARS:
-
thiobarbituric acid reactive substances
- TNF-α:
-
tumor necrosis factor-alpha
References
Carvalho CR, de Paula Pinto Schettino G, Maranhao B, Bethlem EP: Hyperoxia and lung disease. Curr Opin Pulm Med 1998, 4:300–304.
Bailey TC, Cavanagh C, Mehta S, Lewis JF, Veldhuizen RA: Sepsis and hyperoxia effects on the pulmonary surfactant system in wild-type and iNOS knockout mice. Eur Respir J 2002, 20:177–182.
Capellier G, Maupoil V, Boussat S, Laurent E, Neidhardt A: Oxygen toxicity and tolerance. Minerva Anestesiol 1999, 65:388–392.
Klein J: Normobaric pulmonary oxygen toxicity. Anesth Analg 1990, 70:195–207.
Chabot F, Mitchell JA, Gutteridge JM, Evans TW: Reactive oxygen species in acute lung injury. Eur Respir J 1998, 11:745–757.
Crapo JD, Tierney DF: Superoxide dismutase and pulmonary oxygen toxicity. Am J Physiol 1974, 226:1401–1407.
Stogner SW, Payne DK: Oxygen toxicity. Ann Pharmacother 1992, 26:1554–1562.
Feeney L, Berman ER: Oxygen toxicity: membrane damage by free radicals. Invest Ophthalmol 1976, 15:789–792.
Jamieson D, Chance B, Cadenas E, Boveris A: The relation of free radical production to hyperoxia. Annu Rev Physiol 1986, 48:703–719.
Turanlahti M, Pesonen E, Lassus P, Andersson S: Nitric oxide and hyperoxia in oxidative lung injury. Acta Paediatr 2000, 89:966–970.
Barazzone C, Tacchini-Cottier F, Vesin C, Rochat AF, Piguet PF: Hyperoxia induces platelet activation and lung sequestration: an event dependent on tumor necrosis factor-alpha and CD11a. Am J Respir Cell Mol Biol 1996, 15:107–114.
Ben-Ari J, Makhoul IR, Dorio RJ, Buckley S, Warburton D, Walker SM: Cytokine response during hyperoxia: sequential production of pulmonary tumor necrosis factor and interleukin-6 in neonatal rats. Isr Med Assoc J 2000, 2:365–369.
Tsan MF, White JE, Michelsen PB, Wong GH: Pulmonary O 2 toxicity: role of endogenous tumor necrosis factor. Exp Lung Res 1995, 21:589–597.
al-Ali MK, Howarth PH: Nitric oxide and the respiratory system in health and disease. Respir Med 1998, 92:701–715.
Kerwin JF Jr, Lancaster JR Jr, Feldman PL: Nitric oxide: a new paradigm for second messengers. J Med Chem 1995, 38:4343–4362.
Pryor WA, Squadrito GL: The chemistry of peroxynitrite: a product from the reaction of nitric oxide with superoxide. Am J Physiol 1995, 268:L699-L722.
Radi R, Beckman JS, Bush KM, Freeman BA: Peroxynitrite-induced membrane lipid peroxidation: the cytotoxic potential of superoxide and nitric oxide. Arch Biochem Biophys 1991, 288:481–487.
Lyons CR: The role of nitric oxide in inflammation. Adv Immunol 1995, 60:323–371.
Nathan C, Xie QW: Nitric oxide synthases: roles, tolls, and controls. Cell 1994, 78:915–918.
Gaston B, Drazen JM, Loscalzo J, Stamler JS: The biology of nitrogen oxides in the airways. Am J Respir Crit Care Med 1994, 149:538–551.
Fischmann TO, Hruza A, Niu XD, Fossetta JD, Lunn CA, Dolphin E, Prongay AJ, Reichert P, Lundell DJ, Narula SK, Weber PC: Structural characterization of nitric oxide synthase isoforms reveals striking active-site conservation. Nat Struct Biol 1999, 6:233–242.
Schulz C, Gillissen A, Schultze-Werninghaus G: Inducible nitric oxide synthase in pulmonary inflammatory processes. Pneumologie 1998, 52:340–349.
Ermert M, Ruppert C, Gunther A, Duncker HR, Seeger W, Ermert L: Cell-specific nitric oxide synthase isoenzyme expression and regulation in response to endotoxin in intact rat lungs. Lab Invest 2002, 82:425–441.
Hickey MJ, Sharkey KA, Sihota EG, Reinhardt PH, MacMicking JD, Nathan C, Kubes P: Inducible nitric oxide synthase-deficient mice have enhanced leukocyte-endothelium interactions in endotoxemia. FASEB J 1997, 11:955–964.
Hollenberg SM, Broussard M, Osman J, Parrillo JE: Increased micro-vascular reactivity and improved mortality in septic mice lacking inducible nitric oxide synthase. Circ Res 2000, 86:774–778.
Dorger M, Allmeling AM, Kiefmann R, Munzing S, Messmer K, Krombach F: Early inflammatory response to asbestos exposure in rat and hamster lungs: role of inducible nitric oxide synthase. Toxicol Appl Pharmacol 2002, 181:93–105.
Dorger M, Allmeling AM, Kiefmann R, Schropp A, Krombach F: Dual role of inducible nitric oxide synthase in acute asbestos-induced lung injury. Free Radic Biol Med 2002, 33:491–501.
Steudel W, Watanabe M, Dikranian K, Jacobson M, Jones RC: Expression of nitric oxide synthase isoforms (NOS II and NOS III) in adult rat lung in hyperoxic pulmonary hypertension. Cell Tissue Res 1999, 295:317–329.
Ermert M, Ruppert C, Gunther A, Duncker HR, Seeger W, Ermert L: Cell-specific nitric oxide synthase-isoenzyme expression and regulation in response to endotoxin in intact rat lungs. Lab Invest 2002, 82:425–441.
Cucchiaro G, Tatum AH, Brown MC, Camporesi EM, Daucher JW, Hakim TS: Inducible nitric oxide synthase in the lung and exhaled nitric oxide after hyperoxia. Am J Physiol 1999, 277:L636-L644.
Kobayashi H, Hataishi R, Mitsufuji H, Tanaka M, Jacobson M, Tomita T, Zapol WM, Jones RC: Antiinflammatory properties of inducible nitric oxide synthase in acute hyperoxic lung injury. Am J Respir Cell Mol Biol 2001, 24:390–397.
Arkovitz MS, Szabo C, Garcia VF, Wong HR, Wispe JR: Differential effects of hyperoxia on the inducible and constitutive isoforms of nitric oxide synthase in the lung. Shock 1997, 7:345–350.
Haddad IY, Zhu S, Crow J, Barefield E, Gadilhe T, Matalon S: Inhibition of alveolar type II cell ATP and surfactant synthesis by nitric oxide. Am J Physiol 1996, 270:L898-L906.
Pepperl S, Dorger M, Ringel F, Kupatt C, Krombach F: Hyperoxia upregulates the NO pathway in alveolar macrophages in vitro: role of AP-1 and NF-kappa B. Am J Physiol Lung Cell Mol Physiol 2001, 280:L905-L913.
Shea LM, Beehler C, Schwartz M, Shenkar R, Tuder R, Abraham E: Hyperoxia activates NF-kappaB and increases TNF-alpha and IFN-gamma gene expression in mouse pulmonary lymphocytes. J Immunol 1996, 157:3902–3908.
D'Angio CT, Finkelstein JN: Oxygen regulation of gene expression: a study in opposites. Mol Genet Metab 2000, 71:371–380.
Haddad JJ: Antioxidant and prooxidant mechanisms in the regulation of redox (Y)-sensitive transcription factors. Cell Signal 2002, 14:879–897.
Lee PJ, Choi AMK: Pathways of cell signaling in hyperoxia. Free Radic Biol Med 2003, 35:341–350.
Lowenstein CJ, Alley EW, Raval P, Snowman AM, Snyder SH, Russell SW, Murphy WJ: Macrophage nitric oxide synthase gene: two upstream regions mediate induction by interferon gamma and lipopolysaccharide. Proc Natl Acad Sci U S A 1993, 90:9730–9734.
Nunokawa Y, Oikawa S, Tanaka S: Human inducible nitric oxide synthase gene is transcriptionally regulated by nuclear factor-kappa B dependent mechanism. Biochem Biophys Res Commun 1996, 223:347–352.
MacMicking JD, Nathan C, Hom G, Chartrain N, Fletcher DS, Trumbauer M, Stevens K, Xie QW, Sokol K, Hutchinson N, Chen H, Mudgett JS: Altered responses to bacterial infection and endotoxic shock in mice lacking inducible nitric oxide synthase. Cell 1995, 81:641–650.
Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC: Measurement of protein using bicinchoninic acid. Anal Biochem 1985, 150:76–85.
Dignam JD, Lebovitz RM, Roeder RG: Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res 1983,11(1):475–1489.
Thiery J, Teupser D, Walli AK, Ivandic B, Nebendahl K, Stein O, Stein Y, Seidel D: Study of causes underlying the low atherosclerotic response to dietary hypercholesterolemia in a selected strain of rabbits. Atherosclerosis 1996, 121:63–73.
Dedhia HV, Ma JY, Vallyathan V, Dalal NS, Banks D, Flink EB, Billie M, Barger MW, Castranova V: Exposure of rats to hyperoxia: alteration of lavagate parameters and macrophage function. J Toxicol Environ Health 1993, 40:1–13.
Lefer DJ, Jones SP, Girod WG, Baines A, Grisham MB, Cockrell AS, Huang PL, Scalia R: Leukocyte-endothelial cell interactions in nitric oxide synthase-deficient mice. Am J Physiol 1999, 276:H1943-H1950.
Auten RL, Whorton MH, Nicholas MS: Blocking neutrophil influx reduces DNA damage in hyperoxia-exposed newborn rat lung. Am J Respir Cell Mol Biol 2002, 26:391–397.
Kleeberger SR, Reddy SP, Zhang LY, Cho HY, Jedlicka AE: Toll-like receptor 4 mediates ozone-induced murine lung hyperpermeability via inducible nitric oxide synthase. Am J Physiol Lung Cell Mol Physiol 2001, 280:L326-L333.
Eiserich JP, Hristova M, Cross CE, Jones AD, Freeman BA, Halliwell B, van der Vliet A: Formation of nitric oxide-derived inflammatory oxidants by myeloperoxidase in neutrophils. Nature 1998, 391:393–397.
Kozlov AV, Sobhian B, Duvigneau C, Gemeiner M, Nohl H, Redl H, Bahrami S: Organ specific formation of nitrosyl complexes under intestinal ischemia/reperfusion in rats involves NOS-independent mechanism(s). Shock 2001, 15:366–371.
Desmarquest P, Chadelat K, Corroyer S, Cazals V, Clement A: Effect of hyperoxia on human macrophage cytokine response. Respir Med 1998, 92:951–960.
Horinouchi H, Wang CC, Shepherd KE, Jones R: TNF alpha gene and protein expression in alveolar macrophages in acute and chronic hyperoxia-induced lung injury. Am J Respir Cell Mol Biol 1996, 14:548–555.
Sass G, Koerber K, Bang R, Guehring H, Tiegs G: Inducible nitric oxide synthase is critical for immune-mediated liver injury in mice. J Clin Invest 2001, 107:439–447.
Chen F, Kuhn DC, Sun SC, Gaydos LJ, Demers LM: Dependence and reversal of nitric oxide production on NF-kappa B in silica and lipopolysaccharide-induced macrophages. Biochem Biophys Res Commun 1995, 214:839–846.
Kupatt C, Weber C, Wolf A, Becker BF, Smith TW, Kelly RA: Nitric oxide attenuates reoxygenation-induced ICAM-1 expression in coronary microvascular endothelium: role of NF-kappa B. J Mol Cell Cardiol 1997, 29:2599–2609.
Janssen YM, Matalon S, Mossman BT: Differential induction of c-fos, c-jun, and apoptosis in lung epithelial cells exposed to ROS or RNS. Am J Physiol 1997, 273:L789-L796.
Acknowledgements
The authors gratefully acknowledge the excellent technical assistance of Mrs. A.-M. Allmeling, Mrs. A. Schropp, and Mrs. E. Ronft.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
AKH carried out the hyperoxic model, subsequent cytological and biochemical analyses, and writing and preparation of the manuscript. MD and FK participated in the direction of the study as well as in writing and preparation of the manuscript. CK carried out the electrophoretic mobility shift assays. The data presented in this paper are part of the doctoral thesis of AKH. All authors read and approved the final manuscript.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Hesse, AK., Dörger, M., Kupatt, C. et al. Proinflammatory role of inducible nitric oxide synthase in acute hyperoxic lung injury. Respir Res 5, 11 (2004). https://doi.org/10.1186/1465-9921-5-11
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1465-9921-5-11