
Abstract
Lung cancer is the number one cause of cancer-related death in the western world. Its incidence is highly correlated with cigarette smoking, and about 10% of long-term smokers will eventually be diagnosed with lung cancer, underscoring the need for strengthened anti-tobacco policies. Among the 10% of patients who develop lung cancer without a smoking history, the environmental or inherited causes of lung cancer are usually unclear. There is no validated screening method for lung cancer even in high-risk populations and the overall five-year survival has not changed significantly in the last 20 years. However, major progress has been made in the understanding of the disease and we are beginning to see this knowledge translated into the clinic.
In this review, we will summarize the current state of knowledge regarding the cascade of events associated with lung cancer development. From subclinical DNA damage to overt invasive disease, the mechanisms leading to clinically and molecularly heterogeneous tumors are being unraveled. These lesions allow cells to escape the normal regulation of cell division, apoptosis and invasion. While all subtypes of non-small cell lung cancer have historically been treated the same, stage-for-stage, recent technological advances have allowed a better understanding of the molecular classification of the disease and provide hypotheses for molecular early detection and targeted therapeutic strategies.
Similar content being viewed by others
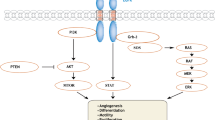


Introduction
The pathogenesis of lung cancer involves the accumulation of multiple molecular abnormalities over a long period of time [1, 2]. Genomic instability is universally found during accumulation of these hits [3]. The alterations can happen at the level of gene silencing through methylation, DNA sequence changes, DNA segment amplification or deletion or whole chromosome gains or losses. These changes occur early in normal-appearing tissues that do not have the characteristics of cancer cells. Microdissection of lesions of the bronchial epithelium as well as of invasive tumors has provided purified tissue for the analysis of point mutations [4], chromosomal deletions [5], microsatellite instability [6, 7] and DNA methylation patterns [8].
The most common early genetic alterations in non-small cell lung cancer involve loss of genomic regions of chromosomes 3p and 9p, deletions of chromosomal arm on 5p and mutations of p53 and K-ras [9]. Loss of chromosomal regions on chromosomes 3p and 9p have been recognized as early events [10] and identified in preinvasive lesions and in the normal appearing epithelium of smokers [11, 12]. In contrast, p53 and K-ras mutations have been seen primarily in later stages of preneoplasia or frank invasive lesions [9]. Amplification of large regions on the q arm of chromosome 3 has been characterized in invasive carcinomas [13] only recently in preinvasive lesions [14].
The historical focus of much of this research has been to identify and study the role of specific genetic abnormalities in tumor cells related to chromosomal abnormalities, inactivation of specific tumor suppressor genes, the activation of specific oncogenes, the expression of hormone receptors and growth factor production associated with the development of cancer. More recently, the contribution of stromal interactions, angiogenesis, apoptosis, and epigenetic phenomena such as posttranslational modification of critical genes has been the subject of intense research. The recent completion of the first draft of the human genome sequence [15] and the availability of high throughput technologies (e.g. microarrays) have prompted investigators to propose studies to discover common genetic abnormalities in both pre- and invasive lung cancers and to test these markers for their potential use in early detection strategies. In this paper we will review the genetic basis of lung cancer progression using a stepwise approach from point mutation to invasion and address its therapeutic implications.
Early events in oncogenesis
Mutations
In the last 20 years somatic mutations have been identified and associated with the development of cancer. These mutations, involving tumor suppressor genes or oncogenes, may or may not be rate-limiting events. Epidemiological data support that groups of cells accumulate several key mutations [16]. The model of the mutator phenotype proposed by Loeb suggests that cells develop a predisposition for mutations early on [3]. This phenotype may be hereditary, yet the key genes remain to be discovered. In the lung, DNA damage can fail to be repaired, resulting in misincorporated nucleotides and therefore mutations. Spontaneous errors of replication attributed to DNA polymerase occur at a rate of 1/10,000 to 1/100,000 base pairs depending on the polymerase. These intrinsic mutations may be an important component underlying genomic instability and eventually tumor growth. We will illustrate this point by commenting on 3 classical examples: k-ras, p53 and p16.
K-ras mutations are most commonly seen in 30% of adenocarcinomas of the lung [17] but much less frequently in other subtypes. K-ras, once mutated (most frequently codon 12 G-T transversions), can transform airway epithelial cells [18, 19] by activating the ERK-MAP kinase pathway. Because K-ras mutation is found early in alveolar atypical hyperplasia, a presumed precursor lesion to adenocarcinomas [20], this may be an important step in the genesis of this subtype of lung cancer. Mutant ras transgenic mice develop adenocarcinomas of the lung as well, supporting this hypothesis.
p53 is a prototype tumor suppressor gene that is the most common genetic lesion in human cancers [21] and is thus well suited for analysis of the mutational spectrum in human cancers. p53 mutations are most commonly seen in squamous carcinoma and small-cell carcinoma of the lung. Mutations predominantly represent G to T transversions consistent with causation by bulky DNA adducts such as the polycyclic hydrocarbons frequently found in the lungs of smokers [22]. The p53 tumor suppressor gene is mutated in over two thirds of lung cancers [23]. When mutated, p53 can function as an oncogene and accumulate in the cytoplasm [24]. Mutated p53 exhibits a prolonged half-life and can thus be found to be overexpressed in about 50% of lung cancers by immunohistochemistry [25]. Although not consistently associated with prognostic significance, there is little doubt that p53 mutations play a key role in tumor development by dysregulation of cell-cycle control and apoptosis.
p16, a tumor suppressor gene and critical member of the Rb pathway, is inactivated in over 40% of NSCLCs. Previous studies have demonstrated that point mutations, loss of heterozygosity on 9p21, or hypermethylation of the gene provide alternate mechanisms of inactivation in 30–50% of NSCLCs [26]. Tumors arising in smokers are found to more frequently harbor point mutations or homozygous deletions as the mechanism of loss of p16 function [27]. The relationship between tobacco and the loss of p16 points to new mechanisms involving smoking in the pathogenesis of lung cancer.
Mutagens
Cigarette smoking is a major risk factor for 85% of lung cancers. Approximately one in ten life-smokers will develop lung cancer, suggesting individual differences in susceptibility [28]. The susceptibility to lung cancer is being approached by molecular epidemiology and identifying links between genes involved in DNA repair, polymorphisms in the cytochrome p450 enzymes and the metabolizing capability of glutathione s-transferase or acetylation [29, 30]
The majority of lung cancers are diagnosed among ex-smokers [31]. This suggests that the accumulation of molecular damage during cigarette exposure has set a cascade of events in motion that leads to the diagnosis of cancer often decades after smoking cessation. Risk factors for lung cancer from smoking (first publicly recognized in the 1964 Report of the U.S. Surgeon General), include total consumption, age at initiation, and years of smoking. Other risk factors include occupational and environmental exposure (asbestos, uranium, radiation), diet (vitamin A, vitamin E, cholesterol), and host (familial aggregation) and genetic factors. Some of the components of cigarette smoke implicated in lung cancer are now recognized. Cigarette smoking is a complex mixture and includes substances that are responsible for DNA adduct formation such as polycyclic aromatic hydrocarbons (PAH), aromatic amines, and tobacco-specific nitrosamines (NKK). These form DNA adducts that may escape normal adduct repair mechanisms and result in heritable alterations in DNA sequence. The resulting conversion of G-C base pairs to T-A leads to activation of the K-ras oncogene and inactivation of the p53 tumor suppressor gene [32]. The activated form of benzopyrene (BaP) is BPDE and can cause DNA adducts, and, in addition to point mutations, can also lead to single strand chromatid breaks that are more frequent in lung cancers [33]. One of the concerning facts in this process is that people who start smoking at young ages seem to be have greater amounts of permanent DNA alterations than smokers who start smoking at an older age [34].
Chromosomal changes
Cancer cells are characterized not only by mutations but also by a series of chromosomal aberrations including deletions and amplifications [35]. The chromosomal regions with frequent losses are found in regions coding for essential tumor suppressor genes and DNA repair genes that may be involved in the pathogenesis of several tumor types [36]. Large areas of deletions (e.g. chromosome 3p, 9p) or amplifications (e.g. 1q, 3q) are commonly seen across the genome of lung cancer. Higher rates of chromosomal changes as determined by loss of heterozygosity (LOH) and CGH have been found in SqCa than in adenocarcinoma of the lung [37, 38].
The most common alterations involve loss of regions of chromosomes 3p21 and 9p21, deletions of chromosomal arm on 5q21 and mutations of p53 associated with LOH on 17p and K-ras point mutations [9]. Interestingly, loss of chromosomal regions on chromosomes 3p and 9p have been recognized as early events [10] and identified in preinvasive lesions and in the normal appearing epithelium of smokers [11, 12]. In contrast, p53 and K-ras mutations have been seen in a high percentage of later stages progression and in early invasive lesions [9].
LOH at chromosome 3p14 was evaluated in smokers and ex-smokers and found to be more frequent in current smokers (22/25 cases) than in former smokers (5/11 cases), a high frequency that correlated with a high metaplasia index [12]. This implies that not only are these chromosomal changes frequent in normal appearing bronchial epithelia but that cells with these changes may regress after smoking cessation and be replaced by cells without this damage. The dynamics of this process is very poorly understood at this time and represents an interesting area of future research.
Lung cancer allelotypes have been investigated in detail and have recently identified new regions of allelic loss using high throughput technologies [39]. Interestingly, differences between smokers and non-smokers have shown LOH on chr. 9 and 17 targets for p16 and p53, respectively [27]. LOH and chromosomal gain is less prevalent at all sites in cancer from non-smokers [27].
Patterns of chromosomal copy number abnormalities in squamous carcinomas of the lung using CGH analysis have been published recently [40–42] and show particularly common amplified regions on chromosomal arms 1q, 3q, 5p, 8q, 11q, 12p, 17q and 20q. Among many areas of genomic abnormality, amplification of chromosomal region 3q26 was found to be the most prevalent abnormality in squamous carcinoma of the lung followed by a deletion of chromosome 3p. Limitations of chromosome-based CGH include its relatively poor genomic resolution (~10–20 MB) [43, 44], lack of sensitivity for detection of aberrations involving megabase sized regions, inability to provide quantitative information about the magnitudes of genome copy number and the insensitivity of CGH to detect aberrations such as translocations that do not alter copy number. Most of these limitations can be overcome by viewing the chromosomes as the framework onto which information is mapped with high-resolution arrays of cloned probes.
Accumulation of specific chromosomal abnormalities has been correlated with clinical and pathological data in NSCLC. Chromosomal abnormalities have been recently correlated with clinical outcome for a variety of cancers [45–47], but often the genes responsible for the observed biology are unknown or only partly known. As mentioned above, 3q amplification is a common finding among many squamous carcinomas of non-lung origin. In particular, amplification of that region is seen in squamous carcinoma of the head and neck [48], esophageal cancers [49], and cervical cancers including cervical dysplasia [50]. In our recent study in NSCLC, among many amplified genes found in chromosome 3q26 (Figure 1), some are candidate oncogenes (phosphatidylinositol-3 kinase catalytic subunit, PIK3CA) or are described to be involved in tumor progression including the somatostatin gene (SST), p63 (p53 homolog gene), telomerase RNA component gene (hTER), and neutral endopeptidase (NEP) [13]. Human cytogenetic methods such as fluorescence in situ hybridization (FISH) are particularly useful in analyses of genomic organization, and copy number in individual cells and are applicable to tissue microarrays. Rather than look at the individual impact of isolated changes, we have begun efforts to "cluster" changes into groups of changes associated with a clinical feature. In an effort to find patterns associated with lung cancer histological subtypes based on array CGH profiling, we first identified 50 clones (most of which were on chr. 3) that best correlated with histological subtype using correlation and permutation analysis. Hierarchical clustering showed a clear pattern of gains and losses for squamous carcinoma, while the pattern for adenocarcinoma was less distinct (Figure 2). We then used an automatic classification method to assign tumor profiles to histological subtypes using a subset of 20 clones. The K-nearest-neighbor classification method correctly assigned 32/37 samples (87%) to proper histological subtype. The best multi-gene model found had a leave-one-out accuracy of 89.2%. Gene copy numbers as measured by array CGH are, collectively, an excellent indicator of histological subtype [51]. These data support the hypothesis that clusters of genes or groups of biomarkers may be more useful than single markers have been in the past as diagnostic, prognostic or predictive markers.

Array comparative genomic hybridization on a squamous carcinoma of the lung. A. array CGH profile on a squamous carcinoma of the lung labeled with Cy3 against normal DNA with Cy5. Each data point in presented mean (n = 4) ± coefficient of variance (CV=STD/Mean). B. View of Chromosome 3 array CGH profile on the same squamous carcinoma of the lung showing the size of the amplicon.

Hierarchical clustering analysis of NSCLCs using array comparative genomic hybridization. Cluster analysis using the 50 BAC clones closely correlated with histological subtype allowed accurate discrimination between SqCa and AdCa. K-nearest-neighbor classification was used to formally test the ability to predict subtype from array CGH profile. Cross-validation yielded 24/27 (89%) correct histological classification. Green squares: increase in copy number of a specific BAC clone, red squares: decrease in copy number.
Specific translocation is another chromosomal abnormality, but it is much less commonly observed in lung cancer than hematologic or mesodermal tumors [52]. Chromosomal translocations modify gene function through the deregulated expression of cellular proto-oncogenes without altering the structure of the protein product or by generating and expressing a chimeric protein with growth-promoting activities. Recently, Dang et al. identified a chromosome 19–15 translocation associated with overexpression of Notch 3 [53]. The authors developed a transgenic mouse model overexpressing Notch 3 causing neonatal mortality with a phenotype suggestive of alveolar cell hyperplasia. These data suggest that Notch3 overexpression prevents epithelial differentiation and this may play a significant role in promoting oncogenesis in a subset of lung cancers [54].
Genomic instability
Genome instability is a fundamental characteristic of cancer initiation and progression. However, our understanding of the time when instability occurs during progression, the rate of instability, and the mechanisms leading to instability is far from complete. Instability can arise from different pathways. In a small fraction of lung tumors, mismatch repair deficiency leads to microsatellite instability at the nucleotide sequence level. In other tumors, abnormal chromosome number (aneuploidy) is the dominant feature [55]. The progressive accumulation of mutations, loss of apoptotic control and regulation of cell proliferation, and the appearance of aneusomy are associated with worsening dysplasia phenotypes and may reflect underlying dysregulation of mechanisms controlling genomic fidelity. Less clear than microsatellite instability is the importance of specific defects in DNA repair in lung cancer. It is known that polymorphisms in DNA repair genes XPD (codon 312 Asp/Asp vs Asp/Asn) have been found to be associated with impaired efficiency of DNA repair and apoptotic function in lung cancer [56]. New techniques, however, are allowing us to assess these changes in individual or small numbers of preneoplastic cells. Copy number changes in single cells can be assessed by FISH probes. Microdissection of dysplastic epithelium has provided purified tissue for the analysis of point mutations [4], chromosomal deletions [5], microsatellite instability [6, 7] and DNA methylation patterns [8]. We thus may be able to ultimately derive a sequential pattern of development for genetic abnormalities in preneoplastic lung epithelium.
Role of viruses in lung tumorigenesis
The understanding of lung cancer molecular approaches has led to the development of transgenic models using viral antigens, including SV40 large T antigen and polyomavirus (PyV) large and middle T antigens that result in a high frequency of tumors. No common respiratory viruses have been conclusively incriminated in the development of lung cancer, but several have been implicated. Human papilloma virus (HPV), for example, has been associated with lung cancer and in particular lung cancer arising in women [57]. Simian Virus 40 has been incriminated in the development of mesothelioma [58]; Epstein Bar Virus (EBV) has been suspected to be involved in the development of papillomas, mesotheliomas and lymphomas of the lung. Many PCR-based assays, however, have attempted to correlate bronchogenic carcinomas with respiratory viruses without success. Recent advances in proteomics may be useful in studying the role of viral infection in airway epithelial cell transformation. The proteomic analysis of tumors may allow the identification of peptide sequences specific to pathogens otherwise ignored in tumorigenesis.
Viruses have also been used (e.g., adenovirus) to facilitate gene entry into cells (adenovirus-mediated gene transfer) or in in vivo gene therapy of human lung cancer using wild-type p53 delivered by retrovirus [59].
Genomic instability causing lung tumorigenesis
A multistep process for clonal evolution
Genetic changes are seen in the transition from normal to intraepithelial cancer to invasive disease. The understanding of the timing of instability during progression, the rate of instability and the mechanisms leading to instability are far from complete. Chronic exposure to carcinogens initiates a process characterized by genetic abnormalities, phenotypic changes and clonal overgrowth throughout the lungs [60]. Measures of genomic instability follow rates of loss of heterozygosity [39] and accumulation of other genomic abnormalities [55]. In the airways, progressively more severe and more frequent abnormalities are seen in preinvasive lesions [61]. The progressive accumulation of genomic abnormalities associated with clonal growth among populations of tumor cells are well described and favor the clonal progression of cancer. Yet cancer remains a rare event if one considers the total number of bronchial epithelial cells and the proliferation rate of patches of clonal abnormalities [62, 63].
While lung cancer originates from one or a few airway epithelial cells, it is clear that exposure of the whole airway mucosa to tobacco smoke could cause the entire bronchial tree to be at increased risk of developing lung cancer, leading to the concept of field cancerization. Field cancerization was first proposed in the fifties [64] and its molecular correlates later confirmed in the airways of human smokers [65, 66]. Field cancerization is also demonstrated by the elevated Ki-67 labeling index in the airways of smokers at more than one site [67]. Although the risk of developing lung cancer increases with the presence of such preinvasive lesions, no one has identified the molecular determinants of preinvasive lesions that may predict irreversible progression to lung cancer.
Carcinogenesis in the airways has proved to be multistep and multifocal and yet clonal in nature. Multiple lines of evidence support the concept of clonal progression of tumors. First, at the chromosomal level, abnormalities found in invasive tumors and their metastases are extremely highly correlated [68, 69]. Similarly, allelic losses or microsatellite abnormalities found to be in preinvasive lesions are found in similar frequencies in invasive lesions [7, 63]. The issue remains complex as a small fraction of tumors appear to be truly independent synchronous primaries and different p53 mutations have been found in synchronous preinvasive lesions [70]. The prolonged, multistep nature of lung cancer development makes this disease process potentially amenable to chemopreventive interventions that should be optimally applied in the earliest preinvasive phases.
Significance of genomic instability in lung tumorigenesis
Some preinvasive lesions are committed to develop into invasive cancer [71, 72]. One critical question that remains is the identification of that specific subset of the plethora of genetic changes in a given lesion that predisposes that lesion to develop into frank cancer. The literature suggests that the number of molecular abnormalities accumulated in the epithelium underlies tumor progression independent of light microscopically observable morphological abnormalities [4, 73]. This observation raises the possibility that genomic instability itself may be independently predictive of tumor progression. Consistent with this hypothesis, the relationship between clonal chromosome alterations and various clinical parameters was evaluated in 70 patients with non-small cell lung cancer [47]. An increased number of marker chromosomes were observed in patients having a higher number of packs of cigarettes smoked over years.
Epigenetic alterations of gene expression in lung cancer
Gene function loss can be mediated by deletion of large chromosomal regions or by inactivation of gene function from genetic mutation, or due to epigenetic modifications of DNA such as promoter hypermethylation or histone deacetylation.
DNA adducts
One marker for significant carcinogen exposure is the level of DNA adducts in normal DNA. DNA adducts are covalent modifications of the DNA that result from exposure to specific activated carcinogens. In addition to being markers of carcinogen exposure, it is possible that these adducts may directly alter regulation of transcription of tumor suppressor or oncogenes [74]. The distribution of benzo[a]pyrene diol epoxide (BPDE) adducts along exons of the p53 gene in BPDE-treated HeLa cells and bronchial epithelial cells has been mapped at nucleotide resolution [22]. Cigarette smokers have higher adduct levels than non-smokers. Because DNA adduct levels in tumor tissue and in blood lymphocytes have been associated with lung cancer [75, 76] and because these levels correlate with daily or lifetime cigarette consumption and do not reverse after smoking cessation [77], DNA adducts have been proposed as potential biomarkers of risk for lung cancer.
In an attempt to identify risk factors associated with the level of DNA adduct accumulation, Wiencke et al. studied DNA adducts in current and former smokers and found that in current smokers the most important variable was the number of cigarettes smoked per day. In contrast, they found that in ex-smokers, the most important variable was age at initiation [34]. Mechanisms responsible for the relationship between DNA adduct levels and age of initiation are unknown, and the relative contribution of decreased adduct removal by DNA repair or cell turnover or increased adduct formation at younger ages is yet to be determined. Prospective study is needed to follow current and ex-smokers over time to determine the value of adduct levels in risk assessment.
DNA adducts have been associated with smoking status and shown to be more prevalent among women. In a matched case-control study nested within the prospective Physicians' Health Study, there was an increased level of DNA adducts in active smokers who developed lung cancer as compared to controls; a finding that was not found among former or non smokers [78]. Women smokers may be at higher risk of developing lung cancer for a given tobacco exposure and women also seem to accumulate aromatic/hydrophobic DNA adducts at a faster rate then men [79]. DNA adduct levels were higher in women even when corrected for smoking dose packs of cigarettes smoked either per day or over years.
Methylation
Among epigenetic alterations, gain of methylation in normally unmethylated CpG islands around gene transcription start sites is an increasingly recognized and important means of altered gene expression in tumors [80]. The genes affected include over half of the tumor suppressor genes that cause familial cancers when mutated in the germline, and the selective advantage for genetic and epigenetic dysfunction in these genes is very similar in sporadic cases. In contrast to genetic mutations that require two hits to inhibit both alleles, aberrant methylation is a dynamic process over multiple division cycles and may cause increasing degrees of gene function loss by increasing the density of methylation on promoter regions. "CpG islands," the targets of DNA methyltransferase, are associated with the transcription start sites in almost half of human genes [81]. Dense methylation of cytosines within CpG islands causes heritable gene silencing [82]. Aberrant methylation can begin very early in tumor progression by causing loss of cell cycle control (p16) [83], loss of mismatch repair function (MLH1) [84] and loss of cell-cell interaction (E-cadherin). The exact mechanism by which hypermethylation may cause tumor progression is still unknown. In fact, there is still debate as to whether methylation is a result rather than a cause of gene function loss [85]. Promoter region hypermethylation has been proposed as an excellent tumor marker. In lung cancer, common methylated loci were found in both tumor and sputum DNA and were detected in the sputum for up to 3 years before the diagnosis of cancer [86].
Acetylation
The dynamics of chromatin formation suggest that the association of DNA methylation and histone deacetylation may cause silencing of hypermethylated genes in tumors. During transcription, chromatin unfolds and allows ribosomal access to the DNA. Acetylation of histone tails on the nucleosome is associated with chromatin unfolding and increased regional transcriptional activity. Histone deacetylases (HDACs) modulate chromatin structure by regulating acetylation of core histone proteins. Deacetylation of histones is thus associated with compacting the DNA and transcriptional repression. In lung cancer cell lines, for example, de-acetylation of histone 3 correlated with retinoic acid refractoriness, a phenomenon related to RARbeta promoter methylation in a subset of cell lines [87]. Inhibitors of HDACs have already shown to decrease the level of a series of oncoproteins [88] suggesting a potential role as antitumor therapeutic agents.
From genetic abnormalities to biomarkers for lung cancer
Lung cancer is a heterogeneous disease. The specific genetic abnormalities mentioned above have thus far proven to be of limited use individually as biomarkers for lung cancer. However, the completion of the first draft of the human genome sequence [15] and the availability of high throughput technologies (e.g. microarray) have prompted us to look in an unbiased way for complex patterns of genetic abnormalities that may be better associated with both pre- and invasive lung cancers and potential markers for use in early detection strategies.
Genomic arrays
DNA amplification and deletion in lung cancers of various histological subtypes have been analyzed by genomic approaches. We recently published the results of such analysis in a series of 37 NSCLCs [13]. With this technique, we demonstrated substantial genomic differences between squamous carcinomas and adenocarcinomas that are consistent with earlier chromosome based comparative genomic hybridization studies [40–42]. The significant difference in the total number of abnormalities between squamous carcinomas and adenocarcinomas suggests that they may differ in the level of genome instability and/or in the mechanisms by which they progress. Chromosome 3q is a common area of chromosomal gain in a variety of solid tumors. When early lesions are treated, they are known to prevent progression to invasive cancer. As discussed above, particularly common were amplified regions on chromosomal arms 1q, 3q, 5p, 8q, 11q, 12p, 17q and 20q, but gene amplification in chromosomal region 3q26 was the most prevalent abnormality. Among many amplified genes found in this region in a variety of solid tumors, some have been called potential candidate oncogenes (phosphatidylinositol-3 kinase catalytic subunit, PIK3CA) or genes suspected to be involved in tumor progression including the somatostatin gene (SST), p63 (p53 homologue gene), telomerase RNA component gene (TERC) and neutral endopeptidase gene (NEP). These patterns may ultimately be more predictive than analysis of expression of any single genes.
Expression arrays
RNA expression patterns may be more functionally relevant than DNA copy number changes, as most of these copy number changes affect cellular behavior via altered expression of included genes. The microarray technology developed in the mid 90's offers the hope that a genetic fingerprint of these tumors can be developed associated with clinical features. Beyond the need for better classification of lung cancers, this technical revolution opens a window of understanding to the world of tumor behavior (disease progression, recurrence, response to therapy) as well as to the mechanisms of tumor development. Tumor expression profiles are also influenced by the surrounding non-malignant cells. The combination of tumor and cell line profiling allows for the study of the regulatory role of both entities [89].
Efforts in classifying lung cancers based on microarray analysis revealed subclasses of adenocarcinomas. Selected genes allow the discrimination between primary lung cancer and metastasis of extrapulmonary sites [90]. Studies of expression profiles of adenocarcinomas of the lung using different chips commercially available [90] or custom arrays [91, 92] identified different classes of tumors with some overlap. Four classes of adenocarcinomas were found to have specific prognosis and molecular signature. These were characterized respectively 1) by expression of cell cycle or proliferation genes, 2) by expression of neuroendocrine markers, 3) by expression of markers of alveolar origin, and 4) by expression of ODC or glutathione S-transferase [91]. The neuroendocrine subclass was found to have outcome significantly worse than the others. The hope is that these subclass differences will point towards new molecular therapeutic opportunities for these subsets. Interestingly, when applied to neuroendocrine tumors, cDNA microarrays found poor correlations between genes expressed in carcinoid and SCLC [93], tumors that may be morphologically similar but that behave very different clinically.
Protein profiling
Recent advances in protein profiling have suggested a poor correlation between gene expression and protein expression. Perhaps more significantly, it is now well established that protein activity is often highly regulated by post-translational modifications such as proteolysis and phosphorylation. Neither protein expression levels nor post-translational modification can be assessed by genomic or cDNA microarray technologies, prompting interest in evaluation of protein expression, commonly referred to as "proteomics".
Investigators, including those at our institution, have attempted to use several proteomic methods of analysis, including 2Dgel and IHC, to identify biomarkers in tumors [94–97] in body fluids such as bronchoalveolar lavage [98] of patients with or without cancer. We recently acquired experience in this method for profiling of proteins in cancer tissue [99]. We applied MALDI-MS to 79 surgically resected lung cancers and 14 normal tissues. Software written by Dr. Jason Moore at Vanderbilt allows assignment of protein peaks in the mass spectral data across samples into unique "bins" corresponding to unique peptide species with correction for multiply charged ions. Hierarchical clustering of the resulting data has allowed the identification of patterns distinguishing between tumor and normal as well as histological subgroups. For example, to identify proteomic patterns that distinguish primary NSCLC from metastases to the lung, we compared protein expression profiles obtained from 34 primary NSCLCs with those from 7 other types of lung metastatic tumors, including 5 metastases to the lung from other sites and 2 lung metastases from previously resected NSCLC in the training cohort. We identified 24 MS signals that could discriminate all of the primary NSCLC from non-primary NSCLC in the training cohort, and were able to perfectly classify blinded samples in a test cohort [100]. Proteomic patterns from primary tumors with prognostic discriminatory power were identified as well and are potentially very useful in the clinical management of lung cancer. Although requiring prospective validation, these data bring proof of concept to an approach that may be found to be very powerful at selecting surgical candidates and other therapeutic strategies based on novel biological targets.
Identification of biomarkers
Biomarkers are needed to identify patients at high risk for lung cancer and to identify surrogate endpoints for response to chemoprevention strategies.
Despite the societal need for the early diagnosis of lung cancer, no role for biomarkers has yet been established for decision-making in intraepithelial neoplasia of the lung. Technical procedures such as tissue processing, use of antibody reagents and data interpretation need to be developed and standardized. A comprehensive and integrated approach linking laboratory findings of IEN of the lung with clinical features holds the potential to identify clinically relevant genetic and protein markers of carcinogenesis.
The number of potential lung cancer-related genes is rapidly growing. Once identified, genes and proteins may be tested in large populations of patients by immunohistochemical or cytogenetic techniques on tissue microarrays [100]. This high throughput method allows the screening of hundreds of lung cancer samples on a single glass slide and will allow retrospective analysis of material stored with associated clinical outcome. The arrays typically comprise core biopsies 0.6 mm in diameter of different tumors and uninvolved lung from the same individuals retrieved from the pathology archives of various institutions [101] (Figure 3). A firmer understanding of the relationship of relevant protein and genetic markers to clinical and pathologic status could lead to more accurate estimates of the anatomic extent of disease, risk of recurrence, and most effective intervention.

Tissue microarrays (TMAs) of lung cancer. TMAs are comprised of core biopsies of 0.6 mm in diameter of different tumors and of uninvolved lung from the same individuals. We retrieved 240 NSCLC tissue blocks from the pathological archives of Vanderbilt University between 1989 and 2001 and arrayed them in triplicate onto 4 separate TMAs. Tissue microarrays allow high throughput analysis of molecular markers identified in squamous lung neoplasia.
From Genetic abnormalities to early detection and new therapies
The identification of early molecular events such as chromosomal gain or loss that predicts tumor development suggests that early detection of lung cancer could be approached by means of molecular analysis. Sputum sample analysis for DNA methylation or chromosomal abnormalities by FISH may represent approaches suitable for early detection. The analysis of sputum, bronchial biopsies of preinvasive lesions using new detection methods such as fluorescence bronchoscopy [102], as well as exhaled breath condensate for tumor metabolites may be shown to be efficient ways of assessing high risk individuals. Early detection by low dose computed tomography scanning is being evaluated prospectively with the National Lung Cancer Screening Trial in 50,000 smokers. The addition of molecular studies may significantly increase the sensitivity and specificity of this new strategy for early detection.
Several therapeutic approaches to cancer have been developed to reduce undesirable expression of gene product or otherwise inhibit its function: (1) gene therapy (e.g. Adenovirus-p53) gene-specific ribozymes, which are able to break down specific RNA sequences, or with antisense oligonucleotides, (2) small molecule inhibition of receptor tyrosine kinases, (3) inhibition of p21(ras) farnesylation either by inhibition of farnesyl transferase or synthesis inhibition of farnesyl moieties, and (4) specific antibody approaches (e.g. anti-HER2 or anti-VEGF). We will touch on a couple of these approaches below.
Specific molecular targets
p53
Recently several phase I studies have evaluated the safety, biological effect and different routes of administration of adenoviral-mediated p53 gene therapy in various tumor types. These studies indicate that adenovirus-mediated p53 gene therapy and introduction of wild-type p53 into tumor cells represents a potentially valuable tool for the therapy of many types of human cancers [103] mainly by causing cell-cycle arrest or apoptosis [104, 105]. When injected intra-tumorally, wt-p53 has shown to be expressed in patients with p53 mutations and 3/7 patients showed regression of tumor size [106]. Using the wild-type p53 recombinant adenovirus, the same group of investigators showed in phase I trial that 16/25 had stabilization of disease and 2 had partial remissions [107]. One of the major limitations of the intra-tumoral approach is the inefficient delivery of genes of interest within the tumor mass. We have shown that intra-alveolar delivery of the gene in patients with bronchioloalveolar carcinoma led to objective responses.
GFR antagonists
Several epithelial tumors express EGFR with and without EGFR amplification [108]. This EGFR overexpression is associated with increased ligand production and hyperactive receptor function. About a third or more of NSCLC showed overexpressed EFGR [109]. Overexpression of EFGR was also associated with poor prognosis of patients with NSCLC [110]. Low-grade bronchial preinvasive lesions have also been shown to overexpress EGFR [111]. EGFR expression has been found to be elevated in metaplastic biopsies when compared to normal biopsies in active smokers [112] and that when co-expressed with p53 may predict squamous cell carcinoma development. Interruption of this autocrine pathway with receptor antibodies (extracellular domain of the protein) or tyrosine kinase inhibitors (competition with the kinase ATP binding site) can cause tumor regression [113, 114]. ZD1839, IRESSA and OSI-774 (Tarceva) are potent and specific inhibitors of the tyrosine kinase moiety of EGFR. Response rates in heavily pretreated patients with NSCLC vary between 10–18% in the IDEAL trials [115, 116], which may seem low but is actually far higher than any standard chemotherapy and represents a major benefit for these low-toxicity oral agents. Studies are proposed to investigate the value of EGFR inhibition in combination therapy, in earlier stage NSCLC and in lung cancer chemoprevention (STOP trials, SPORE Trial of Lung Cancer Prevention). Such chemoprevention trials with molecular and morphologic (preinvasive lesions) surrogate endpoints may suggest reversibility of lesions. However, the rate of spontaneous regression of these preinvasive lesions is, as yet, poorly characterized.
Kras: Farnesyl transferase inhibitor Zarnestra
K-ras was one of the first oncogenes implicated in human cancer. Development of retroviral vectors containing anitsense K-ras constructs or inhibitors of ras function may reduce proliferation or tumorigenicity. Farnesyltransferase enzyme activity is required to transfer farnesyl isoprenoid to the Ras c-terminus to anchor it to the cell membrane. This step is critical for Ras activation as an oncogene. The ras protein is known to undergo a series of post-translational modifications at the c-terminal CAAX motif, which forms a thioether bond of p21 ras with farnesyl and ties it to the plasma membrane [117]. At the cell surface, ras relays growth regulatory signals from receptor tyrosine kinases to various pathways of cell signal transduction. Unfortunately the currently available inhibitors work best with activated H-ras, a rare finding in lung cancer rather than the more common K-ras activation. Also not well explained is the observation that antitumor activity is very poorly correlated with measurable activation of any of the ras genes. However, several farnesyltransferase inhibitors are currently being tested in the clinic. R115777-Zarnestra is also being proposed in the clinic in a secondary chemoprevention trial. This trial is essentially based on the efficacy of FTI-276 on established lung adenomas (considered to be premalignant lesions of the lung) from A/J mice exposed to 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone, a tobacco-related carcinogen [118]. Analysis of the tumors showed a 60% reduction in tumor multiplicity and a 42% reduction in tumor incidence as well as a significant reduction in tumor volume (approximately 58%).
COX-2 inhibition
Cyclooxygenase-2 (COX-2) is an inducible enzyme that catalyzes the production of prostanoids. COX-2 can activate carcinogens in tobacco smoke [119], and COX-2 expression may play a role in angiogenesis by correlating with VEGF levels [120]. In addition, COX-2 activity may have a role in inhibiting apoptosis and modulating immune responses [121]. While nonsteroidal anti-inflammatory drugs have shown to reduce the risk of colorectal cancer, no such evidence yet exists in lung cancer. COX-2 inhibition has proven to reduce lung cancer cell growth in vitro [122]. In vivo, COX-2 has shown to cause persistent remission in patients otherwise refractory to lung cancer. COX-2 overexpression is a marker of poor prognosis in early stage NSCLC [123, 124]. COX-2 inhibitors are being evaluated in combination therapy for chemoprevention and therapy for lung cancer.
Other targeted strategies
Other targets include antibodies against VEGF ligand, EGFr or HER2 and inhibition of proteosome activity to counteract NFκB activation. All of these are currently in large scale clinical trials. Markers identified as being overexpressed in lung cancers represent potential immunotherapy targets even if no significant function can be found for the marker protein. An example is the recent identification of frequent overexpression of the cancer testis antigens from the microarray studies [125]. These genes are already being tested as vaccine targets in melanoma, and are only recently recognized as being overexpressed in the majority of non-small cell lung cancers.
Conclusions
A large number of genetic pathways associated with cancer development are being discovered at a rapid pace. The clinical impact of this recent knowledge on disease management is still relatively small, but real and growing. Little progress has been made in lung cancer chemoprevention, yet preventing, inhibiting and reversing the preneoplastic changes leading to cancer may ultimately prove a much more tractable goal than treating advanced disease. The slow process of carcinogenesis makes this period an open window for chemoprevention so that the intervention occurs when genetic instability is still controllable.
Abbreviations
- CGH:
-
comparative genomic hybridization
- LOH:
-
loss of heterozygosity
- FISH:
-
fluorescence in situ hybridization
- TMA:
-
tissue microarray
- NSCLC:
-
non small cell lung cancer
References
Knudson A. G., Jr.: Genetics of human cancer. Genetics 1975, 79 Suppl:305–316.
Peto R, Roe FJ, Lee PN, Levy L, Clack J: Cancer and ageing in mice and men. Br J Cancer 1975, 32:411–426.
Loeb LA: Mutator phenotype may be required for multistage carcinogenesis. Cancer Res 1991, 51:3075–3079.
Chung GT, Sundaresan V, Hasleton P, Rudd R, Taylor R, Rabbitts PH: Sequential molecular genetic changes in lung cancer development. Oncogene 1995, 11:2591–2598.
Sundaresan V, Ganly P, Hasleton P, Rudd R, Sinha G, Bleehen NM, Rabbitts P: p53 and chromosome 3 abnormalities, characteristic of malignant lung tumours, are detectable in preinvasive lesions of the bronchus. Oncogene 1992, 7:1989–1997.
Miozzo M, Sozzi G, Musso K, Pilotti S, Incarbone M, Pastorino U, Pierotti MA: Microsatellite alterations in bronchial and sputum specimens of lung cancer patients. Cancer Res 1996, 56:2285–2288.
Mao L, Lee DJ, Tockman MS, Erozan YS, Askin F, Sidransky D: Microsatellite alterations as clonal markers for the detection of human cancer. Proc Natl Acad Sci U S A 1994, 91:9871–9875.
Belinsky SA, Nikula KJ, Baylin SB, Issa JP: Increased cytosine DNA-methyltransferase activity is target-cell- specific and an early event in lung cancer. Proc Natl Acad Sci U S A 1996, 93:4045–4050.
Thiberville L, Payne P, Vielkinds J, LeRiche J, Horsman D, Nouvet G, Palcic B, Lam S: Evidence of cumulative gene losses with progression of premalignant epithelial lesions to carcinoma of the bronchus. Cancer Res 1995, 55:5133–5139.
Sundaresan V, Heppell-Parton A, Coleman N, Miozzo M, Sozzi G, Ball R, Cary N, Hasleton P, Fowler W, Rabbitts P: Somatic genetic changes in lung cancer and precancerous lesions. Ann Oncol 1995, 6:27–31; discussion 31–2.
Wistuba ,II, Lam S, Behrens C, Virmani AK, Fong KM, LeRiche J, Samet JM, Srivastava S, Minna JD, Gazdar AF: Molecular damage in the bronchial epithelium of current and former smokers. J Natl Cancer Inst 1997, 89:1366–1373.
Mao L, Lee JS, Kurie JM, Fan YH, Lippman SM, Lee JJ, Ro JY, Broxson A, Yu R, Morice RC, Kemp BL, Khuri FR, Walsh GL, Hittelman WN, Hong WK: Clonal genetic alterations in the lungs of current and former smokers. J Natl Cancer Inst 1997, 89:857–862.
Massion PP, Kuo WL, Stokoe D, Olshen AB, Treseler PA, Chin K, Chen C, Polikoff D, Jain AN, Pinkel D, Albertson DG, Jablons DM, Gray JW: Genomic copy number analysis of non-small cell lung cancer using array comparative genomic hybridization: implications of the phosphatidylinositol 3-kinase pathway. Cancer Res 2002, 62:3636–3640.
Massion PP, Taflan PM, Rahman SMJ, Yildiz P, Shyr Y, Edgerton ME, Westfall MD, Roberts JR, Pietenpol JA, Carbone DP, Gonzalez AL: Significance of p63 amplification and overexpression in lung cancer development and prognosis. Cancer Res 2003, 63:7113–7121.
Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, Gocayne JD, Amanatides P, Ballew RM, Huson DH, Wortman JR, Zhang Q, Kodira CD, Zheng XH, Chen L, Skupski M, Subramanian G, Thomas PD, Zhang J, Gabor Miklos GL, Nelson C, Broder S, Clark AG, Nadeau J, McKusick VA, Zinder N, Levine AJ, Roberts RJ, Simon M, Slayman C, Hunkapiller M, Bolanos R, Delcher A, Dew I, Fasulo D, Flanigan M, Florea L, Halpern A, Hannenhalli S, Kravitz S, Levy S, Mobarry C, Reinert K, Remington K, Abu-Threideh J, Beasley E, Biddick K, Bonazzi V, Brandon R, Cargill M, Chandramouliswaran I, Charlab R, Chaturvedi K, Deng Z, Di Francesco V, Dunn P, Eilbeck K, Evangelista C, Gabrielian AE, Gan W, Ge W, Gong F, Gu Z, Guan P, Heiman TJ, Higgins ME, Ji RR, Ke Z, Ketchum KA, Lai Z, Lei Y, Li Z, Li J, Liang Y, Lin X, Lu F, Merkulov GV, Milshina N, Moore HM, Naik AK, Narayan VA, Neelam B, Nusskern D, Rusch DB, Salzberg S, Shao W, Shue B, Sun J, Wang Z, Wang A, Wang X, Wang J, Wei M, Wides R, Xiao C, Yan C, Yao A, Ye J, Zhan M, Zhang W, Zhang H, Zhao Q, Zheng L, Zhong F, Zhong W, Zhu S, Zhao S, Gilbert D, Baumhueter S, Spier G, Carter C, Cravchik A, Woodage T, Ali F, An H, Awe A, Baldwin D, Baden H, Barnstead M, Barrow I, Beeson K, Busam D, Carver A, Center A, Cheng ML, Curry L, Danaher S, Davenport L, Desilets R, Dietz S, Dodson K, Doup L, Ferriera S, Garg N, Gluecksmann A, Hart B, Haynes J, Haynes C, Heiner C, Hladun S, Hostin D, Houck J, Howland T, Ibegwam C, Johnson J, Kalush F, Kline L, Koduru S, Love A, Mann F, May D, McCawley S, McIntosh T, McMullen I, Moy M, Moy L, Murphy B, Nelson K, Pfannkoch C, Pratts E, Puri V, Qureshi H, Reardon M, Rodriguez R, Rogers YH, Romblad D, Ruhfel B, Scott R, Sitter C, Smallwood M, Stewart E, Strong R, Suh E, Thomas R, Tint NN, Tse S, Vech C, Wang G, Wetter J, Williams S, Williams M, Windsor S, Winn-Deen E, Wolfe K, Zaveri J, Zaveri K, Abril JF, Guigo R, Campbell MJ, Sjolander KV, Karlak B, Kejariwal A, Mi H, Lazareva B, Hatton T, Narechania A, Diemer K, Muruganujan A, Guo N, Sato S, Bafna V, Istrail S, Lippert R, Schwartz R, Walenz B, Yooseph S, Allen D, Basu A, Baxendale J, Blick L, Caminha M, Carnes-Stine J, Caulk P, Chiang YH, Coyne M, Dahlke C, Mays A, Dombroski M, Donnelly M, Ely D, Esparham S, Fosler C, Gire H, Glanowski S, Glasser K, Glodek A, Gorokhov M, Graham K, Gropman B, Harris M, Heil J, Henderson S, Hoover J, Jennings D, Jordan C, Jordan J, Kasha J, Kagan L, Kraft C, Levitsky A, Lewis M, Liu X, Lopez J, Ma D, Majoros W, McDaniel J, Murphy S, Newman M, Nguyen T, Nguyen N, Nodell M, Pan S, Peck J, Peterson M, Rowe W, Sanders R, Scott J, Simpson M, Smith T, Sprague A, Stockwell T, Turner R, Venter E, Wang M, Wen M, Wu D, Wu M, Xia A, Zandieh A, Zhu X: The sequence of the human genome. Science 2001, 291:1304–1351.
Steen HB: The origin of oncogenic mutations: where is the primary damage? Carcinogenesis 2000, 21:1773–1776.
Westra WH, Slebos RJ, Offerhaus GJ, Goodman SN, Evers SG, Kensler TW, Askin FB, Rodenhuis S, Hruban RH: K-ras oncogene activation in lung adenocarcinomas from former smokers. Evidence that K-ras mutations are an early and irreversible event in the development of adenocarcinoma of the lung. Cancer 1993, 72:432–438.
Niklinski J, Niklinska W, Laudanski J, Chyczewska E, Chyczewski L: Prognostic molecular markers in non-small cell lung cancer. Lung Cancer 2001, 34 Suppl 2:S53–8.
Lacal JC, Srivastava SK, Anderson PS, Aaronson SA: Ras p21 proteins with high or low GTPase activity can efficiently transform NIH/3T3 cells. Cell 1986, 44:609–617.
Cooper CA, Carby FA, Bubb VJ, Lamb D, Kerr KM, Wyllie AH: The pattern of K-ras mutation in pulmonary adenocarcinoma defines a new pathway of tumour development in the human lung. J Pathol 1997, 181:401–404.
Harris CC: 1995 Deichmann Lecture--p53 tumor suppressor gene: at the crossroads of molecular carcinogenesis, molecular epidemiology and cancer risk assessment. Toxicol Lett 1995, 82–83:1–7.
Denissenko MF, Pao A, Tang M, Pfeifer GP: Preferential formation of benzo[a]pyrene adducts at lung cancer mutational hotspots in P53. Science 1996, 274:430–432.
Bennett WP, Colby TV, Travis WD, Borkowski A, Jones RT, Lane DP, Metcalf RA, Samet JM, Takeshima Y, Gu JR, et al.: p53 protein accumulates frequently in early bronchial neoplasia. Cancer Res 1993, 53:4817–4822.
Stewart ZA, Pietenpol JA: p53 Signaling and cell cycle checkpoints. Chem Res Toxicol 2001, 14:243–263.
Carbone DP, Mitsudomi T, Chiba I, Piantadosi S, Rusch V, Nowak JA, McIntire D, Slamon D, Gazdar A, Minna J: p53 immunostaining positivity is associated with reduced survival and is imperfectly correlated with gene mutations in resected non-small cell lung cancer. A preliminary report of LCSG 871. Chest 1994, 106:377S-381S..
Belinsky SA: Role of the cytosine DNA-methyltransferase and p16INK4a genes in the development of mouse lung tumors. Exp Lung Res 1998, 24:463–479.
Sanchez-Cespedes M, Decker PA, Doffek KM, Esteller M, Westra WH, Alawi EA, Herman JG, Demeure MJ, Sidransky D, Ahrendt SA: Increased loss of chromosome 9p21 but not p16 inactivation in primary non-small cell lung cancer from smokers. Cancer Res 2001, 61:2092–2096.
Peto R, Darby S, Deo H, Silcocks P, Whitley E, Doll R: Smoking, smoking cessation, and lung cancer in the UK since 1950: combination of national statistics with two case-control studies. Bmj 2000, 321:323–329.
Wright GS, Gruidl ME: Early detection and prevention of lung cancer [In Process Citation]. Curr Opin Oncol 2000, 12:143–148.
Hecht SS: Cigarette smoking and lung cancer: chemical mechanisms and approaches to prevention. Lancet Oncol 2002, 3:461–469.
Burns DM: Primary prevention, smoking, and smoking cessation: implications for future trends in lung cancer prevention. Cancer 2000, 89:2506–2509.
Hecht SS: Tobacco smoke carcinogens and lung cancer. J Natl Cancer Inst 1999, 91:1194–1210.
Wei Q, Gu J, Cheng L, Bondy ML, Jiang H, Hong WK, Spitz MR: Benzo(a)pyrene diol epoxide-induced chromosomal aberrations and risk of lung cancer. Cancer Res 1996, 56:3975–3979.
Wiencke JK, Thurston SW, Kelsey KT, Varkonyi A, Wain JC, Mark EJ, Christiani DC: Early age at smoking initiation and tobacco carcinogen DNA damage in the lung. J Natl Cancer Inst 1999, 91:614–619.
Mitelman F, Mertens F, Johansson B: A breakpoint map of recurrent chromosomal rearrangements in human neoplasia. Nat Genet 1997, 15 Spec No:417–474.
Knuutila S, Aalto Y, Autio K, Bjorkqvist AM, El-Rifai W, Hemmer S, Huhta T, Kettunen E, Kiuru-Kuhlefelt S, Larramendy ML, Lushnikova T, Monni O, Pere H, Tapper J, Tarkkanen M, Varis A, Wasenius VM, Wolf M, Zhu Y: DNA copy number losses in human neoplasms. Am J Pathol 1999, 155:683–694.
Wistuba ,II, Berry J, Behrens C, Maitra A, Shivapurkar N, Milchgrub S, Mackay B, Minna JD, Gazdar AF: Molecular changes in the bronchial epithelium of patients with small cell lung cancer. Clin Cancer Res 2000, 6:2604–2610.
Sato S, Nakamura Y, Tsuchiya E: Difference of allelotype between squamous cell carcinoma and adenocarcinoma of the lung. Cancer Res 1994, 54:5652–5655.
Girard L, Zochbauer-Muller S, Virmani AK, Gazdar AF, Minna JD: Genome-wide allelotyping of lung cancer identifies new regions of allelic loss, differences between small cell lung cancer and non-small cell lung cancer, and loci clustering. Cancer Res 2000, 60:4894–4906.
Balsara BR, Sonoda G, du Manoir S, Siegfried JM, Gabrielson E, Testa JR: Comparative genomic hybridization analysis detects frequent, often high- level, overrepresentation of DNA sequences at 3q, 5p, 7p, and 8q in human non-small cell lung carcinomas. Cancer Res 1997, 57:2116–2120.
Bjorkqvist AM, Husgafvel-Pursiainen K, Anttila S, Karjalainen A, Tammilehto L, Mattson K, Vainio H, Knuutila S: DNA gains in 3q occur frequently in squamous cell carcinoma of the lung, but not in adenocarcinoma. Genes Chromosomes Cancer 1998, 22:79–82.
Petersen I, Bujard M, Petersen S, Wolf G, Goeze A, Schwendel A, Langreck H, Gellert K, Reichel M, Just K, du Manoir S, Cremer T, Dietel M, Ried T: Patterns of chromosomal imbalances in adenocarcinoma and squamous cell carcinoma of the lung. Cancer Res 1997, 57:2331–2335.
Kallioniemi A, Kallioniemi OP, Piper J, Tanner M, Stokke T, Chen L, Smith HS, Pinkel D, Gray JW, Waldman FM: Detection and mapping of amplified DNA sequences in breast cancer by comparative genomic hybridization. Proc Natl Acad Sci U S A 1994, 91:2156–2160.
Bentz M, Plesch A, Stilgenbauer S, Dohner H, Lichter P: Minimal sizes of deletions detected by comparative genomic hybridization. Genes Chromosomes Cancer 1998, 21:172–175.
Bockmuhl U, Schluns K, Kuchler I, Petersen S, Petersen I: Genetic imbalances with impact on survival in head and neck cancer patients. Am J Pathol 2000, 157:369–375.
Gray JW, Collins C: Genome changes and gene expression in human solid tumors. Carcinogenesis 2000, 21:443–452.
Feder M, Siegfried JM, Balshem A, Litwin S, Keller SM, Liu Z, Testa JR: Clinical relevance of chromosome abnormalities in non-small cell lung cancer. Cancer Genet Cytogenet 1998, 102:25–31.
Yamaguchi K, Wu L, Caballero OL, Hibi K, Trink B, Resto V, Cairns P, Okami K, Koch WM, Sidransky D, Jen J: Frequent gain of the p40/p51/p63 gene locus in primary head and neck squamous cell carcinoma. Int J Cancer 2000, 86:684–689.
Pack SD, Karkera JD, Zhuang Z, Pak ED, Balan KV, Hwu P, Park WS, Pham T, Ault DO, Glaser M, Liotta L, Detera-Wadleigh SD, Wadleigh RG: Molecular cytogenetic fingerprinting of esophageal squamous cell carcinoma by comparative genomic hybridization reveals a consistent pattern of chromosomal alterations. Genes Chromosomes Cancer 1999, 25:160–168.
Heselmeyer K, Schrock E, du Manoir S, Blegen H, Shah K, Steinbeck R, Auer G, Ried T: Gain of chromosome 3q defines the transition from severe dysplasia to invasive carcinoma of the uterine cervix. Proc Natl Acad Sci U S A 1996, 93:479–484.
Aliferis CF, Hardin D, Massion PP: Machine learning models for lung cancer classification using array comparative genomic hybridization. Proc AMIA Symp 2002, 7–11.
Zhou JY, Taguchi T, Siegfried JM, Jhanwar SC, Resau J, Testa JR: Characterization of 9q;15q whole-arm translocation derivatives in non- small cell lung carcinomas by fluorescence in situ hybridization. Cancer Genet Cytogenet 1993, 69:1–6.
Dang TP, Gazdar AF, Virmani AK, Sepetavec T, Hande KR, Minna JD, Roberts JR, Carbone DP: Chromosome 19 translocation, overexpression of Notch3, and human lung cancer. J Natl Cancer Inst 2000, 92:1355–1357.
Dang TP, Eichenberger S, Gonzalez A, Olson S, Carbone DP: Constitutive activation of Notch3 inhibits terminal epithelial differentiation in lungs of transgenic mice. Oncogene 2003, 22:1988–1997.
Lengauer C, Kinzler KW, Vogelstein B: Genetic instability in colorectal cancers. Nature 1997, 386:623–627.
Butkiewicz D, Rusin M, Enewold L, Shields PG, Chorazy M, Harris CC: Genetic polymorphisms in DNA repair genes and risk of lung cancer. Carcinogenesis 2001, 22:593–597.
Cheng YW, Chiou HL, Sheu GT, Hsieh LL, Chen JT, Chen CY, Su JM, Lee H: The association of human papillomavirus 16/18 infection with lung cancer among nonsmoking Taiwanese women. Cancer Res 2001, 61:2799–2803.
Testa JR, Carbone M, Hirvonen A, Khalili K, Krynska B, Linnainmaa K, Pooley FD, Rizzo P, Rusch V, Xiao GH: A multi-institutional study confirms the presence and expression of simian virus 40 in human malignant mesotheliomas. Cancer Res 1998, 58:4505–4509.
Carbone DP, Minna JD: In vivo gene therapy of human lung cancer using wild-type p53 delivered by retrovirus [editorial; comment]. J Natl Cancer Inst 1994, 86:1437–1438.
Auerbach O, Stout AP, Hammond EC, Grafinkel L: Changes in bronchial epithelum in reation to cigraette smoking and in relation to lung cancer. N Engl J Med 1961, 265:253–267.
Hirsch FR, Merrick DT, Franklin WA: Role of biomarkers for early detection of lung cancer and chemoprevention. Eur Respir J 2002, 19:1151–1158.
Park IW, Wistuba ,II, Maitra A, Milchgrub S, Virmani AK, Minna JD, Gazdar AF: Multiple clonal abnormalities in the bronchial epithelium of patients with lung cancer. J Natl Cancer Inst 1999, 91:1863–1868.
Wistuba ,II, Behrens C, Milchgrub S, Bryant D, Hung J, Minna JD, Gazdar AF: Sequential molecular abnormalities are involved in the multistage development of squamous cell lung carcinoma. Oncogene 1999, 18:643–650.
Slaughter DP, Southwick HW, Smejkal W: Field cancerization in oratl stratified squamous epithelium: clinical implications of multicentric origin. Cancer 1953, 6:963–968.
Hittelman WN, Voravud N, Shin DM, Lee JS, Ro JY, Hong WK: Early genetic changes during upper aerodigestive tract tumorigenesis. J Cell Biochem Suppl 1993, 233–236.
Smith AL, Hung J, Walker L, Rogers TE, Vuitch F, Lee E, Gazdar AF: Extensive areas of aneuploidy are present in the respiratory epithelium of lung cancer patients. Br J Cancer 1996, 73:203–209.
Lee JJ, Liu D, Lee JS, Kurie JM, Khuri FR, Ibarguen H, Morice RC, Walsh G, Ro JY, Broxson A, Hong WK, Hittelman WN: Long-term impact of smoking on lung epithelial proliferation in current and former smokers. J Natl Cancer Inst 2001, 93:1081–1088.
Petersen S, Aninat-Meyer M, Schluns K, Gellert K, Dietel M, Petersen I: Chromosomal alterations in the clonal evolution to the metastatic stage of squamous cell carcinomas of the lung. Br J Cancer 2000, 82:65–73.
Levin NA, Brzoska PM, Warnock ML, Gray JW, Christman MF: Identification of novel regions of altered DNA copy number in small cell lung tumors. Genes Chromosomes Cancer 1995, 13:175–185.
Lavieille JP, Gazzeri S, Riva C, Reyt E, Brambilla C, Brambilla E: p53 mutations and p53, Waf-1, Bax and Bcl-2 expression in field cancerization of the head and neck. Anticancer Res 1998, 18:4741–4749.
Venmans BJ, van Boxem TJ, Smit EF, Postmus PE, Sutedja TG: Outcome of bronchial carcinoma in situ. Chest 2000, 117:1572–1576.
Bota S, Auliac JB, Paris C, Metayer J, Sesboue R, Nouvet G, Thiberville L: Follow-up of bronchial precancerous lesions and carcinoma in situ using fluorescence endoscopy. Am J Respir Crit Care Med 2001, 164:1688–1693.
Hung J, Kishimoto Y, Sugio K, Virmani A, McIntire DD, Minna JD, Gazdar AF: Allele-specific chromosome 3p deletions occur at an early stage in the pathogenesis of lung carcinoma. Jama 1995, 273:1908.
Wiencke JK: DNA adduct burden and tobacco carcinogenesis. Oncogene 2002, 21:7376–7391.
Cheng YW, Chen CY, Lin P, Huang KH, Lin TS, Wu MH, Lee H: DNA adduct level in lung tissue may act as a risk biomarker of lung cancer. Eur J Cancer 2000, 36:1381–1388.
Vulimiri SV, Wu X, Baer-Dubowska W, de Andrade M, Detry M, Spitz MR, DiGiovanni J: Analysis of aromatic DNA adducts and 7,8-dihydro-8-oxo- 2'- deoxyguanosine in lymphocyte DNA from a case-control study of lung cancer involving minority populations. Mol Carcinog 2000, 27:34–46.
Phillips DH, Hewer A, Martin CN, Garner RC, King MM: Correlation of DNA adduct levels in human lung with cigarette smoking. Nature 1988, 336:790–792.
Tang D, Phillips DH, Stampfer M, Mooney LA, Hsu Y, Cho S, Tsai WY, Ma J, Cole KJ, She MN, Perera FP: Association between carcinogen-DNA adducts in white blood cells and lung cancer risk in the physicians health study. Cancer Res 2001, 61:6708–6712.
Mollerup S, Ryberg D, Hewer A, Phillips DH, Haugen A: Sex differences in lung CYP1A1 expression and DNA adduct levels among lung cancer patients. Cancer Res 1999, 59:3317–3320.
Burbee DG, Forgacs E, Zochbauer-Muller S, Shivakumar L, Fong K, Gao B, Randle D, Kondo M, Virmani A, Bader S, Sekido Y, Latif F, Milchgrub S, Toyooka S, Gazdar AF, Lerman MI, Zabarovsky E, White M, Minna JD: Epigenetic inactivation of RASSF1A in lung and breast cancers and malignant phenotype suppression. J Natl Cancer Inst 2001, 93:691–699.
Antequera F, Bird A: Number of CpG islands and genes in human and mouse. Proc Natl Acad Sci U S A 1993, 90:11995–11999.
Jones PA, Laird PW: Cancer epigenetics comes of age. Nat Genet 1999, 21:163–167.
Belinsky SA, Nikula KJ, Palmisano WA, Michels R, Saccomanno G, Gabrielson E, Baylin SB, Herman JG: Aberrant methylation of p16(INK4a) is an early event in lung cancer and a potential biomarker for early diagnosis. Proc Natl Acad Sci U S A 1998, 95:11891–11896.
Baylin SB, Belinsky SA, Herman JG: Aberrant methylation of gene promoters in cancer---concepts, misconcepts, and promise. J Natl Cancer Inst 2000, 92:1460–1461.
Baylin S, Bestor TH: Altered methylation patterns in cancer cell genomes: cause or consequence? Cancer Cell 2002, 1:299–305.
Palmisano WA, Divine KK, Saccomanno G, Gilliland FD, Baylin SB, Herman JG, Belinsky SA: Predicting lung cancer by detecting aberrant promoter methylation in sputum. Cancer Res 2000, 60:5954–5958.
Suh YA, Lee HY, Virmani A, Wong J, Mann KK, Miller W. H., Jr., Gazdar A, Kurie JM: Loss of retinoic acid receptor beta gene expression is linked to aberrant histone H3 acetylation in lung cancer cell lines. Cancer Res 2002, 62:3945–3949.
Yu X, Guo ZS, Marcu MG, Neckers L, Nguyen DM, Chen GA, Schrump DS: Modulation of p53, ErbB1, ErbB2, and Raf-1 expression in lung cancer cells by depsipeptide FR901228. J Natl Cancer Inst 2002, 94:504–513.
Virtanen C, Ishikawa Y, Honjoh D, Kimura M, Shimane M, Miyoshi T, Nomura H, Jones MH: Integrated classification of lung tumors and cell lines by expression profiling. Proc Natl Acad Sci U S A 2002, 99:12357–12362.
Bhattacharjee A, Richards WG, Staunton J, Li C, Monti S, Vasa P, Ladd C, Beheshti J, Bueno R, Gillette M, Loda M, Weber G, Mark EJ, Lander ES, Wong W, Johnson BE, Golub TR, Sugarbaker DJ, Meyerson M: Classification of human lung carcinomas by mRNA expression profiling reveals distinct adenocarcinoma subclasses. Proc Natl Acad Sci U S A 2001, 98:13790–13795.
Garber ME, Troyanskaya OG, Schluens K, Petersen S, Thaesler Z, Pacyna-Gengelbach M, van de Rijn M, Rosen GD, Perou CM, Whyte RI, Altman RB, Brown PO, Botstein D, Petersen I: Diversity of gene expression in adenocarcinoma of the lung. Proc Natl Acad Sci U S A 2001, 98:13784–13789.
Beer DG, Kardia SL, Huang CC, Giordano TJ, Levin AM, Misek DE, Lin L, Chen G, Gharib TG, Thomas DG, Lizyness ML, Kuick R, Hayasaka S, Taylor JM, Iannettoni MD, Orringer MB, Hanash S: Gene-expression profiles predict survival of patients with lung adenocarcinoma. Nat Med 2002, 8:816–824.
Anbazhagan R, Tihan T, Bornman DM, Johnston JC, Saltz JH, Weigering A, Piantadosi S, Gabrielson E: Classification of small cell lung cancer and pulmonary carcinoid by gene expression profiles. Cancer Res 1999, 59:5119–5122.
Brichory F, Beer D, Le Naour F, Giordano T, Hanash S: Proteomics-based identification of protein gene product 9.5 as a tumor antigen that induces a humoral immune response in lung cancer. Cancer Res 2001, 61:7908–7912.
Stoeckli M, Chaurand P, Hallahan DE, Caprioli RM: Imaging mass spectrometry: a new technology for the analysis of protein expression in mammalian tissues. Nat Med 2001, 7:493–496.
Celis JE, Celis P, Ostergaard M, Basse B, Lauridsen JB, Ratz G, Rasmussen HH, Orntoft TF, Hein B, Wolf H, Celis A: Proteomics and immunohistochemistry define some of the steps involved in the squamous differentiation of the bladder transitional epithelium: a novel strategy for identifying metaplastic lesions. Cancer Res 1999, 59:3003–3009.
Chaurand P, Stoeckli M, Caprioli RM: Direct profiling of proteins in biological tissue sections by MALDI mass spectrometry. Anal Chem 1999, 71:5263–5270.
Noel-Georis I, Bernard A, Falmagne P, Wattiez R: Database of bronchoalveolar lavage fluid proteins. J Chromatogr B Analyt Technol Biomed Life Sci 2002, 771:221–236.
Yanagisawa K, Shyr Y, Xu BJ, Massion PP, Larsen PH, White BC, Roberts JR, Edgerton M, Gonzalez A, Nadaf S, Moore JH, Caprioli RM, Carbone DP: Proteomic patterns of tumour subsets in non-small-cell lung cancer. Lancet 2003, 362:433–439.
Kononen J, Bubendorf L, Kallioniemi A, Barlund M, Schraml P, Leighton S, Torhorst J, Mihatsch MJ, Sauter G, Kallioniemi OP: Tissue microarrays for high-throughput molecular profiling of tumor specimens [see comments]. Nat Med 1998, 4:844–847.
Massion PP, Gray JW: Molecular Cytogenetic Explorations of Human Genome. Genomic Technologies: Present and Future Evolution of Genomic Technologies (Edited by: Stephen McCormack and David Galas). 2002.
Lam S, Kennedy T, Unger M, Miller YE, Gelmont D, Rusch V, Gipe B, Howard D, LeRiche JC, Coldman A, Gazdar AF: Localization of bronchial intraepithelial neoplastic lesions by fluorescence bronchoscopy. Chest 1998, 113:696–702.
Swisher SG, Roth JA, Komaki R, Gu J, Lee JJ, Hicks M, Ro JY, Hong WK, Merritt JA, Ahrar K, Atkinson NE, Correa AM, Dolormente M, Dreiling L, El-Naggar AK, Fossella F, Francisco R, Glisson B, Grammer S, Herbst R, Huaringa A, Kemp B, Khuri FR, Kurie JM, Liao Z, McDonnell TJ, Morice R, Morello F, Munden R, Papadimitrakopoulou V, Pisters KM, Putnam J. B., Jr., Sarabia AJ, Shelton T, Stevens C, Shin DM, Smythe WR, Vaporciyan AA, Walsh GL, Yin M: Induction of p53-regulated genes and tumor regression in lung cancer patients after intratumoral delivery of adenoviral p53 (INGN 201) and radiation therapy. Clin Cancer Res 2003, 9:93–101.
Nguyen DM, Wiehle SA, Koch PE, Branch C, Yen N, Roth JA, Cristiano RJ: Delivery of the p53 tumor suppressor gene into lung cancer cells by an adenovirus/DNA complex. Cancer Gene Ther 1997, 4:191–198.
Cai DW, Mukhopadhyay T, Liu Y, Fujiwara T, Roth JA: Stable expression of the wild-type p53 gene in human lung cancer cells after retrovirus-mediated gene transfer. Hum Gene Ther 1993, 4:617–624.
Roth JA, Nguyen D, Lawrence DD, Kemp BL, Carrasco CH, Ferson DZ, Hong WK, Komaki R, Lee JJ, Nesbitt JC, Pisters KM, Putnam JB, Schea R, Shin DM, Walsh GL, Dolormente MM, Han CI, Martin FD, Yen N, Xu K, Stephens LC, McDonnell TJ, Mukhopadhyay T, Cai D: Retrovirus-mediated wild-type p53 gene transfer to tumors of patients with lung cancer. Nat Med 1996, 2:985–991.
Swisher SG, Roth JA, Nemunaitis J, Lawrence DD, Kemp BL, Carrasco CH, Connors DG, El-Naggar AK, Fossella F, Glisson BS, Hong WK, Khuri FR, Kurie JM, Lee JJ, Lee JS, Mack M, Merritt JA, Nguyen DM, Nesbitt JC, Perez-Soler R, Pisters KM, Putnam J. B., Jr., Richli WR, Savin M, Waugh MK, et al.: Adenovirus-mediated p53 gene transfer in advanced non-small-cell lung cancer. J Natl Cancer Inst 1999, 91:763–771.
Arteaga CL, Johnson DH: Tyrosine kinase inhibitors-ZD1839 (Iressa). Curr Opin Oncol 2001, 13:491–498.
Brabender J, Danenberg KD, Metzger R, Schneider PM, Park J, Salonga D, Holscher AH, Danenberg PV: Epidermal growth factor receptor and HER2-neu mRNA expression in non- small cell lung cancer Is correlated with survival. Clin Cancer Res 2001, 7:1850–1855.
Salomon DS, Brandt R, Ciardiello F, Normanno N: Epidermal growth factor-related peptides and their receptors in human malignancies. Crit Rev Oncol Hematol 1995, 19:183–232.
Kurie JM, Shin HJ, Lee JS, Morice RC, Ro JY, Lippman SM, Hittelman WN, Yu R, Lee JJ, Hong WK: Increased epidermal growth factor receptor expression in metaplastic bronchial epithelium. Clin Cancer Res 1996, 2:1787–1793.
Rusch V, Klimstra D, Linkov I, Dmitrovsky E: Aberrant expression of p53 or the epidermal growth factor receptor is frequent in early bronchial neoplasia and coexpression precedes squamous cell carcinoma development. Cancer Res 1995, 55:1365–1372.
Woodburn JR: The epidermal growth factor receptor and its inhibition in cancer therapy. Pharmacol Ther 1999, 82:241–250.
Giaccone G: Clinical impact of novel treatment strategies. Oncogene 2002, 21:6970–6981.
Baselga J: The EGFR as a target for anticancer therapy--focus on cetuximab. Eur J Cancer 2001, 37 Suppl 4:S16–22.
Herbst RS: Dose-comparative monotherapy trials of ZD1839 in previously treated non- small cell lung cancer patients. Semin Oncol 2003, 30:30–38.
Adjei AA: Blocking oncogenic Ras signaling for cancer therapy. J Natl Cancer Inst 2001, 93:1062–1074.
Lantry LE, Zhang Z, Yao R, Crist KA, Wang Y, Ohkanda J, Hamilton AD, Sebti SM, Lubet RA, You M: Effect of farnesyltransferase inhibitor FTI-276 on established lung adenomas from A/J mice induced by 4-(methylnitrosamino)-1-(3-pyridyl)-1- butanone. Carcinogenesis 2000, 21:113–116.
Wiese FW, Thompson PA, Kadlubar FF: Carcinogen substrate specificity of human COX-1 and COX-2. Carcinogenesis 2001, 22:5–10.
Gallo O, Franchi A, Magnelli L, Sardi I, Vannacci A, Boddi V, Chiarugi V, Masini E: Cyclooxygenase-2 pathway correlates with VEGF expression in head and neck cancer. Implications for tumor angiogenesis and metastasis. Neoplasia 2001, 3:53–61.
Dannenberg AJ, Altorki NK, Boyle JO, Lin DT, Subbaramaiah K: Inhibition of cyclooxygenase-2: an approach to preventing cancer of the upper aerodigestive tract. Ann N Y Acad Sci 2001, 952:109–115.
Hida T, Leyton J, Makheja AN, Ben-Av P, Hla T, Martinez A, Mulshine J, Malkani S, Chung P, Moody TW: Non-small cell lung cancer cycloxygenase activity and proliferation are inhibited by non-steroidal antiinflammatory drugs. Anticancer Res 1998, 18:775–782.
Kennedy AR, McGundy RB, Little JB: Serial sacrifice study of pathogenesis of 210Po-induced lung tumors in Syrian golden hamsters. Cancer Res 1978, 38:1127–1135.
Achiwa H, Yatabe Y, Hida T, Kuroishi T, Kozaki K, Nakamura S, Ogawa M, Sugiura T, Mitsudomi T, Takahashi T: Prognostic significance of elevated cyclooxygenase 2 expression in primary, resected lung adenocarcinomas. Clin Cancer Res 1999, 5:1001–1005.
Sugita M, Geraci M, Gao B, Powell RL, Hirsch FR, Johnson G, Lapadat R, Gabrielson E, Bremnes R, Bunn PA, Franklin WA: Combined use of oligonucleotide and tissue microarrays identifies cancer/testis antigens as biomarkers in lung carcinoma. Cancer Res 2002, 62:3971–3979.
Acknowledgement
This work was supported by the Vanderbilt Ingram Cancer Center SPORE in Lung Cancer from the National Institutes of Health, the Flight Attendants Medical Research Institute, the Damon Runyon Foundation, the Office of Research and Development, and the Department of Veterans Affairs. The authors thank Tamara Lasakow for editorial assistance with the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Massion, P.P., Carbone, D.P. The molecular basis of lung cancer: molecular abnormalities and therapeutic implications. Respir Res 4, 12 (2003). https://doi.org/10.1186/1465-9921-4-12
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1465-9921-4-12