
Abstract
Background
Asthma is a chronic disease that is characterized by airway hyperresponsiveness and airway remodeling. The underlying mechanisms that mediate the pathological processes are not fully understood. Abl is a non-receptor protein tyrosine kinase that has a role in the regulation of smooth muscle contraction and smooth muscle cell proliferation in vitro. The role of Abl in airway hyperresponsiveness and airway remodeling in vivo is largely unknown.
Methods
To evaluate the role of Abl in asthma pathology, we assessed the expression of Abl in airway tissues from the ovalbumin sensitized and challenged mouse model, and human asthmatic airway smooth muscle cells. In addition, we generated conditional knockout mice in which Abl expression in smooth muscle was disrupted, and then evaluated the effects of Abl conditional knockout on airway resistance, smooth muscle mass, cell proliferation, IL-13 and CCL2 in the mouse model of asthma. Furthermore, we determined the effects of the Abl pharmacological inhibitors imatinib and GNF-5 on these processes in the animal model of asthma.
Results
The expression of Abl was upregulated in airway tissues of the animal model of asthma and in airway smooth muscle cells of patients with severe asthma. Conditional knockout of Abl attenuated airway resistance, smooth muscle mass and staining of proliferating cell nuclear antigen in the airway of mice sensitized and challenged with ovalbumin. Interestingly, conditional knockout of Abl did not affect the levels of IL-13 and CCL2 in bronchoalveolar lavage fluid of animals treated with ovalbumin. However, treatment with imatinib and GNF-5 inhibited the ovalbumin-induced increase in IL-13 and CCL2 as well as airway resistance and smooth muscle growth in animals.
Conclusions
These results suggest that the altered expression of Abl in airway smooth muscle may play a critical role in the development of airway hyperresponsiveness and airway remodeling in asthma. Our findings support the concept that Abl may be a novel target for the development of new therapy to treat asthma.
Similar content being viewed by others
Introduction
Asthma is a chronic airway disease characterized by airway hyperresponsiveness (AHR) and airway remodeling, which lead to impaired respiratory air flow in patients with asthma. However, the underlying mechanisms for the pathological processes are not fully understood.
AHR is largely attributed to hyperreactivity of airway smooth muscle [1]. The contractile properties of human asthmatic airway smooth muscle cells are distinctive from normal human airway smooth muscle cells [2, 3]. In addition, the hyperreactivity of airway smooth muscle tissues occurs in animal models of asthma [4, 5]. Furthermore, increased airway smooth muscle cell proliferation contributes to the progression of airway remodeling in asthma [6, 7]. Airway smooth muscle hyperplasia may facilitate the thickening of the bronchial wall and promote AHR to a variety of stimuli [7, 8].
Abl (Abelson tyrosine kinase, c-Abl) is a non-receptor tyrosine kinase that participates in the regulation of a range of cellular functions including migration and adhesion of nonmuscle cells [9, 10]. Recent studies have implicated Abl in the regulation of vascular smooth muscle contraction in vitro. Contractile activation induces Abl phosphorylation, an indication of Abl activation [10, 11], in smooth muscle. Knockdown of Abl attenuates smooth muscle force development in response to contractile activation [11–13]. Moreover, Abl has been implicated in regulating smooth muscle cell proliferation. Activation of Abl occurs in smooth muscle cells in response to stimulation with growth factors. Silencing of Abl inhibits smooth muscle cell proliferation induced by growth factors [14, 15]. Nevertheless, the role of Abl in asthma pathology in vivo is largely unknown.
In this study, we generated smooth muscle-specific conditional knockout mice, and determined whether smooth muscle-specific knockout of Abl affects AHR and airway remodeling in a mouse model of chronic asthma. Our results suggest that the altered expression of Abl in airway smooth muscle is critical for the development of AHR and airway remodeling.
Methods
Animals and measurement of airway resistance
Abl-lox mice were a gift of Dr. Koleske of Yale University [16]. SM22cre mice were purchased from The Jackson Laboratory. Abl-lox mice (genetic background, 129/Svj) were crossed with SM22cre mice on C57BL/6 background. These mice express Cre recombinase under control of a smooth muscle-specific SM22 promoter. As a consequence, this loxP flanked exon 5 of the abl gene was excised in smooth muscle cells (Figure 1A). The sequences of primers used to identify the genotype were: Primer 1, 5’-CTGTACGTGTCCTCCGAGAG-3’; Primer 2, 5’-CTTCAAGGTCTTCACGGCCA-3’; Primer 3, 5’-GATGTCTCTACAGGGTTAAGATTAAGAGCA-3’; Primer 4, 5’-TGTGCATAGCAGGAAGTCCTCCAGAG-3’; Primer 5, 5’-AGTTAACACACCTCCAGAGTGAGTGCCCT-3’.

Targeting of the abl gene in smooth muscle cells. (A) SM22-Cre construct possesses a SM22 promoter (PSM22) and a Cre coding region. The abl locus containing a floxed version of the Exon5 of abl and the predicted product of Cre-mediated recombination are also shown. Numbered arrows represent PCR primers. (B) Ethidium bromide-stained agarose gel of PCR products amplified from mouse tails of indicated mouse strains. PCR with primers 3 and 5 demonstrate selective deletion of DNA between loxP sites in the presence of Cre recombinase in vivo. Each lane represents tail DNA samples isolated from an individual mouse of the indicated genotype. (C) Extracts of airway smooth muscle cells from Abl-lox and Ablsm−/− mice were analyzed by immunoblotting. Knockout of the Abl protein is verified in airway smooth muscle. Representative blots from three separate experiments are shown.
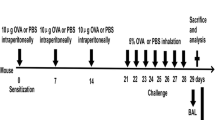
Age- and sex-matched Abl-lox (control) mice and Ablsm−/− (smooth muscle specific knockout of abl gene) mice (6–7 weeks old) were sensitized by intraperitoneal injection of sterile LPS-free ovalbumin (OVA) (Pierce) or phosphate buffered saline (PBS, control) for three weeks, and challenged with intranasal instillations of OVA or PBS twice a week for eight weeks using pre-viously-described protocols [17] with minor modification (Figure 2). To measure airway resistance, mice were anesthetized with an intraperitoneal injection of pentobarbital sodium, tracheotomized, and connected to the FlexiVent system (SCIREQ, Montreal, Canada) on Day 77. Mice were mechanically ventilated at 150 breaths/minute with a tidal volume of 10 ml/kg and a positive end-expiratory pressure (PEEP) of 3.35 cm H2O. Following baseline measurements, mice were challenged with methacholine (MCh) aerosol for 10 seconds at different doses. Airway resistance was measured for each mouse after inhalation of the aerosol. Dose–response curve was then determined. This study was approved by the Institutional Committee of Animal Care and Usage of Albany Medical College.

Sensitization, challenge and treatment protocol for the animal model of chronic asthma. I.P., intraperitoneal injection. I.N., intranasal instillation.
To assess the effects of inhibitors, BALB/c mice (6–7 weeks old) were purchased from The Jackson Laboratory. They were sensitized and challenged by OVA as described above. In addition, animals were intranasally instilled with 10 mg/kg GNF-5 or imatinib, or PBS 1 h before OVA instillation and 5 h after OVA instillation for last three weeks (Figure 2). Airway resistance in these mice was then assessed on Day 77.
Assessment of tracheal ring contraction
Mice were euthanized by injection of pentobarbital (140 mg/kg). A segment of tracheas (4–5 mm in length) was immediately removed and placed in physiological saline solution (PSS) containing 110 mM NaCl, 3.4 mM KCl, 2.4 mM CaCl2, 0.8 mM MgSO4, 25.8 mM NaHCO3, 1.2 mM KH2PO4, and 5.6 mM glucose. The solution was aerated with 95%O2-5%CO2 to maintain a pH of 7.4. Two stainless steel wires were passed through the lumen of tracheal rings. One of the wires was connected to the bottom of organ baths and the other was attached to a Grass force transducer that had been connected to a computer with A/D converter (Grass). Tracheal segments were then placed in PSS at 37°C. Passive tension with 0.5 g was applied to each segment for 60 min. Contractile force in response to various treatments was then measured.
Cell culture
Human airway smooth muscle (HASM) cells were prepared from human bronchi and adjacent tracheas obtained from the International Institute for Advanced Medicine [15]. Human tissues were non-transplantable and consented for research. This study was approved by the Albany Medical College Committee on Research Involving Human Subjects. Briefly, muscle tissues were incubated for 20 min with dissociation solution [130 mM NaCl, 5 mM KCl, 1.0 mM CaCl2, 1.0 mM MgCl2, 10 mM Hepes, 0.25 mM EDTA, 10 mM D-glucose, 10 mM taurine, pH 7, 4.5 mg collagenase (type I), 10 mg papain (type IV), 1 mg/ml BSA and 1 mM dithiothreitol]. All enzymes were purchased from Sigma-Aldrich. The tissues were then washed with Hepes-buffered saline solution (composition in mM: 10 Hepes, 130 NaCl, 5 KCl, 10 glucose, 1 CaCl2, 1 MgCl2, 0.25 EDTA, 10 taurine, pH 7). The cell suspension was mixed with Ham’s F12 medium supplemented with 10% (v/v) fetal bovine serum (FBS) and antibiotics (100 units/ml penicillin, 100 μg/ml streptomycin). Cells were cultured at 37°C in the presence of 5% CO2 in the same medium. The medium was changed every 3–4 days until cells reached confluence, and confluent cells were passaged with trypsin/EDTA solution [15, 18, 19]. Smooth muscle cells within passage 5 were used for the studies.
Immunoblot analysis
Cells were lysed in SDS sample buffer composed of 1.5% dithiothreitol, 2% SDS, 80 mM Tris–HCl (pH 6.8), 10% glycerol and 0.01% bromophenol blue. The lysates were boiled in the buffer for 5 min and separated by SDS-PAGE. Proteins were transferred to a nitrocellulose membrane. The membrane was blocked with bovine serum albumin or milk for 1 h and probed with use of primary antibody followed by horseradish peroxidase-conjugated secondary antibody (Fisher Scientific). Proteins were visualized by enhanced chemiluminescence (Fisher Scientific) using the LAS-4000 Fuji Image System. Abl antibody was purchased from BD Biosciences and Santa Cruz Biotechnology. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) antibody was purchased from Fitzgerald (Acton, MA). The levels of proteins were quantified by scanning densitometry of immunoblots (Fuji Multigauge Software). The luminescent signals from all immunoblots were within the linear range.
Immunohistochemistry
Mouse lungs were placed in frozen tissue-embedding medium (Neg 52, Richard-Allen Scientific) and cryosectioned using Cryostats (Richard-Allen Scientific). Tissue sections were fixed for 15 min in 4% paraformaldehyde, and were then washed three times in PBS buffer followed by permeabilization with 0.2% Triton X-100 dissolved in PBS for 5 min. These tissues were incubated with α-smooth muscle actin antibody (Sigma) or proliferating cell nuclear antigen (PCNA) antibody (Thermo Scientific) followed by appropriate secondary antibody conjugated to Alexa-488 or Alex-543 (Molecular Probes/Life Technologies). The sections were also counterstained with 4',6-diamidino-2-phenylindole to visualize the nucleus. The samples were viewed and digitally captured using a Leica microscope system (MDI 6000). All immunohistochemical measurements were performed by using the NIH ImageJ software.
Analysis of airway inflammation
Lungs from sacrificed mice were lavaged three times with 1 mL sterile Hanks balanced salt solution (HBSS) containing 3 mM EDTA. Bronchoalveolar lavage fluid (BALF) was collected after centrifugation and, the supernatant was removed and frozen at −80°C for cytokine/chemokine measurements (See below). The cell pellet was resuspended in HBSS, and total number of inflammatory cells in the BALF was counted by using a hemocytometer. Differential cell counts (macrophages, neutrophils, lymphocytes, and eosinophils) were performed by counting 100 cells from cytospin preparations stained with DiffQuick stain. The levels of IL-13 and CCL2 in the BALF were determined using ELISA kits (R&D systems) according to the manufacturer’s instructions.
Statistical analysis
All statistical analysis was performed using Prism 4 software (GraphPad Software, San Diego, CA). Comparison among multiple groups was performed by one-way analysis of variance followed by Tukey’s multiple comparison test. Differences between two groups were analyzed by Student-Newman-Keuls test or Dunn's method. Values of n refer to the number of experiments used to obtain each value. P < 0.05 was considered to be significant.
Results
Abl is required for airway smooth muscle contraction
Our previous studies demonstrate that Abl regulates vascular smooth muscle contraction [11–13]. To determine the role of Abl in airway smooth muscle, we generated SM22creAbl-lox mice, a mouse model with smooth muscle cell–specific disruption of the abl gene (Figure 1A). Genotyping and immunoblot analysis verified knockout of Abl in airway smooth muscle (Figure 1B and C). Previous studies by others demonstrate that SM22 is expressed in airway smooth muscle tissues [20–22], which suggests that SM22 promoter is functional in airway smooth muscle. Our results are consistent with these studies.
Contractile responses of mouse tracheal rings to ACh stimulation were compared between Ablsm−/− (smooth muscle knockout of Abl) mice and Abl-lox (control) mice. As shown in Figure 3A, contractile responses of mouse tracheal rings to ACh were lower in Ablsm−/− mice than in Abl-lox mice, which was dose-dependent.

Abl is required for airway smooth muscle contraction. (A) Tracheal rings of Ablsm−/− mice and Abl-lox (control) mice were treated with difference concentration of acetylcholine (ACh). Dose–response of these rings was then evaluated. Contractile force is normalized to maximal force of tracheal rings from Abl-lox mice. Values are mean ± SE (n = 9–10). * Significantly lower active force in Ablsm−/− mice than in Abl-lox at corresponding doses (P < 0.01). (B) Tracheal rings of Abl-lox mice were pretreated with or without the Abl inhibitors imatinib or GNF-5 (10 μM, 10 min). ACh dose–response of tissues was then determined. Pretreatment with imatinib or GNF-5 attenuated ACh-induced contraction in vitro (n = 15–18, * P < 0.01). (C) Tracheal rings of Abl-lox mice were precontracted with 100 μM ACh. Different concentrations of imatinib or GNF-5 were then imposed to assess the relaxation response. Treatment with the pharmacological inhibitors induced relaxation of tracheal segments precontracted by ACh (n = 17–19).
We also evaluated acute effects of the Abl pharmacological inhibitors imatinib (Gleevec, STI-571) [11, 23] and GNF-5 [24] on airway smooth muscle contraction. Treatment of mouse tracheal rings with imatinib significantly attenuated force development induced by ACh (Figure 3B). Likewise, GNF-5 had inhibitory effects on contraction of tracheal segments with slightly stronger potency (Figure 3B). Furthermore, treatment with imatinib or GNF-5 induced relaxation of tracheal rings precontracted by ACh (Figure 3C).
The expression of Abl is upregulated in asthmatic airway smooth muscle
We evaluated the expression of Abl in airway tissues of OVA-sensitized and challenged mice, a well-recognized animal model mimicking allergen-induced asthma in humans [17]. As shown in Figure 4A, the amount of Abl was elevated in airway tissues of OVA-treated mice compared to naïve animals. However, the levels of GAPDH were similar in OVA-treated mice and naïve mice. The ratio of Abl/GAPDH in airway tissues was higher in OVA-treated mice than in naïve mice (Figure 4B). To validate this finding in human asthma, we assessed Abl expression in HASM cells from normal subjects and patients with severe asthma. The level of Abl was higher in asthmatic cells than in normal cells. The ratios of Abl/GAPDH in asthmatic cells were significantly higher as compared to normal cells (Figure 4A and B).

The expression of Abl is higher in asthmatic airways tissues/cells. (A) The upregulation of Abl expression in the airways of ovalbumin (OVA)-sensitized and challenged mice and human airway smooth muscle (HASM) cells of patients with severe asthma. Blots of tissue/cell extracts were probed with use of antibodies against Abl and GAPDH. 1, tracheal extracts of naïve mice; 2, tracheal extracts of OVA-sensitized and challenged mice; 3, normal HASM cell extracts; 4, asthmatic HASM cell extracts. (B) For quantification analysis, Abl/GAPDH ratios in OVA-treated animals or asthmatic HASM cells are normalized to corresponding control tissues or cells (*P < 0.05).
Conditional knockout of Abl in smooth muscle attenuates OVA-sensitized airway resistance and contraction of tracheal rings
We used a chronic asthma animal model [17] to determine whether Abl in smooth muscle is involved in AHR. Briefly, Abl-lox and Ablsm−/− mice were sensitized by OVA for three weeks and challenged by OVA for eight weeks (Figure 2). Airway resistance in response to methacholine (MCh) inhalation was measured using the FlexiVent system. OVA sensitization and challenge induced a higher response to MCh inhalation in Abl-lox mice as compared to Abl-lox mice treated with PBS. However, airway resistance induced by MCh was lower in Ablsm−/− mice sensitized and challenged by OVA than in Abl-lox mice treated with OVA. The airway resistance was also lower in naïve Ablsm−/− mice than in naïve Abl-lox mice (Figure 5A).

Abl knockout attenuates airway resistance and contraction of tracheal rings in the animal model of asthma. (A) Abl-lox and Ablsm−/− mice were sensitized and challenged by OVA. Airway resistance (RAW) in these mice was evaluated using the FlexiVent system. RAW in Abl-lox mice treated with OVA was higher than in Abl-lox mice treated with PBS, verifying the development of AHR in the mouse model. RAW in Ablsm−/− mice treated with OVA was reduced compared to Abl-lox mice treated with OVA (n = 10–14, *P < 0.05). (B) The contractile response of tracheal rings from these mice was determined by using an in vitro organ bath system. Contractile force is normalized to maximal force of rings from OVA-treated Abl-lox mice. Contractile force of OVA-sensitized and challenged Abl-lox mice was increased as compared to naïve Abl-lox mice. OVA-induced airway smooth muscle contraction was reduced in Ablsm−/− mice (n = 10–14, *P < 0.05).
We also assessed the effects of Abl knockout on airway smooth muscle hyperreactivity in vitro. Contractile force in isolated tracheal rings from OVA-treated Abl-lox mice was greater compared to naïve Abl-lox mice. However, active force of isolated tracheal rings from OVA-treated Ablsm−/− mice was reduced compared to OVA-treated Abl-lox mice. Contractile response of tracheal rings from naïve Ablsm−/− mice was also lower compared to naïve Abl-lox mice (Figure 5B).
Pharmacological inhibition of Abl diminishes AHR and smooth muscle hyperreactivity
We also evaluated the effects of the Abl inhibitors imatinib and GNF-5 on AHR in asthmatic animals. The OVA sensitization and challenge increased airway resistance in BALB/c mice as compared to BALB/c mice treated with PBS (Figure 6A). In contrast, the OVA-induced increase in airway resistance was reduced in the animals treated with imatinib or GNF-5 (Figure 6A). In addition, treatment with imatinib or GNF-5 inhibited the ACh-induced contraction in isolated mouse tracheal rings of OVA-sensitized and challenged mice (Figure 6B).

Effects of imatinib and GNF-5 on airway resistance and contractile response of tracheal rings from OVA-sensitized and challenged mice. (A) BALB/c mice were sensitized and challenged with OVA in the presence of imatinib, GNF-5 or vehicle (V). Airway resistance in these mice was then measured. Intranasal instillation of imatinib and GNF-5 inhibited RAW in mice sensitized and challenged by OVA. *Significantly lower airway resistance in animals treated with imatinib or GNF-5 compared to animals treated with vehicle (P < 0.05, n = 7–8). (B) Treatment with imatinib and GNF-5 attenuated the OVA-sensitized tracheal contraction in vitro. Contractile force is normalized to maximal force of rings from OVA- and vehicle-treated mice. *Significantly lower tracheal contraction from animals treated with imatinib or GNF-5 compared to vehicle-treated animals (P < 0.05, n = 7–8).
We noticed that airway resistance in response to MCh inhalation was slightly higher in BALB/c mice than in Abl-lox mice sensitized and challenged by OVA (Figure 5A and Figure 6A). This is not surprising because BALB/c mouse strain is known to have skewed Th2 response compared to other mouse strains [25, 26].
Conditional knockout of Abl inhibits airway smooth muscle growth in the animal model of asthma
To determine the role of Abl in the remodeling of airway smooth muscle, we assessed whether conditional knockout of Abl in smooth muscle affects the allergen-induced airway smooth muscle mass by determining the area of α-smooth muscle actin (a smooth muscle marker) staining in the airways of Abl-lox and Ablsm−/− mice sensitized and challenged with OVA.
The area of α-smooth muscle actin staining in the airways of Abl-lox mice treated with OVA was greater than that in Abl-lox mice treated with PBS, as evidenced by immunofluorescent analysis. In contrast, the area of actin staining in the airways of Ablsm−/− mice treated with OVA was reduced as compared to Abl-lox mice treated with OVA (Figure 7A & B). These results suggest that conditional knockout of Abl is able to attenuate the allergen-induced increase in airway smooth muscle mass. In addition, the fluorescent intensity of α-smooth muscle actin staining was higher in Abl-lox mice treated with OVA compared to naïve Abl-lox mice, suggesting higher α-smooth muscle actin expression in the remodeled airway in asthmatic models [6]. Furthermore, we determined the effects of imatinib or GNF-5 on airway smooth muscle growth. Treatment with the inhibitors attenuated an increase in airway smooth muscle mass in BALB/c mice sensitized and challenged by OVA (Figure 7B).

Abl knockout inhibits airway remodeling in the animal model of asthma. (A) Representative images illustrating the effects of Abl knockout on α-actin and PCNA staining. Abl-lox and Ablsm−/− mice were sensitized and challenged with OVA. Lung sections of the mice were immunostained for α-actin (red) and PCNA (green). The sections were also counterstained with use of 4',6-diamidino-2-phenylindole to visualize the nucleus (blue). (B) Actin positive area in Ablsm−/− mice treated with OVA was lower compared to Abl-lox mice exposed to OVA (n = 6–7, *P < 0.05). Treatment with imatinib or GNF-5 inhibited the OVA-induced increase in actin positive area from BALB/c mice (n = 6–7, *P < 0.05). C, control animals. (C) Fluorescent intensity of PCNA colocalized with actin in the airways in Ablsm−/− mice treated with OVA was lower than that in Abl-lox mice exposed to OVA (n= 6–7, *P < 0.05). Treatment with imatinib or GNF-5 inhibited the OVA-induced increase in PCNA staining from BALB/c mice (n = 6–7, *P < 0.05). C, control animals.
We also evaluated whether the knockout of Abl influences allergen-induced airway smooth muscle cell proliferation. Proliferating cell nuclear antigen (PCNA) is a critical protein that is expressed by proliferating cells in S phase of the cell cycle. Thus, it has been widely used as a marker of cell proliferation in the airways [8, 27]. The fluorescent intensity of PCNA colocalized with α-smooth muscle actin was greater in the airways of Abl-lox mice treated with OVA compared with Abl-lox mice treated with PBS. However, the intensity of PCNA costained with actin in the airways of Ablsm−/− mice treated with OVA was lower than that in the airways of Abl-lox mice treated with OVA (Figure 7A and C). Moreover, treatment with the Abl inhibitors imatinib or GNF-5 diminished the fluorescent intensity of PCNA in BALB/c mice treated with OVA (Figure 7C). These results indicate that Abl has a role in the allergen-induced airway smooth muscle proliferation.
Effects of conditional knockout of Abl and Abl inhibitors on airway inflammation in the animal model of asthma
As a consequence of allergic sensitization and challenge, inflammatory cells enter into the lungs and cytokine/chemokine levels are increased in the bronchoalveolar space of asthmatic patients and animal models, which are characteristic features of allergic airway inflammation [7, 28, 29]. To determine whether the smooth muscle-specific depletion of Abl affects recruitment of inflammatory cells, we determined total and differential cell counts of BALF in lungs of naïve and OVA-treated Abl-lox mice and Ablsm−/− mice.
OVA sensitization and challenge increased the numbers of total and differential cells in the lungs of Abl-lox mice. However, the allergen-induced increase in cell numbers in the lungs in Ablsm−/− mice was similar to that in Abl-lox mice (Figure 8A and B).

Effects of Abl knockout, imatinib and GNF-5 on total and differential cell counts in BALF. Total (A) and differential (B) cell numbers in BALF of Ablsm−/− mice were similar to those of Abl-lox mice when treated with PBS or OVA (n = 6–7). However, treatment with imatinib or GNF-5 inhibited the OVA-induced increase in total (C) and differential (D) cell numbers in BALF of BALB/c mice (n = 7, *P < 0.05).
We also evaluated the effects of the Abl inhibitors imatinib and GNF-5 on cell counts of BALF from mice treated with PBS or OVA. OVA sensitization and challenge increased total and differential cell counts of BALF from BALB/c mice. Treatment with imatinib and GNF-5 reduced the OVA-induced increase in inflammatory cell numbers (Figure 8C and D).
To determine the role of Abl in smooth muscle in the production of cytokine and chemokine, we evaluated the level of IL-13 and CCL2 (representative cytokine and chemokine in asthma pathology) [5, 30, 31] in the BALF in lungs of naïve and OVA-treated Abl-lox and Ablsm−/− mice. OVA sensitization and challenge increased the level of IL-13 and CCL2 in the BALF of Abl-lox mice. In addition, the allergen-induced increase in IL-13 and CCL2 in the lungs of Ablsm−/− mice was similar to those in Abl-lox mice (Figure 9A and B). However, treatment with imatinib and GNF-5 diminished the OVA-induced increase in IL-13 and CCL2 in the lungs of BALB/c mice (Figure 9C and D).

Effects of Abl knockout, imatinib and GNF-5 on the levels of IL-13 and CCL2 in BALF. (A) The level of IL-13 in BALF of Ablsm−/− mice was similar to those of Abl-lox mice treated with PBS or OVA (n = 7). (B) Abl conditional knockout did not affect the concentration of CCL2 in BALF (n = 6). (C) The OVA-induced increase in IL-13 level in BALF from BALB/c mice was attenuated by treatment with imatinib or GNF-5 (n= 7, *P < 0.05). (D) Treatment with imatinib or GNF-5 inhibited the OVA-induced enhancement of CCL2 in BALF from BALB/c mice (n = 6, *P < 0.05).
Discussion
Abl is a non-receptor tyrosine kinase that has a role in regulating smooth muscle contraction and smooth muscle cell proliferation in vitro. The role of Abl in the pathogenesis of asthma in vivo is largely unknown. In this study, Abl expression is upregulated in asthmatic airways. More importantly, conditional knockout of Abl in smooth muscle inhibits airway resistance and airway smooth muscle growth in the animal model of chronic asthma. The results suggest that Abl plays a critical role in the progression of AHR and airway remodeling in chronic asthma.
Our previous studies demonstrate that Abl is essential for vascular smooth muscle force development [10–13]. In this report, conditional knockout of Abl in smooth muscle diminished contractile response of tracheal rings. Moreover, acute inhibition of Abl by the pharmacological agents attenuated contraction in tracheal rings. The results suggest that Abl is necessary for airway smooth muscle contraction. Abl may regulate the functional states of several proteins including Crk-associated substrate and Abi1, which in turn regulate actin dynamics and smooth muscle contraction [11–13, 32, 33].
AHR largely stems from hyperreactivity of airway smooth muscle [1]. The pathological mechanisms that mediate airway smooth muscle hyperreactivity and AHR in asthma are not completely elucidated. Th2 cytokines including IL-13 has been implicated in smooth muscle hypercontractility and AHR [34–36]. In this study, the expression of Abl was upregulated in airway tissues of the animal model of asthma as well as in smooth muscle cells of patients with severe asthma. Furthermore, conditional knockout of Abl in smooth muscle attenuated airway smooth muscle hyperreactivity in vitro and airway resistance in mice sensitized and challenged by the allergen. To rule out the potential effects by compensation in genetically-modified mice, we also determined the acute effects of the Abl pharmacological inhibitors imatinib [11, 23] and GNF-5 [24] on airway resistance in vivo and airway smooth muscle hyperreactivity in vitro. Treatment with the inhibitors also diminished the OVA-sensitized airway resistance in vivo and tracheal contraction in vitro. The results suggest that Abl has a critical role in the development of AHR in asthma.
Airway remodeling is a characteristic feature of severe asthma. In addition to fibrosis, enhanced deposition of extracellular matrix protein, epithelial injury and airway smooth muscle hypertrophy, proliferation of airway smooth muscle cells markedly contributes to the pathogenesis of airway remodeling [6, 8, 29, 37]. Our recent studies demonstrate that Abl is required for smooth muscle cell proliferation in in vitro studies [14, 15]. Abl may modulate cell proliferation by affecting actin polymerization and the Raf-1/MEK/ERK1/2 pathway [14, 15].
Growth factors such as epidermal growth factor and platelet-derived growth factor have been implicated in the progression of airway remodeling [7]. In this report, smooth muscle mass in the airways was reduced in conditional knockout mice sensitized and challenged by ovalbumin. In addition, the cell proliferation marker PCNA was also diminished in conditional knockout mice treated with the allergen. Moreover, treatment with the pharmacological inhibitors had similar effects. Thus, the increased expression of Abl in smooth muscle may contribute to the development of airway remodeling in chronic asthma.
In response to allergic sensitization and challenge, inflammatory cells enter into the lungs and cytokine/chemokine levels are increased in the bronchoalveolar space of asthmatic patients and animal models [4, 30, 34]. Because airway smooth muscle cells have ability to secret cytokines in vitro[28], we assessed whether Abl knockout in smooth muscle affects airway inflammation. Conditional knockout of Abl did not affect the increase in inflammatory cell numbers, IL-13 and CCL2 in animals sensitized and challenged by the allergen. The results lead us to suggest that Abl expression in smooth muscle does not modulate inflammatory cell infiltration and production of IL-13 and CCL2 in asthma.
On the contrary, treatment with imatinib and GNF-5 reduced the OVA-induced increase in inflammatory cell numbers, and levels of IL-13 and CCL2. The results suggest that global inhibition of Abl diminishes airway inflammation in chronic asthma, which is consistent with the findings that Abl may regulate migration and synthetic functions of immune cells in vitro[38–41].
Currently, β2 agonists are widely used to treat asthma. β2 agonists reduce symptoms of airway obstruction by inducing airway smooth muscle relaxation. However, this therapy has various limitations including β2 adrenergic receptor desensitization [42]. In this study, we demonstrate that Abl in smooth muscle has a critical role in the pathogenesis of AHR and airway remodeling. Furthermore, global inhibition of Abl by pharmacological agents attenuates airway inflammation. Thus, our findings support the concept that Abl may be a novel target for the development of new therapy to treat asthma.
Conclusions
Abl is a non-receptor tyrosine kinase that has a role in regulating smooth muscle contraction and smooth muscle cell proliferation in vitro. The role of Abl in asthma pathogenesis in vivo is not well elucidated. Our present results suggest that the altered expression of Abl in smooth muscle plays a critical role in the progression of AHR and airway remodeling in chronic asthma. Furthermore, global inhibition of Abl attenuates airway inflammation. Thus, Abl may be a novel target for the development of new therapy to treat asthma.
Abbreviations
- Abl:
-
Abelson tyrosine kinase
- ACh:
-
acetylcholine
- AHR:
-
airway hyperresponsiveness
- BALF:
-
Bronchoaleolar lavage fluid
- CCL2:
-
Chemokine (C-C motif) ligand 2
- GAPDH:
-
Glyceraldehyde 3-phosphate dehydrogenase
- GNF-5:
-
bcr-Abl inhibitor III
- HASM:
-
Human airway smooth muscle
- IL-13:
-
Interleukin-13
- MCh:
-
Methacholine
- OVA:
-
Ovalbumin.
References
Amrani Y, Tliba O, Deshpande DA, Walseth TF, Kannan MS, Panettieri RA: Bronchial hyperresponsiveness: insights into new signaling molecules. Curr Opin Pharmacol. 2004, 4: 230-234. 10.1016/j.coph.2004.02.004.
Ma XF, Cheng ZQ, Kong H, Wang Y, Unruh H, Stephens NL, Laviolette M: Changes in biophysical and biochemical properties of single bronchial smooth muscle cells from asthmatic subjects. Am J Physiol Lung Cell Mol Physiol. 2002, 283: L1181-L1189.
Bjorck T, Gustafsson LE, Dahlen SE: Isolated bronchi from asthmatics are hyperresponsive to adenosine, which apparently acts indirectly by liberation of leukotrienes and histamine. Am Rev Respir Dis. 1992, 145: 1087-1091. 10.1164/ajrccm/145.5.1087.
Guedes AG, Paulin J, Rivero-Nava L, Kita H, Lund FE, Kannan MS: CD38-deficient mice have reduced airway hyperresponsiveness following IL-13 challenge. Am J Physiol Lung Cell Mol Physiol. 2006, 291: L1286-L1293. 10.1152/ajplung.00187.2006.
Guedes AG, Jude JA, Paulin J, Kita H, Lund FE, Kannan MS: Role of CD38 in TNF-alpha-induced airway hyperresponsiveness. Am J Physiol Lung Cell Mol Physiol. 2008, 294: L290-L299.
Bentley JK, Deng H, Linn MJ, Lei J, Dokshin GA, Fingar DC, Bitar KN, Henderson WR, Hershenson MB: Airway smooth muscle hyperplasia and hypertrophy correlate with glycogen synthase kinase-3(beta) phosphorylation in a mouse model of asthma. Am J Physiol Lung Cell Mol Physiol. 2009, 296: L176-L184.
Ammit AJ, Panettieri RA: Airway smooth muscle cell hyperplasia: a therapeutic target in airway remodeling in asthma?. ProgCell Cycle Res. 2003, 5: 49-57.
Dekkers BG, Bos IS, Gosens R, Halayko AJ, Zaagsma J, Meurs H: The integrin-blocking peptide RGDS inhibits airway smooth muscle remodeling in a guinea pig model of allergic asthma. Am J Respir Crit Care Med. 2010, 181: 556-565. 10.1164/rccm.200907-1065OC.
Hu H, Bliss JM, Wang Y, Colicelli J: RIN1 is an ABL tyrosine kinase activator and a regulator of epithelial-cell adhesion and migration. Curr Biol. 2005, 15: 815-823. 10.1016/j.cub.2005.03.049.
Wang JY: Controlling Abl: auto-inhibition and co-inhibition?. Nat Cell Biol. 2004, 6: 3-7. 10.1038/ncb0104-3.
Jia L, Tang DD: Abl activation regulates the dissociation of CAS from cytoskeletal vimentin by modulating CAS phosphorylation in smooth muscle. AJP - Cell Physiol. 2010, 299: C630-C637. 10.1152/ajpcell.00095.2010.
Chen S, Wang R, Li QF, Tang DD: Abl knockout differentially affects p130 Crk-associated substrate, vinculin, and paxillin in blood vessels of mice. Am J Physiol Heart Circ Physiol. 2009, 297: H533-H539. 10.1152/ajpheart.00237.2009.
Anfinogenova Y, Wang R, Li QF, Spinelli AM, Tang DD: Abl silencing inhibits CAS-Mediated process and constriction in resistance arteries. Circ Res. 2007, 101: 420-428. 10.1161/CIRCRESAHA.107.156463.
Jia L, Wang R, Tang DD: Abl regulates smooth muscle cell proliferation by modulating actin dynamics and ERK1/2 activation. Am J Physiol Cell Physiol. 2012, 302: C1026-C1034. 10.1152/ajpcell.00373.2011.
Wang R, Mercaitis OP, Jia L, Panettieri RA, Tang DD: Raf-1, actin dynamics and Abl in human airway smooth muscle cells. Am J Respir Cell Mol Biol. 2013, 48: 172-178. 10.1165/rcmb.2012-0315OC.
Moresco EMY, Donaldson S, Williamson A, Koleske AJ: Integrin-mediated dendrite branch maintenance requires Abelson (Ab1) family kinases. J Neurosci. 2005, 25: 6105-6118. 10.1523/JNEUROSCI.1432-05.2005.
Wegmann M, Goggel R, Sel S, Sel S, Erb KJ, Kalkbrenner F, Renz H, Garn H: Effects of a low-molecular-weight CCR-3 antagonist on chronic experimental asthma. Am J Respir Cell Mol Biol. 2007, 36: 61-67. 10.1165/rcmb.2006-0188OC.
Li QF, Spinelli AM, Tang DD: Cdc42GAP, reactive oxygen species, and the vimentin network. AJP - Cell Physiol. 2009, 297: C299-C309. 10.1152/ajpcell.00037.2009.
Li QF, Spinelli AM, Wang R, Anfinogenova Y, Singer HA, Tang DD: Critical role of Vimentin Phosphorylation at Ser-56 by p21-activated Kinase in Vimentin Cytoskeleton signaling. J Biol Chem. 2006, 281: 34716-34724. 10.1074/jbc.M607715200.
Shardonofsky FR, Moore J, Schwartz RJ, Boriek AM: Airways in smooth muscle alpha-actin null mice experience a compensatory mechanism that modulates their contractile response. J Appl Physiol. 2012, 112: 898-903. 10.1152/japplphysiol.00417.2011.
Li L, Miano JM, Cserjesi P, Olson EN: SM22 alpha, a marker of adult smooth muscle, is expressed in multiple myogenic lineages during embryogenesis. Circ Res. 1996, 78: 188-195. 10.1161/01.RES.78.2.188.
Camoretti-Mercado B, Forsythe SM, LeBeau MM, Espinosa R, Vieira JE, Halayko AJ, Willadsen S, Kurtz B, Ober C, Evans GA, et al: Expression and cytogenetic localization of the human SM22 gene (TAGLN). Genomics. 1998, 49: 452-457. 10.1006/geno.1998.5267.
Daniels CE, Wilkes MC, Edens M, Kottom TJ, Murphy SJ, Limper AH, Leof EB: Imatinib mesylate inhibits the profibrogenic activity of TGF-beta and prevents bleomycin-mediated lung fibrosis. JClinInvest. 2004, 114: 1308-1316.
Zhang J, Adrian FJ, Jahnke W, Cowan-Jacob SW, Li AG, Iacob RE, Sim T, Powers J, Dierks C, Sun F, et al: Targeting Bcr-Abl by combining allosteric with ATP-binding-site inhibitors. Nature. 2010, 463: 501-506. 10.1038/nature08675.
Whitehead GS, Wang T, DeGraff LM, Card JW, Lira SA, Graham GJ, Cook DN: The chemokine receptor D6 has opposing effects on allergic inflammation and airway reactivity. Am J Respir Crit Care Med. 2007, 175: 243-249. 10.1164/rccm.200606-839OC.
Gueders MM, Paulissen G, Crahay C, Quesada-Calvo F, Hacha J, Van Hove C, Tournoy K, Louis R, Foidart JM, Noel A, Cataldo DD: Mouse models of asthma: a comparison between C57BL/6 and BALB/c strains regarding bronchial responsiveness, inflammation, and cytokine production. Inflamm Res. 2009, 58: 845-854. 10.1007/s00011-009-0054-2.
Tormanen KR, Uller L, Persson CG, Erjefalt JS: Allergen exposure of mouse airways evokes remodeling of both bronchi and large pulmonary vessels. Am J Respir Crit Care Med. 2005, 171: 19-25. 10.1164/rccm.200406-698OC.
Howarth PH, Knox AJ, Amrani Y, Tliba O, Panettieri RA, Johnson M: Synthetic responses in airway smooth muscle. J Allergy ClinImmunol. 2004, 114: S32-S50. 10.1016/j.jaci.2004.04.041.
Lazaar AL, Panettieri RA: Airway smooth muscle: a modulator of airway remodeling in asthma. J Allergy ClinImmunol. 2005, 116: 488-495. 10.1016/j.jaci.2005.06.030.
Wills-Karp M: Interleukin-13 in asthma pathogenesis. ImmunolRev. 2004, 202: 175-190.
Lazaar AL, Panettieri RA: Airway smooth muscle as a regulator of immune responses and bronchomotor tone. ClinChest Med. 2006, 27: 53-69. vi
Tang DD, Anfinogenova Y: Physiologic properties and regulation of the actin cytoskeleton in vascular smooth muscle. J Cardiovasc Pharmacol Ther. 2008, 13: 130-140. 10.1177/1074248407313737.
Wang T, Cleary RA, Wang R, Tang DD: Role of the adapter protein Abi1 in actin-associated signaling and smooth muscle contraction. J Biol Chem. 2013, 288: 20713-20722. 10.1074/jbc.M112.439877.
Zimmermann N, Hershey GK, Foster PS, Rothenberg ME: Chemokines in asthma: cooperative interaction between chemokines and IL-13. J Allergy ClinImmunol. 2003, 111: 227-242. 10.1067/mai.2003.139.
Walter DM, McIntire JJ, Berry G, McKenzie AN, Donaldson DD, DeKruyff RH, Umetsu DT: Critical role for IL-13 in the development of allergen-induced airway hyperreactivity. JImmunol. 2001, 167: 4668-4675.
Tliba O, Deshpande D, Chen H, Van BC, Kannan M, Panettieri RA, Amrani Y: IL-13 enhances agonist-evoked calcium signals and contractile responses in airway smooth muscle. BrJ Pharmacol. 2003, 140: 1159-1162. 10.1038/sj.bjp.0705558.
Orsini MJ, Krymskaya VP, Eszterhas AJ, Benovic JL, Panettieri RA, Penn RB: MAPK superfamily activation in human airway smooth muscle: mitogenesis requires prolonged p42/p44 activation. Am J Physiol. 1999, 277: L479-L488.
Brightbill H, Schlissel MS: The effects of c-Abl mutation on developing B cell differentiation and survival. IntImmunol. 2009, 21: 575-585.
Huang Y, Comiskey EO, Dupree RS, Li S, Koleske AJ, Burkhardt JK: The c-Abl tyrosine kinase regulates actin remodeling at the immune synapse. Blood. 2008, 112: 111-119. 10.1182/blood-2007-10-118232.
Silberman I, Sionov RV, Zuckerman V, Haupt S, Goldberg Z, Strasser A, Ben-Sasson ZS, Baniyash M, Koleske AJ, Haupt Y: T cell survival and function requires the c-Abl tyrosine kinase. Cell Cycle. 2008, 7: 3847-3857. 10.4161/cc.7.24.7267.
Baruzzi A, Berton G: The tyrosine kinase Abl is a component of macrophage podosomes and is required for podosome formation and function. Eur J Immunol. 2012, 42: 2720-2726. 10.1002/eji.201142270.
Penn RB: Embracing emerging paradigms of G protein-coupled receptor agonism and signaling to address airway smooth muscle pathobiology in asthma. Naunyn Schmiedebergs Arch Pharmacol. 2008, 378: 149-169. 10.1007/s00210-008-0263-1.
Acknowledgments
This work was supported by NHLBI Grants HL-110951 and HL-113208 from the National Institutes of Health (to Dale D. Tang). We thank Orion P. Mercaitis for technical support. We thank Dr. Anthony Koleske of Yale University for providing Abl-floxed mice.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
RAC performed animal studies. RW carried out cell culture studies and participated in animal studies and immunohistochemical studies. TW performed in vitro contraction studies. DDT designed the experiments, analyzed data and wrote the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Cleary, R.A., Wang, R., Wang, T. et al. Role of Abl in airway hyperresponsiveness and airway remodeling. Respir Res 14, 105 (2013). https://doi.org/10.1186/1465-9921-14-105
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1465-9921-14-105