
Abstract
The interaction between prM and E proteins in flavivirus-infected cells is a major driving force for the assembly of flavivirus particles. We used site-directed mutagenesis to study the potential role of the transmembrane domains of the prM proteins of Japanese encephalitis virus (JEV) in prM-E heterodimerization as well as subviral particle formation. Alanine insertion scanning mutagenesis within the GXXXG motif in the first transmembrane segment of JEV prM protein affected the prM-E heterodimerization; its specificity was confirmed by replacing the two glycines of the GXXXG motif with alanine, leucine and valine. The GXXXG motif was found to be conserved in the JEV serocomplex viruses but not other flavivirus groups. These mutants with alanine inserted in the two prM transmembrane segments all impaired subviral particle formation in cell cultures. The prM transmembrane domains of JEV may play importation roles in prM-E heterodimerization and viral particle assembly.
Similar content being viewed by others


Background
Japanese encephalitis virus (JEV) is a small enveloped positive-strand RNA virus that belongs to the genus Flavivirus of the family Flaviviridae [1, 2]. The RNA genome of all flaviviruses contain sequences that code for three structural protein genes (capsid C, membrane precursor prM, and envelope E) and seven non-structural protein genes (NS1, NS2A, NS2B, NS3, NS4A, NS4B, NS5), as well as flanking un-translated regions [1, 2]. The flavivirus assembly process includes (i) interaction of prM and E proteins by heterodimer formation in the endoplasmic reticulum (ER), (ii) encapsulation of the genomic RNA by the C protein and enclosure by cell membrane-derived lipid bilayers containing prM and E proteins to form immature virions, and (iii) cleavage of the prM protein to M protein by furin or a furin-like protease in the trans-Golgi network (TGN) to release viral particles. Subviral particles (SPs) that do not contain genomic RNA and protein C have been found in flavivirus-infected cells [3]. Co-expression of prM and E envelope proteins resulted in the formation and secretion of SPs in cell cultures for tick-borne encephalitis virus (TBEV) [4], dengue virus (DENV) [5], JEV [6, 7], Murray Valley encephalitis virus (MVEV) [8], St. Louis encephalitis virus (SLEV) [9], and West Nile virus (WNV) [10]. The interaction of prM and E proteins in flavivirus-infected cells is a major driving force of the assembly of virus although assembly mechanisms of the Flaviviridae are only very incompletely understood.
The prM and E envelope proteins are type I transmembrane (TM) proteins and both contain stem and anchor regions at their C-terminal ends [11] as illustrated in Fig. 1 The stem region of the prM protein contains one helix domain (prM-H), and that of the E protein has two helix domains (E-H1, E-H2). The anchor regions of the prM and E proteins both contain two separate anchor domains (prM-TM1, prM-TM2, E-TM1, E-TM2). The stem and anchor regions of the prM and E proteins of DENV have both been predicted to include two alpha-helices [12]. High-resolution cryo-EM images of DENV show that the prM-H domain is partially buried in the outer lipid leaflet while the E-H1 and E-H2 domains are either angled or lie flat on the outer lipid leaflet [12]. The TM anchor regions of prM and E proteins (prM-TM1, prM-TM2, E-TM1, E-TM2) all form anti-parallel coiled-coil helices and do not penetrate the lipid membranes to come in contact with nucleocapsids [12]. In TBEV, the E-TM1 and E-H2 domains substantially influenced the stability of the prM-E interaction but did not affect the prM-mediated intracellular transport or secretion of soluble E protein according to C-terminal deletion analysis [13]. However, alanine insertion scanning mutagenesis in the anchor regions of prM and E of yellow fever virus (YFV) did not affect the prM-E interaction, but did inhibit SP formation [14]. Replacement of the stem and anchor regions of the E protein of DENV with that of JEV promoted SP production [15] but the E-H1 domain of JEV did not influence the SP secretion of DENV in CHO cells [16]. The E-TM2 domain of TBEV was demonstrated to be associated with virus particle formation, acting as a signal peptide for NS1 protein [17]. To our knowledge, most reported studies have focused on the stem and anchor regions of the E protein, and very little information is available for the prM protein.

Schematic dagram of flavivirus prM and E protein where the stem and anchor regions located at their C-terminal ends.
In this work, the two anchor domains of prM protein (prM-TM1, prM-TM2) were studied using alanine insertion scanning mutagenesis to disrupt the helix-helix formation, and their roles in prM-E interaction and SP formation were characterized. Our earlier work has demonstrated that intracellular formation of the prM-E complex of JEV envelope proteins can be detected using baculovirus coexpression of prM and E in trans, and that the His-99 of prM significantly influences the prM-E interaction [18]. This paper reports that the GXXXG motif in the prM-TM1 domain influences the formation of prM-E heterodimers in infected cells. All of the mutants with alanine inserted in the prM-TM1 or prM-TM2 segment inhibited SP formation in culture supernatants, suggesting that the two TM segments of prM may play important roles for JEV particle assembly.
Methods
Cell lines and viruses
Spodoptera frugiperda Sf9 cells were grown at 28°C in TNM-FH medium (Sigma) supplemented with 10% fetal bovine serum (FBS). Hybridoma cells for production of the monoclonal antibodies (mAbs) 5B1 (anti-JEV prM) and E3.3 (anti-JEV E) were grown at 37°C in ISCOVE's modified Dulbecco's medium (Invitrogen) supplemented with 10% FBS. The JEV strain CH2195LA [GenBank:AF221499] [19, 20] and recombinant baculoviruses were produced and propagated in Sf9 cells.
Construction of recombinant baculoviruses
Plasmid pUC18-prME containing nucleotides 414 to 2477 from JEV virus strain CH2195LA was the parent construct used for alanine-insertion mutants, substitution mutants and wild type controls. Alanine-insertion mutants at appropriate positions of prM were constructed using overlapping PCR. Substitution mutants at position 142 and 146 of prM were also constructed using overlapping PCR. All constructs contained BamHI and EcoRI restriction sites for ligation to the pBlueBac4 baculovirus vector (Invitrogen). The constructed recombinant pBlueBac4 plasmids were co-transfected with a linearized baculovirus Bac-N-Blue DNA (Invitrogen) to Sf9 cells treated with Cellfectin (Invitrogen) and incubated for 5-7 days. Recombinant baculoviruses were obtained from the blue-stained plaques formed in the infected Sf9 cells overlaid with agarose medium that contained 5-bromo-4-chloro-3-indoyl-β-D-galactoside (X-gal). Three plaque purifications were performed to obtain the recombinant baculoviruses that were used in this study.
Western blotting
Sf9 cells co-infected with recombinant baculoviruses were harvested and analyzed with 12% SDS-PAGE under reducing condition. Infected cell lysates were electrically transferred onto the nitro-cellulose membranes, blocked by 5% skim milk, and reacted with mAbs 5B1 and E3.3, respectively. As an internal control, the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was parallel used and detected by anti-GAPDH mAb (SignalChem). All of these samples were then reacted with anti-mouse IgG conjugated to peroxidase (KPL) and visualized using ECL (PerkinElmer).
RNA extraction and complementary DNA preparation by RT
The co-infected Sf9 cells at 3 days post infection were harvested and immediately extracted for total RNAs using RNeasy mini kit (Qiagen) according to the protocol of the supplier, and subsequently quantified with a Nano-Drop ND-1000 spectrophotometer (NanoDrop Technologies). RNA purity was estimated by the absorbance ratio A260/A280. The calculated ratios were in the range of 1.8 to 1.9 from the indicated samples and indicate high purity RNAs. One microgram total RNA was reverse transcribed using the M-MLV Reverse Transcriptase (Promega) with 250 ng random hexamer primers (Invitrogen) according to the manufacturer's instructions, without an RNase inhibitor in a final volume of 20 μL. The mixture was incubated for one hour at 37°C.
Quantitative real-time PCR
Quantitative real-time PCR was performed in white LightCycler 480 Multiwell Plate 96 plates (Roche) in a final reaction volume of 10 μL. For detection of prM mRNA transcripts [GenBank:AF221499]: Forward primer 5'- aggaatcctggctacgcttt -3', reverse primer 5'-cgttgttactgccaagcatc -3' and specific probe 5'-ggcggcgg -3'. For detection of E mRNA transcripts [GenBank:AF221499]: Forward primer 5'- gaaggggagcattgacacat -3', reverse primer 5'-gattgttctcccaatcgcttt-3' and specific probe 5'-ttctcctg -3'. For detection of GAPDH mRNA transcripts [GenBank:NM_080369]: Forward primer 5'-aagggaatcctgggctacac-3', reverse primer 5'-aatgggtgtcgctgaagaag-3' and specific probe 5'-ggaggtgg -3'. According to the primer optimization matrix, varying amounts of the forward and reverse primer (Invitrogen) of 100 nmol/L, 200 nmol/L, and 300 nmol/L were mixed in 1× LightCycler 480 Probes Master (Roche) containing 100 nmol/L and 200 nmol/L of the human Universal Probe Library probe (Roche), respectively, and 1.0 μL of cDNA as template (1:20, taken from one appropriate source described above, but consistent throughout a single experiment). The initial denaturation (95°C, 10 minutes) was followed by 45 cycles of 10 seconds at 95°C, 30 seconds at 60°C, and a final cooling step at 40°C for 10 seconds. Each primer concentration combination was analyzed in duplicate for each cDNA source used.
PNGaseF and EndoH glycosidase treatment of glycoprotein
Sf9 cells infected by recombinant baculoviruses were harvested and lysed using a lysis buffer for 3 hours at 4°C. Then, 10X glycoprotein denaturing buffer (New England BioLab) was added to the cell lysate to a 1X concentration and the whole was boiled at 95°C for 5 minutes. Subsequently, 14 μL of denatured sample was mixed with 2 μL 10X G7 buffer, 2 μL 10% NP-40, and 2 μL PNGaseF or EndoH to yield a 20 μL reaction mixture. Finally, the reaction mixtures were left to stand at 37°C for 3 hours to allow deglycosylation.
Confocal laser scanning microscopic analysis for subcellular localization
Infected Sf9 cells that had been grown on 15 mm glass coverslips for 24 hour post infection (hpi) were washed three times in TBS (140 mM NaCl; 25 mM Tris-HCl, pH 6.1) incubated in a medium that contained 10 nM DiIC13(3) (an ER-staining dye, Molecular Probes) for 30 min, and finally fixed with 3.7% paraformaldehyde for 1 h. The cells were then permeabilized for 1 h with TBS containing 0.1% Triton X-100. The expression of JEV proteins in these cells was revealed by staining with the anti-E mAb E3.3 or anti-prM mAb 5B1 followed by goat anti-mouse Alexa Fluor 647 IgG (Molecular Probes). The subcellular localizations in these cells were examined by confocal laser scanning microscopy (Zeiss LSM 500) using a 100x 1.3 oil objective.
Metabolic labeling and sucrose gradient sedimentation analysis
Subconfluent monolayers of Sf9 cells grown in 6-well plates were infected with recombinant baculoviruses at multiplicity of infection (MOI) = 5. At 24 hpi, the TNM-FH medium was removed and replaced with TNM-FH medium that contained 0.01% normal methionine (Met) and 0.02-0.10 mCi/mL of [35S]methionine/cysteine. Baculovirus-infected Sf9 cells were treated with a lysis buffer (50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 2 mM EDTA, 1% Triton X-100 and 1 mM phenylmethylsufonylfluoride), and then pre-cleared using protein A Sepharose (Pharmacia). Each purified cell lysate was layered on a 3-60% (wt/wt) sucrose gradient in gradient buffer (50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 2 mM EDTA, 0.5% Triton X-100, 1 mM phenylmethylsufonylfluoride). The gradients were centrifuged in a Hitachi RPS40ST rotor at 38,000 rpm and 15°C for 22 h. After centrifugation, each fraction was collected, immunoprecipitated using mAb E3.3 (anti-JEV E protein) and incubated for 5 h at 4°C; then protein A Sepharose slurry was added and the system was incubated for another 16 h under the same conditions. The immunoprecipitates were separated by centrifugation at 2500 rpm for 3 min and the pellets thus formed were washed twice with incubation buffer (63 mM Tris-HCl [pH 6.8]). The precipitated material was solubilized by heating (95°C for 5 min) with a reducing-electrophoresis sample buffer (50 mM Tris-HCl pH6.8, 2% SDS, 0.1% bromophenol blue, 5% 2-mercatoethanol, and 10% glycerol) and further analyzed by SDS-PAGE (12%) and fluorography. At least two independent experiments were performed to obtain the reproducible results.
Enzyme-Linked Immunosorbent Assay (ELISA) for quantification of E protein
Samples from each fraction were diluted in coating buffer (0.1 M Na2CO3 [pH 9.6]) and used to coat 96-well microplates (Corning, Costar 9018) by overnight incubation at 4°C. Following incubation, the samples were blocked using 3% bovine serum albumin (BSA) for 2 h at room temperature, and the solid phase was then reacted with mAb E3.3 (anti-E monoclonal antibody; 1:500 dilution) for 2 h at room temperature. The bound antibodies were detected following incubation with the anti-mouse IgG conjugated to peroxidase (KPL; 1:2000 dilution) for 1 h at room temperature. The ELISA products were developed using a chromogen solution containing 2, 2'-azino-di-(3-ethylbenzthiazoline-6-sulfonate; ABTS) and hydrogen peroxide and their absorbance values were measured at 405 nm. The levels of E proteins were calculated from the absorbance values by comparison with a standard curve established with purified domain III, which also reacted with mAb E3.3 at a known protein concentration [18]. The ELISA data were further normalized with the relative total E protein expression level, which was achieved by treating the baculovirus-infected Sf9 cells with 10 μg/ml Brefeldin A (Merck), a fungal macrolide antibiotic which disassembly the golgi compartment and blocks the secretion. The relative total E protein expression level was determined by the intensity of Western blotting bands with software Gel-Pro Analyzer (Media Cybernetics).
Transmission electronic microscopy with immuno-gold labeling
The culture supernatants of the baculovirus-infected Sf9 cells (MOI = 2.5) or JEV-infected Sf9 cells at 5 dpi were centrifuged, precipitated using 10% polyethylene glycol (PEG)- 8,000, and then resuspended in 1 ml TN buffer (10 mM Tris-HCl pH7.5, 100 mM NaCl). The suspensions were subjected to centrifugation in a continuous sucrose gradient (3-60% wt/wt) at 38,000 rpm for 24 h at 4°C (RPS40ST rotor, HITACHI 85P-72 ultracentrifuge). The virus particles were collected from the fractions in 13 tubes with volumes of 0.85 mL for transmission electron microscopy (TEM). A single-droplet negative staining procedure was used for TEM visualization. Sample droplets of 30 μL were absorbed for 10 min onto Formvar carbon-coated copper grids (200 mesh; Agar scientific), washed in distilled water, and then stained with 2% uranyl acetate for 1 min. After drying in air, the negatively-stained samples were labeled with anti-prM mAb 5B1 or anti-E mAb E3.3 for 1 h, and then with anti-mouse IgG conjugated with 5 nm gold nanoparticles for 1 hr. The prepared grids were examined in a HITACHI H-7500 TEM at 100 kV.
Results
Expression of prM proteins with inserted alanine in co-infected Sf9 cells
The prM anchor region of the JEV 2195 strain contains the prM-TM1 (residues prM131-147) and prM-TM2 (residues prM153-167) segments [7] (Fig. 2); these two segments can form helix-helix complexes in lipid bilayers. Since alanine insertion scanning mutagenesis is typically used to identify the residues critical for helix-helix interactions [21, 22], we constructed a series of prM mutants with alanine inserted in the prM-TM1 and prM-TM2 segments including prM139, prM143, prM147, prM157, prM161, and prM165 (Fig. 2). The positions of alanine insertion in the prM-TM1 and prM-TM2 segments were adopted from a previous study on helix-helix interactions in the anchor region of the YFV prM protein [14]. In this study, we investigated the potential role of the TM domains of the JEV prM protein in prM-E heterodimerization as well as in SP formation. The analyses were conducted using a baculovirus expression system with prM and E expressed in trans. The signal peptide of E protein is located at the C-terminal end of prM protein, approximately in the prM-TM2 region. Use of the prM and E coexpression in trans provides the advantage to study the prM TM domains without affecting the signal peptide of E protein for E protein expression, translocation, and the interactions with prM as a heterodimeric complex formation as we reported previously [7].

Positions of the alanine insertion mutations in the two transmembrane regions (TM1 and TM2) of prM protein of JEV CH2195LA strain. A series of mutants was constructed; Bac-prM, Bac-prM A139, Bac-prM A143, Bac-prM A147, Bac-prM A157, Bac-prM A161 and Bac-prM A165. Arrows indicate the positions of alanine insertion.
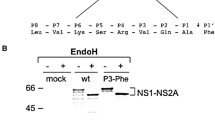
To demonstrate that the prM proteins with inserted alanine did not influence the expression or subcellular localization of the prM-E interaction, Sf9 cells were co-infected with Bac-prM (wild-type and mutants) and Bac-E at MOI = 5. These alanine-inserted mutations in the prM-TM1 and prM-TM2 segments did not affect the total expression of E and prM proteins in the co-infected Sf9 cells (Fig. 3A). The mRNA transcript levels of prM and E of the prM mutants in the co-infected Sf9 cells were similar as measured by using RT-quantitative real time PCR methods (Fig. 3B). Furthermore, the cell lysates of the co-infected Sf9 cells treated with PNGaseF or EndoH enzymes did not show differences in their glycoprotein patterns in SDS-PAGE gels, suggesting these alanine-inserted prM mutants also did not affect the intracellular localization of mutant prM proteins (Fig. 4). The intracellular localization of the alanine-inserted prM proteins was further confirmed using confocal laser scanning microscopy. Sf9 cells that were infected with Bac-prM (wild type) or Bac-prM (alanine-insertion mutants) were doubly stained with a prM-specific MAb and DiC, an ER-specific dye. The doubly stained images obtained by confocal laser scanning microscopy indicated that the intracellular distributions of prM and the prM proteins with inserted alanine almost overlapped with the ER marker (Fig. 5). Thus, none of the prM mutants with inserted alanine influenced the expression or subcellular localization of the prM protein in the co-infected Sf9 cells.

(A) The total expression of prM and E proteins in Sf9 cells co-infected with Bac-prM (wild type and mutants A139, A143, A147, A157, A161, A165) and Bac-E. GAPDH was measured as an internal control; (B) The mRNA transcript levels of prM and E in the co-infected Sf9 cells measured by real-time RT-qPCR. The wt prM/E expression level was taken as 100%.

Treatment of recombinant prM with endoglycosidase PNGaseF and EndoH. The prM mutants with inserted alanine were treated with PNGaseF (P) and EndoH (E) to analyze the presence and composition of their N-linked glycans.

Observation of subcellular localization using confocal laser scanning microscopy. The subcellular location of wild type and mutant prM was visualized by double-staining of the infected Sf9 cells. The ER compartment was stained with 10 nM DiIC13(3), and the prM proteins were stained with mAb 5B1, followed by goat anti-mouse Alexa Fluor 647 IgG. Photographs were taken with appropriate excitation laser wavelengths and merged to reveal the co-localization of prM and ER compartment.
Effects of prM proteins with inserted alanine on prM-E heterodimerization in co-infected Sf9 cells
To investigate the alanine-inserted prM mutants affecting prM-E heterodmerization, Sf9 cells co-infected with Bac-prM (wild type or alanine-insertion mutants) and Bac-E were radiolabelled with [35S]methionine-cysteine medium for 24 h, purified using sucrose gradient sedimentation, and immunoprecipitated by an E-specific mAb. The results in SDS-PAGE gels showed that the two reactive bands (corresponding to prM of 21 kDa and E of 56 kDa) were present in sucrose gradient fractions 5 and 6 of the co-infected Sf9 cells with Bac-prM (wild type) (Fig. 6A), Bac-prM A139 (Fig. 6B), Bac-prM A143 (Fig. 6C), Bac-prM A147 (Fig. 6D), Bac-prM A157 (Fig. 6E), Bac-prM A161 (Fig. 6F) and Bac-prM A165 (Fig. 6G). However, the intensity of binding to the prM-E complex by some prM mutants with inserted alanine differed from the wild type prM. For instance, a smaller amount of E protein was precipitated when coexpressed with prM A143, prM A147, or prM A165, compared to those of E protein when coexpressed with the WT or other mutant prM proteins. To characterize these mutations in the prM-TM1 and prM-TM2 that affected the association between E and prM, the percentage of heterodimerization of prM to E was taken as the prM/E ratio. The prM/E ratio was measured for the stable prM-E complex in each sucrose gradient fractionation to minimize the differential precipitation efficiency in each sample. Also a side-by-side comparison of the total levels of E and prM prior to precipitation was also conducted where we did not find differences of prM and E expression for the WT and mutants (data not shown). The prM/E ratios in the sucrose gradient fractions 4 to 6 were 100% (Bac-prM), 86% (Bac-prM A139), 51% (Bac-prM A143), 61% (Bac-prM A147), 77% (Bac-prM A157), 75% (Bac-prM A161), and 87% (Bac-prM A165) (Fig. 7). The Bac-prM A143 mutant most reduced the formation of prM-E heterodimers in infected cells.

Sucrose gradient sedimentation analysis of alanine-insertion mutagenesis of the anchor region of prM protein. Sf9 cells were coinfected with Bac-E and Bac-prM mutants. Cell lysates labeled with [35S] were applied for centrifugation in a 3 to 60% (wt/wt) sucrose gradient. Each fraction was immunoprecipitated with monoclonal antibody E3.3. The immune complexes were analyzed by reducing SDS-PAGE and fluorography. (A) Sf9 cells coinfected with Bac-prM and Bac-E; (B) Sf9 cells coinfected with prM A139 and Bac-E; (C) Sf9 cells coinfected with prM A143 and Bac-E; (D) Sf9 cells coinfected with prM A147 and Bac-E; (E) Sf9 cells coinfected with prM A157 and Bac-E; (E) Sf9 cells coinfected with prM A157 and Bac-E; (F) Sf9 cells coinfected with prM A161 and Bac-E; (G) Sf9 cells coinfected with prM A165 and Bac-E. The data presented in this figure are three independent experiments.

prM-E heterodimerization of prM mutants with inserted alanine as a percentage of the binding intensity of wild type prM. Each prM to E ratio was calculated. The wild type prM/E ratio was taken as 100% in evaluating the mutant prM-E binding affinity. Each data represents the average of the experiments for a single prM mutant and the error is standard deviation. The positions of the alanine insertions in the TM region of prM are indicated.
Effects of prM proteins with inserted alanine on SP formation in co-infected Sf9 cells
To further evaluate the SP formation affected by the alanine-inserted prM mutants, culture supernatants were collected from Sf9 cells that were co-infected with Bac-prM (wild type or alanine-insertion mutants) and Bac-E, purified using sucrose gradient sedimentation, and the E protein concentration in each sucrose fraction was measured by ELISA. The results showed that much less SP was formed in Sf9 cells that were infected with any of the prM mutants with inserted alanine than wild type Bac-prM (Fig. 8A-G). The release of SP from Sf9 cells that were co-infected with the wild-type Bac-prM (Fig. 8A) was verified by TEM using immuno-gold labeling with the anti-E mAb E3.3 (Fig. 8H) and the anti-prM mAb 5B1 (Fig. 8I). Quantification of the SP release from the co-infected Sf9 cells was determined by calculating the underneath area of the E protein concentrations. The results were further normalized to the total E protein expression of the same co-infected cells treated with brefeldin A to prevent SP release (Fig. 8J). Thus, the percentage of SP release in the co-infected cells was calculated from the underneath area of sucrose gradient fractions of culture supernatants of the co-infected Sf9 cells and the value was normalized to the total E protein contents measured in brefeldin A-treated Sf9 cells. The results showed the percentage of SP release dropped from 100% (Bac-prM) to 27% (Bac-prM A139), 22% (Bac-prM A143), 26% (Bac-prM A147), 29% (Bac-prM A157), 34% (Bac-prM A161), and 39% (Bac-prM A165) (Fig. 9). All the prM mutants with inserted alanine resulted in significant impairment for the formation of SP in baculovirus-infected Sf9 cells, suggesting that the two TM domains of prM may play important roles for the formation and release of SP.

Alanine-insertion mutagenesis of the TM regions of prM protein: effect on SP formation. Recombinant SPs secreted from Sf9 cells co-infected with with Bac-E and (A) Bac-prM; (B) Bac-prM A139; (C) Bac-prM A143; (D) Bac-prM A147; (E) Bac-prM A157; (F) Bac-prM A161, and (G) Bac-prM A165 were quantified by ELISA. The size and shape of the secreted recombinant SP were observed by TEM using immuno-gold labeling with (H) mAb E3.3 and (I) and mAb 5B1. (J) The total E protein in Sf9 cells was determined in the presence of Brefeldin A, a fungal a fungal macrolide antibiotic which blocks E protein secretion. GAPDH was taken as an internal loading control.

Quantification of the SP release was determined by calculating the underneath area of the E protein concentrations from fractions 7 to 10 in Fig. 7 A-G. The underneath areas were normalized by the corresponding total amount of E protein in Fig 7J and the percentage of SP release represented the relative value of each prM mutant (Fig. 7B-G) compared to the wild type (Fig. 7A)
Glycine substitution mutagenesis in the prM-TM1 segment
To further characterize the prM A143 mutant that influenced the prM-E interaction, the prM-TM1 sequences were analyzed and a GXXXG motif was identified at residues 142-146 (G142WMLG146S147) (see Fig. 1). The GXXXG motif contains amino acids that are hydrophobic and commonly present in many TM proteins [23]. Therefore the glycine at residues 142 and/or 146 were replaced with alanine, leucine or valine, respectively, to demonstrate the specificity of the GXXXG motif (Bac-prM G142A, Bac-prM G142L, Bac-prM G142V, Bac-prM G146A, Bac-prM G146L, Bac-prM G146V) (Fig. 10). Similarly, these GXXXG mutants did not affect the total expression of E and prM proteins in the co-infected Sf9 cells (Fig. 11A). The mRNA transcript levels of prM and E of these GXXXG mutants in the co-infected Sf9 cells were around the same as measured by using RT-quantitative real time PCR methods (Fig. 11B). However, the percentage of heterodimerization of prM to E for characterization of the mutations that affected the association between E and prM revealed the glycine substitutes at 142 and 146 by alanine, leucine or valine reduced by approximately 50%, except the mutant G142A giving a 25% loss (Fig. 12). Therefore, both glycine residues in the GXXXG-motif of the prM-TM1 segment are equally important to the formation of a stable prM-E complex.

Characterization of the GXXXG motif of the TM1 region of the prM protein by glycine-substitution mutagenesis. Glycine residues at 142 and 146 were substituted by alanine, leucine, or valine to investigate the relationship between the GXXXG motif and the prM-E binding affinity.

(A) The total expression of prM and E proteins in Sf9 cells co-infected with Bac-prM (wild type and mutants G142A, G142L, G142V, G146A, G146L, G146V) and Bac-E. GAPDH was measured as an internal control; (B) The mRNA transcript levels of prM and E in the co-infected Sf9 cells measured by real-time RT-qPCR. The wt prM/E expression level was taken as 100%.

The percentage of the binding intensity of wild type prM. Each prM to E ratio was calculated where wild type prM/E ratio was taken as 100% in evaluating the glycine-substituted mutant prM-E binding affinity. Each data represents the average of the experiments for a single prM mutant and the error is standard deviation.
Sequence alignment analysis of TM regions of prM proteins of different flavivirus groups
The amino acid sequence alignments of the prM TM regions of 5 groups of flaviviruses, including JEV sero-complex viruses (JEV, KUNV, MVEV, SLEV, WNV), four-serotype dengue viruses (DENV-1, DENV-2, DENV-3, DENV-4), TBEV, and YFV were analyzed to determine whether the GXXXG motif was conserved. These sequences were aligned and analyzed using the Showalign program from Jemboss on the Jemboss server http://bioinfo.life.nthu.edu.tw/bioinfo.htm. Additional file 1 summarizes the results, which show that the amino acid sequences of the prM-TM1 are nearly identical except amino acid 140 in three strains of JEV but vary among other JEV serocomplex viruses, DENV-1, DENV-2, DENV-3, DENV-4, TBEV, and YFV. However, the GXXXG motif is present in all JEV serocomplex viruses.
Discussion
The flavivirus assembly process is driven mainly by the interactions between two envelope proteins prM and E. Co-expression of these two proteins in cultured cells can yield recombinant SPs. In this work, alanine insertion and site-directed mutagenesis were employed to study the influence of the TM anchor region of prM protein on the formation of prM-E heterodimers and SP production. The results show that the GXXXG motif present in the prM-TM1 domain is the element that affects the formation of prM-E heterodimers. Both TM segments of the prM protein are crucial for SP formation and release.
Alanine-insertion mutagenesis was employed to disrupt the TM helical structures in the TM anchor regions of prM protein (prM-TM1 and prM-TM2) in experiments. Six prM mutants with inserted alanine (prM139A, prM143A, prM147A, prM157A, prM161A, prM165A) were used; the insertion sites were chosen according to a study of alanine insertion mutagenesis of the TM proteins of YFV [14]. This insertion mutagenesis has been applied for identifying critical segments of TM region involved in helix-helix interactions [21, 22, 24]. These mutated proteins may also have subtle alternations in protein stability, expression, folding, and translocation. Baculovirus insect cell expression has been widely used to express complex proteins particularly requiring post-translational modifications [25]. In this study we employed a baculovirus coexpression system with prM and E expressed in trans where alanine insertion and substitution mutagenesis of prM-TM1 and prM-TM2 fragments were conducted to study the interactions of prM and E proteins in co-infected Sf9 cells. Our results show that a series of alaine inserted prM mutants neither affected the mRNA and protein expression levels of prM nor the subcellular localization of the prM protein in Sf9 cells. However, whether these prM mutants may alter anchorage of mutant prM proteins into membranes or cause a conformational change of the extracellular domain of prM are still required for further confirmation. Among these alanine-inserted prM mutants, the prM-143A mutant within the GXXXG motif in the prM-TM1 segment markedly reduced the formation of the prM-E heterodimers by up to 50%. This result is consistent with our earlier study using C-terminal truncation, which showed a reduction of the formation of the prM-E complex by approximately 40% for the TM1 of prM protein, as compared to the complete abolishment of the interactions by the prM His-99 mutation [7].
The impairment of SP formation but a modest disruption of prM-E heterodimerization by all prM mutants with inserted alanine was observed in our study. For example, the mutant prM165 exhibited an interaction profile nearly identical to wild type in the prM-E heterodimerization (Fig. 7), yet had a significant effect on SP release (Fig. 9). Conversely, mutants with a more "dramatic" reduction in the interaction between prM and E displayed similar reductions in the release of SP. Mutations in the two prM TM segments may increase the heterogeneity of SP, as evidenced by the broader peak of E protein in these mutants (Fig. 8B-G). The broaden fractions of SPs from prM mutants may come from E protein itself to form an oligomer and release into culture medium through envelopment by vesicles. However, the total E protein contents measured in the same infected cells treated with Brefeldin A, a fungal macrolide antibiotic to disassembly the Golgi compartment and block the SP secretion, had similar levels among the wild type and prM mutants (Fig. 8J). These results indicate that the assembly, budding and secretion of SP are not only determined by the interactions among the prM and E enveloped proteins but also several post-assembly events such as budding and secretion. The newly synthesized prM and E enveloped proteins can associate to form homo- or hetero-dimeric/oligomeric complexes which can not be incorporated into an icosahedral lattice for budding into the ER lumen. The final stage of SP release in the infected cells still requires a rapid transport along the compartments of the secretory pathway. It was reported that some of the prM mutations located outside the TM region in DENV and TBE hardly affected the prM-E heterodimerization but greatly inhibited the SP secretion [26, 27]. The SP release through envelop budding is driven by multimerization of the envelope proteins prM and E to create an icosahedral lattice which is composed by 60 asymmetric trimers of prM-E heterodimers [28]. Then the prM-E heterodimers, transient contacts between prM TM domains occurred during the budding process, dissociate due to the major rearrangement into the E:E dimers during the maturation process of the particles after cleavage of prM by a furin in the trans Golgi network [29]. Further investigation of SP budding and secretion particularly in late-step secretory pathways are required to understand the detailed mechanisms of flavivirus assembly, exit, and maturation.
The GXXXG motif in the prM-TM1 segment of the JEV 2195LA strain was further studied using site-specific mutagenesis to demonstrate its specificity for helix-helix interaction in the TM anchor regions. Replacing the glycine residues in the GXXXG motif with three other small amino acids (alanine, leucine and valine), supporting extensive interhelical van der Waals interactions, indicated that the two glycine residues were equally important to the formation of prM-E heterodimers (Fig. 12). This site-specific mutagenesis result is consistent with the previously reported effect of the GXXXG motif on the formation of the E1:E2 heterdimer in hepatitis C virus and Semliki Forest virus [24, 30, 31]. However, virus assembly depends on a replacement by leucine of the conserved glycine residues in the GXXXG motif in Semliki Forest virus [32]. Thus, the importance of the conserved glycine residues in the TM anchor regions may vary with the variations in amino acid sequence among virus species.
The topology of the TM regions of enveloped proteins of enveloped viruses has been recently revealed for the enveloped protein heterodimerization and the SP formation, but with inconclusive results [14, 24, 31, 33, 34]. In an earlier study on YFV, the alanine insertions did not affect prM-E heterodimerization but greatly impaired the SP release of YFV [13]. Alanine insertion within the center of the TMDs of E1 or E2 or in the N-terminal part of the TMD of E1 dramatically affected for 60-90% reduction of hepatitis C virus envelope glycoproteins [24]. Even the E1-E2 heterodimerization was not affected, some mutants still showed reduced HCVpp infectivity [31]. The differences among these viruses may be due to their similar topology of TM region of envelope proteins. The TM regions of prM and E proteins of flaviviruses are potentially longer than their counterpart in HCV [13, 33]. Further sequence alignment analysis of the anchor regions of prM proteins of various flaviviruses, shown in Additional file 1, indicates that the LGXXLG motif is present in the TM1 region of JEV and MVE; the IGXXLG motif in KUNV, SLEV and WNV; the LAXXIG motif in DENV-1 to DENV-4; and the IAXXVG motif in the YFV. As revealed by a statistical analysis of the anchor regions of TM proteins, most of the amino acid patterns of small residues (Gly, Ala and Ser) at i and i+4 are associated with large aliphatic residues (Ile, Val and Leu) at neighboring positions (i.e. i+/-1 and i+/-2) in the TM anchor regions [35]. Sequence alignment analysis of the anchor regions of prM proteins of various flaviviruses indicates that the prM-TM1 domain of YFV contains AYLVG residues rather than the GXXXG motif. Furthermore, the connecting segment between prM-TM1 and prM-TM2 domains of JEV prM proteins has four hydrophilic residues and one charged residue (NNGQR). Replacing the arginine (R) residue with alanine in the prM connected segment did not affect the prM-E heterodimerization and SP formation (data not shown). The data on SP inhibition by prM insertion mutants in the two TM segments but not the connecting segment suggest that the prM-TM1 and prM-TM2 domains may play importation roles in the formation and release of the JEV particle assembly.
Conclusion
In this study, we characterized the involvement of JEV prM TM1 and TM2 region in the prM-E heterodimerization and the assembly of subviral particles. Furthermore, we identified a conserved GXXXG motif in the prM TM1 region of the JEV serocomplex viruses, which may play an important role in the early biosynthesis process of the virus.
References
Burke D: Flaviviruses. Fields virology. Edited by: Knipe DM, Howley P. 2001, 1043-1125. 4
Linderbach B: Flaviviridae: the viruses and their replication. Fields virology, Lippincott-Raven, Philadelphia, Pa. Edited by: Knipe DM, Howley P. 2001, 991-1041. 4
Russel P: The Togaviruses. 1980, Academic Press, New York, NY, 503-529.
Allison SL, Stadler K, Mandl CW, Kunz C, Heinz FX: Synthesis and secretion of recombinant tick-borne encephalitis virus protein E in soluble and particulate form. J Virol. 1995, 69: 5816-5820.
Konishi E, Fujii A: Dengue type 2 virus subviral extracellular particles produced by a stably transfected mammalian cell line and their evaluation for a subunit vaccine. Vaccine. 2002, 20: 1058-1067. 10.1016/S0264-410X(01)00446-7.
Konishi E, Fujii A, Mason PW: Generation and characterization of a mammalian cell line continuously expressing Japanese encephalitis virus subviral particles. J Virol. 2001, 75: 2204-2212. 10.1128/JVI.75.5.2204-2212.2001.
Lin YJ, Wu SC: Histidine at residue 99 and the transmembrane region of the precursor membrane prM protein are important for the prM-E heterodimeric complex formation of Japanese encephalitis virus. J Virol. 2005, 79: 8535-8544. 10.1128/JVI.79.13.8535-8544.2005.
Kroeger MA, McMinn PC: Murray Valley encephalitis virus recombinant subviral particles protect mice from lethal challenge with virulent wild-type virus. Arch Virol. 2002, 147: 1155-1172. 10.1007/s00705-002-0809-3.
Purdy DE, Noga AJ, Chang GJ: Noninfectious recombinant antigen for detection of St. Louis encephalitis virus-specific antibodies in serum by enzyme-linked immunosorbent assay. J Clin Microbiol. 2004, 42: 4709-4717. 10.1128/JCM.42.10.4709-4717.2004.
Davis BS, Chang GJ, Cropp B, Roehrig JT, Martin DA, Mitchell CJ, Bowen R, Bunning ML: West Nile virus recombinant DNA vaccine protects mouse and horse from virus challenge and expresses in vitro a noninfectious recombinant antigen that can be used in enzyme-linked immunosorbent assays. J Virol. 2001, 75: 4040-4047. 10.1128/JVI.75.9.4040-4047.2001.
Mukhopadhyay S, Kuhn RJ, Rossmann MG: A structural perspective of the flavivirus life cycle. Nat Rev Microbiol. 2005, 3: 13-22. 10.1038/nrmicro1067.
Zhang W, Chipman PR, Corver J, Johnson PR, Zhang Y, Mukhopadhyay S, Baker TS, Strauss JH, Rossmann MG, Kuhn RJ: Visualization of membrane protein domains by cryo-electron microscopy of dengue virus. Nat Struct Biol. 2003, 10: 907-912. 10.1038/nsb990.
Allison SL, Stiasny K, Stadler K, Mandl CW, Heinz FX: Mapping of functional elements in the stem-anchor region of tick-borne encephalitis virus envelope protein E. J Virol. 1999, 73: 5605-5612.
Op De Beeck A, Molenkamp R, Caron M, Younes Ben A, Bredenbeek P, Dubuisson J: Role of the transmembrane domains of prM and E proteins in the formation of yellow fever virus envelope. J Virol. 2003, 77: 813-820. 10.1128/JVI.77.2.813-820.2003.
Chang GJ, Hunt AR, Holmes DA, Springfield T, Chiueh TS, Roehrig JT, Gubler DJ: Enhancing biosynthesis and secretion of premembrane and envelope proteins by the chimeric plasmid of dengue virus type 2 and Japanese encephalitis virus. Virology. 2003, 306: 170-180. 10.1016/S0042-6822(02)00028-4.
Purdy DE, Chang GJ: Secretion of noninfectious dengue virus-like particles and identification of amino acids in the stem region involved in intracellular retention of envelope protein. Virology. 2005, 333: 239-250. 10.1016/j.virol.2004.12.036.
Orlinger KK, Hoenninger VM, Kofler RM, Mandl CW: Construction and mutagenesis of an artificial bicistronic tick-borne encephalitis virus genome reveals an essential function of the second transmembrane region of protein e in flavivirus assembly. J Virol. 2006, 80: 12197-12208. 10.1128/JVI.01540-06.
Lin CW, Wu SC: A functional epitope determinant on domain III of the Japanese encephalitis virus envelope protein interacted with neutralizing-antibody combining sites. J Virol. 2003, 77: 2600-2606. 10.1128/JVI.77.4.2600-2606.2003.
Wu SC, Lian WC, Hsu LC, Liau MY: Japanese encephalitis virus antigenic variants with characteristic differences in neutralization resistance and mouse virulence. Virus Res. 1997, 51: 173-181. 10.1016/S0168-1702(97)00098-1.
Wu SC, Lee SC: Complete nucleotide sequence and cell-line multiplication pattern of the attenuated variant CH2195LA of Japanese encephalitis virus. Virus Res. 2001, 73: 91-102. 10.1016/S0168-1702(00)00235-5.
Braun P, Persson B, Kaback HR, von Heijne G: Alanine insertion scanning mutagenesis of lactose permease transmembrane helices. J Biol Chem. 1997, 272: 29566-29571. 10.1074/jbc.272.47.29566.
Mingarro I, Whitley P, Lemmon MA, von Heijne G: Ala-insertion scanning mutagenesis of the glycophorin A transmembrane helix: a rapid way to map helix-helix interactions in integral membrane proteins. Protein Sci. 1996, 5: 1339-1341. 10.1002/pro.5560050712.
Ulmschneider MB, Sansom MS: Amino acid distributions in integral membrane protein structures. Biochim Biophys Acta. 2001, 1512: 1-14. 10.1016/S0005-2736(01)00299-1.
Op De Beeck A, Montserret R, Duvet S, Cocquerel L, Cacan R, Barberot B, Le Maire M, Penin F, Dubuisson J: The transmembrane domains of hepatitis C virus envelope glycoproteins E1 and E2 play a major role in heterodimerization. J Biol Chem. 2000, 275: 31428-31437. 10.1074/jbc.M003003200.
Altmann F, Staudacher E, Wilson IB, Marz L: Insect cells as hosts for the expression of recombinant glycoproteins. Glycoconj J. 1999, 16: 109-123. 10.1023/A:1026488408951.
Pryor MJ, Azzola L, Wright PJ, Davidson AD: Histidine 39 in the dengue virus type 2 M protein has an important role in virus assembly. J Gen Virol. 2004, 85: 3627-3636. 10.1099/vir.0.80283-0.
Yoshii K, Konno A, Goto A, Nio J, Obara M, Ueki T, Hayasaka D, Mizutani T, Kariwa H, Takashima I: Single point mutation in tick-borne encephalitis virus prM protein induces a reduction of virus particle secretion. J Gen Virol. 2004, 85: 3049-3058. 10.1099/vir.0.80169-0.
Ferlenghi I, Clarke M, Ruttan T, Allison SL, Schalich J, Heinz FX, Harrison SC, Rey FA, Fuller SD: Molecular organization of a recombinant subviral particle from tick-borne encephalitis virus. Mol Cell. 2001, 7: 593-602. 10.1016/S1097-2765(01)00206-4.
Stadler K, Allison SL, Schalich J, Heinz FX: Proteolytic activation of tick-borne encephalitis virus by furin. J Virol. 1997, 71: 8475-8481.
Sjoberg M, Garoff H: Interactions between the transmembrane segments of the alphavirus E1 and E2 proteins play a role in virus budding and fusion. J Virol. 2003, 77: 3441-3450. 10.1128/JVI.77.6.3441-3450.2003.
Ciczora Y, Callens N, Penin F, Pecheur EI, Dubuisson J: Transmembrane domains of hepatitis C virus envelope glycoproteins: residues involved in E1E2 heterodimerization and involvement of these domains in virus entry. J Virol. 2007, 81: 2372-2381. 10.1128/JVI.02198-06.
Liao M, Kielian M: The conserved glycine residues in the transmembrane domain of the Semliki Forest virus fusion protein are not required for assembly and fusion. Virology. 2005, 332: 430-437. 10.1016/j.virol.2004.11.035.
Cocquerel L, Wychowski C, Minner F, Penin F, Dubuisson J: Charged residues in the transmembrane domains of hepatitis C virus glycoproteins play a major role in the processing, subcellular localization, and assembly of these envelope proteins. J Virol. 2000, 74: 3623-3633. 10.1128/JVI.74.8.3623-3633.2000.
Ronecker S, Zimmer G, Herrler G, Greiser-Wilke I, Grummer B: Formation of bovine viral diarrhea virus E1-E2 heterodimers is essential for virus entry and depends on charged residues in the transmembrane domains. J Gen Virol. 2008, 89: 2114-2121. 10.1099/vir.0.2008/001792-0.
Senes A, Gerstein M, Engelman DM: Statistical analysis of amino acid patterns in transmembrane helices: the GxxxG motif occurs frequently and in association with beta-branched residues at neighboring positions. J Mol Biol. 2000, 296: 921-936. 10.1006/jmbi.1999.3488.
Acknowledgements
This research work was supported by the National Health Research Institutes (NHRI-EX97-9534SI) and the National Science Council (NSC-95-2311-B-007-024-MY3), Taiwan.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
YJL performed the immunoprecipitation and TEM. JGP quantified the mRNA/protein expression level and analyzed the experimental data. SCW designed the experiments and wrote the manuscript. All authors read and approved the final manuscript.
Ying-Ju Lin, Jia-Guan Peng contributed equally to this work.
Electronic supplementary material
12929_2010_153_MOESM1_ESM.DOCX
Additional file 1: Amino acid sequence alignment analysis of TM regions of prM protein (prM130-167) from genus Flavivirus. The numbers of amino acid sequences of 22 flavivirus strains (NCBI and EMBL accession numbers listed) are given (the consensus amino acids symbolized by dashes). Gly142 and Gly146 of the G XXXG motif are marked with a black box. (DOCX 32 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Lin, YJ., Peng, JG. & Wu, SC. Characterization of the GXXXG motif in the first transmembrane segment of Japanese encephalitis virus precursor membrane (prM) protein. J Biomed Sci 17, 39 (2010). https://doi.org/10.1186/1423-0127-17-39
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1423-0127-17-39