
Abstract
Background
We developed and clinically test a new method to manufacture mature dendritic cells for autologous cell therapy of solid and hematological malignancies.
Methods
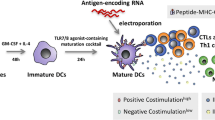
Peripheral blood monocytes are matured into dendritic cells with GM-CSF/IL-4 mixture, and then activated through IL1b, TNFa, IFNa-2A, and Poly(I:C), then viably frozen until use. DC are injected intra-dermally for a total of 6 injections.
Results
High-grade mDC with high pre- and post- freezing yields could be generated. These DC spontaneously produce IL12p70 and they can be further stimulated via CD40. In a patient with advanced pancreatic cancer treated with our mDC formulation, we observed no toxicity but remarkable immune response and objective response in terms of tumor shrinking.
Conclusion
We describe a new method to manufacture GMP-grade dendritic cells for autologous therapeutic cancer vaccines. We also show proof-of-principle efficacy in a patient with advanced pancreatic cancer.
Trial registration
Similar content being viewed by others


Background
We describe a novel, clinical-grade, fully matured monocyte-derived dendritic cells (DC) pulsed with a cocktail of immunogenic tumor peptides, able to induce specific T-cell responses in vitro and in vivo.
DC are the most powerful regulators of immunity: they are essential to initiate and control T-cell activation towards specific antigens. Cancer immunotherapy based on the adoptive transfer of DCs benefit from the ability of these cells to orchestrate cytotoxic lymphocytes and natural killer cells response against tumors [1]. Phase 3 trials with DC have been conducted mostly in patients with melanoma, prostate cancer, glioma, and renal cell cancer [2], although they are applicable to patients suffering from any other malignancy. DC immunotherapy has been shown to be safe in several studies [3,4,5,6,7,8,9,10]. Along with such an optimal safety, which is superior to that of chemotherapies and of many other immunotherapy options, DC can trigger tumor antigen-specific cytotoxic lymphocytes that are able to identify and eliminate cancer cells [11]. The majority of the clinical studies originated from prostate cancer and renal cell carcinoma, which showed that DC adoptive transfer elicited T-cell tumor responses in more than 50% of subjects (and up to 70%) [10]. Despite their safety and demonstrated immunogenicity, DC therapies have also shown poor objective clinical response [12]. A comprehensive meta-analysis on DC therapy for melanoma, prostate cancer, malignant glioma, and renal cell carcinoma demonstrated that the clinical benefit (complete or partial response), afforded by DC-based therapies was statistically significant but small (7–10%) [2]. Limited objective response is generally accompanied by limited prolongation of overall survival, as shown by the phase 3 trial of Sipuleucel-T which nevertheless was deemed sufficient by the FDA for approval in 2010. Median overall survival was prolonged by about 4 months, with only about 5% of the patients showing objective response [13].
It is evident that DC therapies have a great potential for the treatment of cancer, but there is still substantial need to improve their efficacy. The main areas for improvement include DC activation and their Cytotoxic T Lymphocyte (CTL) activation potential. The majority of clinical trials have been conducted with “first-generation” DC, mainly DC-enriched vaccines, such as Sipuleucel-T, where immature, interleukin-4 (IL4) monocyte-derived DC were used [1]. While clinical benefit has been demonstrated with such immature DC, results from the major prostate cancer, melanoma, and glioma trails indicate that mature DC are superior in terms of clinical outcome [10, 14, 15]. Finally, the ability of DC to induce the CTL response is a relevant factor influencing clinical activity [2, 16,17,18]. These are directed against antigens used to load the immature or mature DC in vitro. The effectiveness of DC is therefore influenced by the immunogenicity of the selected antigens, while its safety depends on their expression pattern, which ideally should be restricted to the tumor. We have evaluated a set of antigens belonging to the cancer/testis antigens (CTA) family in solid and hematologic malignancies. Basing on previous studies from our group and others, we have selected a panel of 5 antigens, namely SP17 [19,20,21,22,23,24,25,26,27,28,29,30], AKAP4 [31,32,33], PTTG1 [29, 34], Ropporin-1 [35, 36], and Span-XB [37, 38] as targets for our vaccine. We have identified Tumor-Associated Peptide Antigens (TAPAs) within the structure of the above-mentioned CTAs, predicted to have the highest binding affinity to HLA-A and HLA-B molecules, using the proprietary artificial neural network Diamond™. Positions and sequences of the TAPAs are as follows: SP17 (103–111) ILDSSEEDK, AKAP4 (150–159) YADQVNIDY, PTTG1 (70–79) ATEKSVKTK, Ropporin-1 (113–122) FTEEIEWLK, Span-Xb (86–94) DQMEEEEFI.
With the goal of developing “next generation” DC vaccine, to improve the current “state of the art” DC formulations, we developed BSK01™, an intra-dermal, autologous DC vaccine. We report in vitro, ex vivo, and preliminary results of a phase I clinical study, demonstrating the feasibility of generating a next-generation DC vaccine, its safety, immunogenicity and clinical response in a patient with refractory, metastatic pancreatic cancer.
Methods
Isolation of peripheral blood mononuclear cells PBMCs
Peripheral blood from 3 healthy donors (obtained from the Gulf Coast Regional Blood Center, Houston, TX, USA) was subjected to Ficoll-gradient purification method. PBMCs were counted by the trypan-blue exclusion method, and suspended in CellGro® DC Medium (CellGenix, Newington, NH, USA). 7 × 107 PBMCs were seeded in a T150 flask in a total volume of 35 mL DC-medium.
Generation of immature DC
To enrich the PBMCs cultures in monocytes, after 2-h incubation at 37 °C and 5% CO2, non-adherent cells were removed from the flasks, and adherent cells were cultured in DC-medium supplemented with 800 U/mL GM-CSF and 1000 U/mL IL-4 (both from CellGenix). On days 3 and 6, half culture volume was centrifuged, discarded, and the cell pellet was suspended in fresh DC-medium containing 1600 U/mL GM-CSF and 2000 U/mL IL-4.
iDC phagocytic ability
The endocytosis assay was performed as previously described [39]. Fifty μg/mL of the endocytic tracer (Dextran 10,000 m.w. conjugated with Alexa Fluor 488, Al488-dextran) were added. Endocytosis of the tracer was halted after 30 min by rapid cooling of the cells on ice. The fluorescence intensity of the cells was analyzed by flow cytometry on a FC500 (Beckman-Coulter). Incubation of cells with the endocytic tracer on ice was used as a background control.
Maturation and pulsing
iDC were adjusted at 2 × 106 cells/mL in DC-medium and matured with 800 U/mL GM-CSF, 500 U/mL IL1β, 1000 U/mL TNFa (all from GellGenix), 1000 U/mL IFNα-2A (Humanzyme, Chicago, IL, USA), and 20 μg/mL HMW poly I:C (Invivogen, San Diego, CA, USA). After a 16-h incubation, mDC were pulsed with 10 μg/mL TAPAs: SP17, AKAP4, Ropporin, PTTG1, Span-Xb, or the negative control, Human Actin PepMix™ (Theracode JPT Inc., Acton, MA, USA). After 3 h at 37 °C and 5% CO2, cells were washed with fresh DC-medium, harvested and counted.
Phenotypic characterization of mature DC
mDC were washed with ice-cold DPBS once and suspended in Stain Buffer BSA (BD Pharmingen™, San Jose, CA, USA) at the density of 106/mL. mDC were incubated on ice in the dark with the manufacturer’s recommended amounts of the appropriate specific antibodies or isotype-matched negative controls. FSC/SSC ratio was used to gate the DC population as previously described [40]. Additional file 1: Figure S1, Additional file 2: Figure S2 and Additional file 3: Figure S3 show the purity of the mDC population. The antibodies used were the following: anti-CD1a PE (R&D Systems FAB7076P), anti-CD14 FITC (R&D Systems FAB3832F), anti-CD80 PE (R&D Systems AB140P), anti-CD83 PE (R&D Systems FAB1774P), anti-CD86 FITC (R&D Systems FAB141F), anti-CD58 PE (BD 555921), anti-HLA-DR FITC (R&D Systems FAB4869F). Dead cells were excluded through FSC/SSC gating.
CD40 activation and IL12p70 E.L.I.S.a.
mDC were seeded in 0.5 mL DC-medium and stimulated for 48 h with 0.5 μg/mL Mega-CD40L (Enzo Life Sciences, NY, USA) of vehicle as previously described [41]. Supernatants were collected and IL12p70 concentration was measured using the Human IL-12 p70 Quantikine ELISA Kit (R&D Systems).
Sterility tests
mDC were confirmed to be sterile and endotoxin-free with the following methods. Microbiology cultures were setup to grow and identify bacteria, yeast or mold in the specimen. Enriched and selective media were used to grown bacteria, yeast or mold. The sample was inoculated on blood agar (MacConkey), and Saboraud Dextrose (two plates were incubated at 36 °C and 25 °C). Final results on all plates for bacterial, yeast and mold growth were evaluated after a 7-day incubation period. No growth was observed on any of the plates following a > =7-day incubation period. It was determined that the test article contained less than 10 CFU/mL.
A polymerase chain reaction (PCR) for Mycoplasma was performed using generic primers designed to amplify a conserved region in the 16S of Mycoplasma species. This primer set detects 20 different species of the above agents. 1.5 μg of DNA were amplified in a 26 μL PCR reaction for 35 cycles. Results were negative for Mycoplasma contamination.
mDC supernatants were tested using the Pierce™ LAL Chromogenic Endotoxin Quantitation Kit (Thermo Fisher Scientific, Waltham, MA, USA). Results showed less than 1 EU/mL.
Cryopreserving and thawing mDC
mDC were cryopreserved by centrifuging at 400×g for 10 min at room temperature and suspending the cell pellet in ice-cold, male AB normal human serum (NHS, Sigma-Aldrich) at the density of 2 × 106 cells/mL. One volume of ice-cold NHS supplemented with 20% W/W DMSO was dropwise added and gently mixed. The cell suspension was then loaded into CellSeal Closed-System Cryogenic Vials (1 mL/vial, Cook Regentec Indianapolis, IN). Vials were transferred in isopropanol bath-cryoboxes and allowed to cool at − 80 °C overnight. The following day, vials were transferred in liquid nitrogen for extended storage. mDC were thawed rapidly (~ 5 min) in a 37 °C thermostatic dry bath, then diluted in 10 volumes of DC-medium and counted for viability evaluation.
Clinical study
The safety, immunogenicity, and clinical activity of BSK01™ were evaluated in a Phase I clinical trial (ClinicalTrials.gov Identifier: NCT02705703). Patients were enrolled upon signing informed consent per IRB specifications (Salus central IRB, https://www.salusirb.com). BSK01™ was manufactured as described above and, after passing quality control tests (DC maturation, viability, and bioburden), it was shipped to clinical facilities in liquid nitrogen vapor phase. Cryo-preserved, autologous, matured, and TAPAs-loaded DC were thawed as described above, then immediately injected in the groin area. PBMCs were collected at baseline (before the first dose of BSK01™ was administered), and at different time points during the course of treatment (see results section), to evaluate the immune response by ELISA and flow-cytometry. Imaging studies were performed at baseline and after completion of BSK01™ therapy or until therapy discontinuation. Eligible patients were those whose tumors expressed at least one of the antigenic panel described above. Patients received 3 days of subcutaneous Granulocyte Macrophage Colony Stimulating Factor (GM-CSF) to increase bone marrow production of monocytes, and whole blood was obtained by phlebotomy for the generation of patient-specific BSK01™. Patients also received 5 days of low-dose cyclophosphamide prior to each vaccination with TAPA-pulsed mDCs to decrease T-reg activity. TAPAs-pulsed mDCs were administered at a fixed dose of up to 107 mDCs at least 2 days following cyclophosphamide administration. The vaccination schedule was once every 14 days via subcutaneous (SC) and intradermal (ID) injections (0.5 mL SC and 0.5 mL ID). The specific doses for the two patients described in the Results section were as follows: CRC patient, 6.8 × 106 DC/dose, PanC patient, 107 DC/dose. Low-dose GM-CSF (50 micrograms/day SQ for 5 days) was administered SC around the site of BSK01™ injection, as a vaccine adjuvant.
Results
DC viability and phagocytic ability
Following exposure to GM-CSF and IL-4, iDC were harvested on day 7 and viability was measured by the trypan-blue exclusion method. On average, there were 2.07 × 107 total cells (95% C.I. = ±1.3 × 104), with 99% viability (95% C.I. = ±3%). Percent viability was calculated as \( \frac{number\ of\ viable\ cells}{number\ of\ vible\ cells+ number\ of\ dead\ cells}\ast 100 \). Analysis of the phagocytic potential (Fig. 1a and b) revealed that 96% of the iDC population incorporated Al488 in 30 min, showing efficient endocytic potential. On average, the MFI of cells incubated at 37 °C was 6.8 times higher than that of cells incubated at 4 °C.

Endocytosis assay. a mature DC were incubated with the endocytic tracer (Dextran 10,000 m.w. conjugated with Alexa Fluor 488, Al488-dextran). The amount of Al488-dextran that was taken up by the DC was measured as fluorescence intensity using a FC500 flow-cytometer. The assay was conducted at 4 C as a negative control for endocytosis. b The MFI (Mean Fluorescence Intensity) was measured in triplicate assays at 37 C and 4 C. The average net MFi (37 C – 4 C) +/− 95% C.I. is shown for 3 subjects
Pulsed, mature DC showed high viability, differentiation and activation
Pulsed and matured DC (mDC) were 98.5% viable on average and showed consistent intra- and inter-subject recovery rates (75.8% of pulsed iDC were recovered on average, Fig. 2).

a Pulsed, mature DC yields. The recovery efficiency is depicted as a function of the peptide used in pulsing procedures. The recovery efficiency was measured as a percent of iDC. obtained after maturation and pulsing (mDC) using the formula mDC/iDC × 100. b mDC viability. The viability of pulsed, mDC before and after freezing and thawing was measured using the trypan-blue exclusion method. Histograms show the percent viable cells calculated with the following formula (total cells – trypan blue-positive cells) / (total cells) × 100. Each count was performed in triplicate (from 3 aliquots of the cell suspension), and bars represent the average of the replicates (confidence intervals ranged from +/− 5 to +/− 12% of the mean)
Figure 2b shows the recovery and viability of mDC after freezing and thawing, demonstrating high recovery rates, which were close to 98% on average.
mDC immunophenotype (Fig. 3) confirmed high levels of differentiation (CD80, CD86, HLA-DR) and activation markers (CD1a, CD58, CD83), while the monocyte marker, CD14 was barely detectable. Additional file 1: Figure S1, Additional file 2: Figure S2 and Additional file 3: Figure S3 show representative flow-cytometry plots obtained with SP17-pulsed mDC.

Immunophenotype of pulsed mDC. Cells were characterized using the indicated differentiation and activation markers by flow-cytometry. No statistically significant difference was noted in the percent of positive events for each CD antigen across tested antigens or subjects (all one-way ANOVA p > 0.05)
Figure 4 shows that mDC secreted IL12p70 under basal culture conditions, and that the levels of IL12p70 could be stimulated by engaging CD40 with soluble CD40L. The purity of the mDC population was between 76 and 86% of the total culture. Results were consistent across the different peptides used for pulsing, but subject 3 showed lower amounts of IL12p70 secretion. Since CD1a-positive DC represent the IL12p70-producing population of cultured DC [42], this result is in accordance with the lower frequency of CD1a-positive mDC compared to those measured for subjects 1 and 2.

Production of IL12p70. The DC activation marker, IL12p70, was measured by ELISA with or without CD40L stimulation. Histograms show the average IL12p70 level measured in the mDC medium in triplicate. For each peptide stimulation, a two-way t test (alpha = 0.05) was performed to compare the mean IL12p70 production in vehicle-treated versus CD40L-treated mDC (*, 0.01 < p < 0.05; **, p < 0.01; NS, not significant)
In vivo safety and immunogenicity
At the time this manuscript is submitted, a total of 3 patients with metastatic solid malignancies were treated. Two of them had refractory colorectal cancer (CRC), and one had refractory pancreatic cancer (PanC). One of the CRC patients discontinued the study before the first immunological evaluation was completed. No serious adverse events were reported. Evidence of immunological efficacy was demonstrated in all remaining patients, and evidence of clinical activity, in terms of reduction of tumor burden, was detected in the PanC patient (see below in vivo efficacy discussion). PBMCs were isolated after the third BSK01™ dose and evaluated by ELISA for the production of IP-10 and by flow-cytometry for the up-regulation of the CTL marker, CD137. Figure 5 shows that post-therapy PBMCs, exposed for 48 h to the same peptides used for the patient-specific vaccine, responded to antigen recall by expressing significantly higher amounts of IP-10 and CD137 with respect to PBMCs collected at baseline. Of note, the basal response was non-zero, which is compatible with the hypothesis of the presence of a pre-vaccine, spontaneous immune response, albeit sub-optimal.

TAPA-specific T-cell response. The TAPA-specific activation of PBMCs-derived T-cells was measured by flow-cytometry by evaluating the number of CD8 + CD137+ events per 10,000 PBMCs exposed to the indicated peptides. Results show average (+/− 95% C.I.) out of triplicate measurements, obtained from PBMCs taken at baseline (before the first vaccine dose), and after the third dose of vaccine. For each peptide stimulus, a two-way t test (alpha = 0.05) was performed to compare the mean CD8 + CD137+ numbers or the mean IP-10 levels between baseline and post-third vaccine (**, p < 0.01; ***, p < 0.001; NS, not significant). The horizontal lines in the upper panels indicate the number of total CD8+ event /10,000 cells before the stimulation with the peptides in vitro (regardless of CD137 expression)
In vivo efficacy
A 73-year-old male patient with the history of a locally advanced pancreatic cancer was enrolled in the BSK01™ trial. The patient was treated with neoadjuvant gemcitabine, concurrent 5FU and radiation therapy, and consolidation 5-FU, followed by surgical resection by Whipple and splenectomy in December 2009 with indeterminate margins. The cancer eventually became metastatic and the subject underwent multiple lines of chemotherapy, including FOLFIRINOX, Gemzar and Abraxane, and most recently Onivyde/leucovorin/5FU for progressive liver and lung metastases in August 2017. The patient received 6 BSK01 doses over 12 weeks, demonstrating a tumor antigen specific response by ELISA and flow-cytometry (Fig. 5). Results from imaging after treatment showed an enlargement in the tumor (not considered to be pseudo-progression), and the patient then received a repeat course of Onivyde/leucovorin/5FU with the follow-up scan revealing a decrease in size of the liver and lung metastasis (Fig. 6). Importantly, the patient’s disease has remained stable for over 7 months.

Imaging results. Photographs show the PET (pre-treatment) and CAT (post-treatment) scans, capturing 3 different metastatic sites. The estimated lesions size is indicated underneath each picture
Discussion
The majority of advanced-phase trials of therapeutic DCs are based on mature DC [2]. Meta-analyses in prostate cancer [10], melanoma [14], and malignant glioma [15] have clearly shown the existence of a significant correlation between dendritic cell activation and maturation parameters and survival outcome. Accordingly, although monocyte-derived DC differentiated with GM-CSF and IL-4 have been most widely used in clinical trials, TNFα, IL-1β, IL-6, and PGE2 matured DC have been the object of more recent investigations [2]. In the present study, we have manufactured DC through an innovative cocktail consisting of TNFα, IL-1β, IFNα, and poly I:C. Compared to the aforementioned cocktail, we replaced IL-6 and PGE2 with IFNα, and poly I:C. Although previous studies included PGE2 in the maturation cocktail [43, 44], it was shown that DC matured with poly I:C displayed improved T-cell activation abilities compared with PGE2-matured DC [45]. While IL-6 has been often included in the DC maturation cocktail in a number of clinical trials [2], it was shown to reduce DC migration and it was associated with impaired DC maturation in vivo [46, 47]. On the other hand, DC induced in the presence of IFNα were reported to have remarkably stronger T-cell activation ability than those matured without IFNα [48].
Which DC maturation markers are important for clinical effectiveness is still a matter of debate, and it may depend on tumor characteristics, but CD80, CD83, CD86, and HLA-DR are the most significant. De Vries et al. [14] found that CD83 alone as a DC maturation marker is correlated with improved clinical efficacy of the vaccine in melanoma patients, while Heiser et al. used CD83 as a DC maturation marker for their study in prostate cancer, with a total CD83 expression ~ 11% in DC [49]. HLA-DR and CD86 were found to be good surrogates of DC maturation by Kim et al. in renal cell carcinoma, with about 90% matured DC [50], while Su et al. indicated CD83 and CD86 as maturation markers [51]. Other studies suggest the use of a combination of CD80, CD83, CD86, and HLA-DR, along with CCR7, for a more reliable assessment of DC maturation, for example Waeckerle-Men et al. described that ~ 50% CD83, 90% CD86, 40% CD80, and 80% HLA-DR expression had clinical benefits [52]. We therefore selected CD80, CD83, CD86, and HLA-DR as maturation markers. Moreover, we assessed the expression of the adhesion molecule, CD58 (LFA-3), since it has been shown that the interaction of CD58 with CD2 on T-cells is required for the clustering of DCs and CD4+ T cells [53, 54]. Our mDC maturation and activation protocol resulted in dramatic upregulation of CD1a, CD83, CD80, CD86, HLA-DR, and CD58, together with down-regulation of CD14. Such pattern indicates the successful generation of fully matured DC. Importantly, mDC showed excellent viability and recovery rates after freeze and thawing. The ability to produce IL12p70 under basal condition and to respond to CD40 stimulation are critical indicators of mDC potency [55] shown to correlate with patients’ progression-free survival [56]. The levels of CD83 induction we obtained (over 75%) were superior to those reported by others [14, 49, 52], further supporting potency of BSK01, as CD83 correlates with the immune response [57] and DC potency [58]. Our TAPA-based mDC preparation has several advantages over those described in previous clinical studies, where mature DC were pulsed with tumor lysates or tumor-derived mRNAs which may contain also self-antigens and immune-regulatory factors that can limit the response or can trigger toxicity against non-specific targets, or on a limited set of tumor antigens.
The results from an early stage clinical trial in patients with refractory and metastatic solid malignancies reveled that BSK01™ was well tolerated at the dose of up to 107 DC for a total of six doses, and that it was capable of triggering a peptide-specific immune response.
We report here evidence of a clinical benefit in a patient with refractory pancreatic cancer, and at last follow-up, the patient’s PFS was 7 months. Although we report here the positive clinical response of only 1 case, the correlation between BSK01™ therapy in conjunction with chemotherapy, has the potential to lead to the reduction of refractory metastatic lesions [59, 60]. Lastly, BSK01™ was not associated with any untoward side effects in the 2 patients treated.
Conclusions
BSK01™ represents a significant improvement over first-generation DC vaccines, such as Sipuleucel-T, as shown in Additional file 4: Figure S4. Based on these encouraging results, we plan to expand our experience with BSK01™ in a Phase II clinical trial.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Abbreviations
- CRC:
-
Colorectal cancer
- DC:
-
Dendritic cells
- ELISA:
-
Enzyme-linked immunosorbent assay
- HLA:
-
Human leukocyte antigen
- IL12p70:
-
70-KDa form of interleukin 12
- PanC:
-
Pancreatic cancer
- PBMC:
-
Peripheral blood mononuclear cell
- PFS:
-
Progression-free survival
- TAPA:
-
Tumor-associated peptide antigens
References
Kirkwood JM, Butterfield LH, Tarhini AA, Zarour H, Kalinski P, Ferrone S. Immunotherapy of cancer in 2012. CA Cancer J Clin. 2012;62(5):309–35.
Anguille S, Smits EL, Lion E, van Tendeloo VF, Berneman ZN. Clinical use of dendritic cells for cancer therapy. Lancet Oncol. 2014;15(7):e257–67.
Ai YQ, Cai K, Hu JH, Jiang LW, Gao YR, Zhao H, et al. The clinical effects of dendritic cell vaccines combined with cytokine-induced killer cells intraperitoneal injected on patients with malignant ascites. Int J Clin Exp Med. 2014;7(11):4272–81.
Chodon T, Comin-Anduix B, Chmielowski B, Koya RC, Wu Z, Auerbach M, et al. Adoptive transfer of MART-1 T-cell receptor transgenic lymphocytes and dendritic cell vaccination in patients with metastatic melanoma. Clin Cancer Res. 2014;20(9):2457–65.
Kamigaki T, Matsuda E, Okada S, Naitoh K, Kondo T, Ibe H, et al. Prospective evaluation of safety of immune-cell therapy for patients with various types of advanced cancer. Anticancer Res. 2014;34(8):4601–7.
Olin MR, Low W, McKenna DH, Haines SJ, Dahlheimer T, Nascene D, et al. Vaccination with dendritic cells loaded with allogeneic brain tumor cells for recurrent malignant brain tumors induces a CD4(+)IL17(+) response. J Immunother Cancer. 2014;2:4.
Wang D, Zhang B, Gao H, Ding G, Wu Q, Zhang J, et al. Clinical research of genetically modified dendritic cells in combination with cytokine-induced killer cell treatment in advanced renal cancer. BMC Cancer. 2014;14:251.
Whiteside TL, Ferris RL, Szczepanski M, Tublin M, Kiss J, Johnson R, et al. Dendritic cell-based autologous tumor vaccines for head and neck squamous cell carcinoma: promise vs reality. Head Neck. 2015;38:E494–501.
Zhu H, Yang X, Li J, Ren Y, Zhang T, Zhang C, et al. Immune response, safety, and survival and quality of life outcomes for advanced colorectal cancer patients treated with dendritic cell vaccine and cytokine-induced killer cell therapy. Biomed Res Int. 2014;2014:603871.
Draube A, Klein-Gonzalez N, Mattheus S, Brillant C, Hellmich M, Engert A, et al. Dendritic cell based tumor vaccination in prostate and renal cell cancer: a systematic review and meta-analysis. PLoS One. 2011;6(4):e18801.
Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer. 2012;12(4):265–77.
Huber ML, Haynes L, Parker C, Iversen P. Interdisciplinary critique of sipuleucel-T as immunotherapy in castration-resistant prostate cancer. J Natl Cancer Inst. 2012;104(4):273–9.
Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363(5):411–22.
de Vries IJ, Lesterhuis WJ, Scharenborg NM, Engelen LP, Ruiter DJ, Gerritsen MJ, et al. Maturation of dendritic cells is a prerequisite for inducing immune responses in advanced melanoma patients. Clin Cancer Res. 2003;9(14):5091–100.
Yamanaka R, Homma J, Yajima N, Tsuchiya N, Sano M, Kobayashi T, et al. Clinical evaluation of dendritic cell vaccination for patients with recurrent glioma: results of a clinical phase I/II trial. Clin Cancer Res. 2005;11(11):4160–7.
Carrasco J, Van Pel A, Neyns B, Lethe B, Brasseur F, Renkvist N, et al. Vaccination of a melanoma patient with mature dendritic cells pulsed with MAGE-3 peptides triggers the activity of nonvaccine anti-tumor cells. J Immunol. 2008;180(5):3585–93.
Ribas A, Comin-Anduix B, Chmielowski B, Jalil J, de la Rocha P, McCannel TA, et al. Dendritic cell vaccination combined with CTLA4 blockade in patients with metastatic melanoma. Clin Cancer Res. 2009;15(19):6267–76.
Reyes D, Salazar L, Espinoza E, Pereda C, Castellon E, Valdevenito R, et al. Tumour cell lysate-loaded dendritic cell vaccine induces biochemical and memory immune response in castration-resistant prostate cancer patients. Br J Cancer. 2013;109(6):1488–97.
Bumm K, Grizzi F, Franceschini B, Koch M, Iro H, Wurm J, et al. Sperm protein 17 expression defines 2 subsets of primary esthesioneuroblastoma. Hum Pathol. 2005;36(12):1289–93.
Chiriva-Internati M, Gagliano N, Donetti E, Costa F, Grizzi F, Franceschini B, et al. Sperm protein 17 is expressed in the sperm fibrous sheath. J Transl Med. 2009;7:61.
Chiriva-Internati M, Grizzi F, Weidanz JA, Ferrari R, Yuefei Y, Velez B, et al. A NOD/SCID tumor model for human ovarian cancer that allows tracking of tumor progression through the biomarker Sp17. J Immunol Methods. 2007;321(1–2):86–93.
Chiriva-Internati M, Wang Z, Pochopien S, Salati E, Lim SH. Identification of a sperm protein 17 CTL epitope restricted by HLA-A1. Int J Cancer. 2003;107(5):863–5.
Chiriva-Internati M, Wang Z, Salati E, Bumm K, Barlogie B, Lim SH. Sperm protein 17 (Sp17) is a suitable target for immunotherapy of multiple myeloma. Blood. 2002;100(3):961–5.
Chiriva-Internati M, Wang Z, Salati E, Timmins P, Lim SH. Tumor vaccine for ovarian carcinoma targeting sperm protein 17. Cancer. 2002;94(9):2447–53.
Chiriva-Internati M, Wang Z, Salati E, Wroblewski D, Lim SH. Successful generation of sperm protein 17 (Sp17)-specific cytotoxic T lymphocytes from normal donors: implication for tumour-specific adoptive immunotherapy following allogeneic stem cell transplantation for Sp17-positive multiple myeloma. Scand J Immunol. 2002;56(4):429–33.
Chiriva-Internati M, Wang Z, Xue Y, Bumm K, Hahn AB, Lim SH. Sperm protein 17 (Sp17) in multiple myeloma: opportunity for myeloma-specific donor T cell infusion to enhance graft-versus-myeloma effect without increasing graft-versus-host disease risk. Eur J Immunol. 2001;31(8):2277–83.
Chiriva-Internati M, Yu Y, Mirandola L, Jenkins MR, Chapman C, Cannon M, et al. Cancer testis antigen vaccination affords long-term protection in a murine model of ovarian cancer. PLoS One. 2010;5(5):e10471.
Lim SH, Wang Z, Chiriva-Internati M, Xue Y. Sperm protein 17 is a novel cancer-testis antigen in multiple myeloma. Blood. 2001;97(5):1508–10.
Mirandola L, Figueroa JA, Phan TT, Grizzi F, Kim M, Rahman RL, et al. Novel antigens in non-small cell lung cancer: SP17, AKAP4, and PTTG1 are potential immunotherapeutic targets. Oncotarget. 2015;6(5):2812–26.
Straughn JM Jr, Shaw DR, Guerrero A, Bhoola SM, Racelis A, Wang Z, et al. Expression of sperm protein 17 (Sp17) in ovarian cancer. Int J Cancer. 2004;108(6):805–11.
Chiriva-Internati M, Ferrari R, Yu Y, Hamrick C, Gagliano N, Grizzi F, et al. AKAP-4: a novel cancer testis antigen for multiple myeloma. Br J Haematol. 2008;140(4):465–8.
Chiriva-Internati M, Yu Y, Mirandola L, D’Cunha N, Hardwicke F, Cannon MJ, et al. Identification of AKAP-4 as a new cancer/testis antigen for detection and immunotherapy of prostate cancer. Prostate. 2012;72(1):12–23.
Mirandola L, Yu Y, Jenkins MR, Chiaramonte R, Cobos E, John CM, et al. Tracking human multiple myeloma xenografts in NOD-Rag-1/IL-2 receptor gamma chain-null mice with the novel biomarker AKAP-4. BMC Cancer. 2011;11:394.
Chiriva-Internati M, Ferrari R, Prabhakar M, Yu Y, Baggoni L, Moreno J, et al. The pituitary tumor transforming gene 1 (PTTG-1): an immunological target for multiple myeloma. J Transl Med. 2008;6:15.
Chiriva-Internati M, Mirandola L, Yu Y, Jenkins MR, Gornati R, Bernardini G, et al. Cancer testis antigen, ropporin, is a potential target for multiple myeloma immunotherapy. J Immunother. 2011;34(6):490–9.
Mirandola L, Wade R, Verma R, Pena C, Hosiriluck N, Figueroa JA, et al. Sex-driven differences in immunological responses: challenges and opportunities for the immunotherapies of the third millennium. Int Rev Immunol. 2015;34(2):134–42.
Wang Z, Zhang Y, Liu H, Salati E, Chiriva-Internati M, Lim SH. Gene expression and immunologic consequence of SPAN-Xb in myeloma and other hematologic malignancies. Blood. 2003;101(3):955–60.
Frank C, Hundemer M, Ho AD, Goldschmidt H, Witzens-Harig M. Cellular immune responses against the cancer-testis antigen SPAN-XB in healthy donors and patients with multiple myeloma. Leuk Lymphoma. 2008;49(4):779–85.
Yanagawa Y, Matsumoto M, Togashi H. Enhanced dendritic cell antigen uptake via alpha2 adrenoceptor-mediated PI3K activation following brief exposure to noradrenaline. J Immunol. 2010;185(10):5762–8.
Wen YJ, Barlogie B, Yi Q. Idiotype-specific cytotoxic T lymphocytes in multiple myeloma: evidence for their capacity to lyse autologous primary tumor cells. Blood. 2001;97(6):1750–5.
Zaccard CR, Watkins SC, Kalinski P, Fecek RJ, Yates AL, Salter RD, et al. CD40L induces functional tunneling nanotube networks exclusively in dendritic cells programmed by mediators of type 1 immunity. J Immunol. 2015;194(3):1047–56.
Cernadas M, Lu J, Watts G, Brenner MB. CD1a expression defines an interleukin-12 producing population of human dendritic cells. Clin Exp Immunol. 2009;155(3):523–33.
Jonuleit H, Kuhn U, Muller G, Steinbrink K, Paragnik L, Schmitt E, et al. Pro-inflammatory cytokines and prostaglandins induce maturation of potent immunostimulatory dendritic cells under fetal calf serum-free conditions. Eur J Immunol. 1997;27(12):3135–42.
De Vries IJ, Krooshoop DJ, Scharenborg NM, Lesterhuis WJ, Diepstra JH, Van Muijen GN, et al. Effective migration of antigen-pulsed dendritic cells to lymph nodes in melanoma patients is determined by their maturation state. Cancer Res. 2003;63(1):12–7.
Moller I, Michel K, Frech N, Burger M, Pfeifer D, Frommolt P, et al. Dendritic cell maturation with poly(I:C)-based versus PGE2-based cytokine combinations results in differential functional characteristics relevant to clinical application. J Immunol. 2008;31(5):506–19.
Park SJ, Nakagawa T, Kitamura H, Atsumi T, Kamon H, Sawa S, et al. IL-6 regulates in vivo dendritic cell differentiation through STAT3 activation. J Immunol. 2004;173(6):3844–54.
Hegde S, Pahne J, Smola-Hess S. Novel immunosuppressive properties of interleukin-6 in dendritic cells: inhibition of NF-κB binding activity and CCR7 expression. FASEB J. 2004;18(12):1439–41.
Mailliard RB, Wankowicz-Kalinska A, Cai Q, Wesa A, Hilkens CM, Kapsenberg ML, et al. α-Type-1 polarized dendritic cells: a novel immunization tool with optimized CTL-inducing activity. Cancer Res. 2004;64(17):5934–7.
Heiser A, Coleman D, Dannull J, Yancey D, Maurice MA, Lallas CD, et al. Autologous dendritic cells transfected with prostate-specific antigen RNA stimulate CTL responses against metastatic prostate tumors. J Clin Invest. 2002;109(3):409–17.
Kim JH, Lee Y, Bae Y-S, Kim WS, Kim K, Im HY, et al. Phase I/II study of immunotherapy using autologous tumor lysate-pulsed dendritic cells in patients with metastatic renal cell carcinoma. Clin Immunol. 2007;125(3):257–67.
Su Z, Dannull J, Heiser A, Yancey D, Pruitt S, Madden J, et al. Immunological and clinical responses in metastatic renal cancer patients vaccinated with tumor RNA-transfected dendritic cells. Cancer Res. 2003;63(9):2127–33.
Waeckerle-Men Y, Uetz-von Allmen E, Fopp M, von Moos R, Bohme C, Schmid HP, et al. Dendritic cell-based multi-epitope immunotherapy of hormone-refractory prostate carcinoma. Cancer Immunol Immunother. 2006;55(12):1524–33.
Wang Y, Xiong X, Wu D, Wang X, Wen B. Efficient activation of T cells by human monocyte-derived dendritic cells (HMDCs) pulsed with Coxiella burnetii outer membrane protein Com1 but not by HspB-pulsed HMDCs. BMC Immunol. 2011;12:52.
Aimanianda V, Haensler J, Lacroix-Desmazes S, Kaveri SV, Bayry J. Novel cellular and molecular mechanisms of induction of immune responses by aluminum adjuvants. Trends Pharmacol Sci. 2009;30(6):287–95.
Butterfield LH, Gooding W, Whiteside TL. Development of a potency assay for human dendritic cells: IL-12p70 production. J Immunother. 2008;31(1):89–100.
Stio M, Vanni P, Ferrini PR, Giachetti E, Bosi A, Pinzauti G. Neutral maltase of human granulocytes: localization on the extracytoplasmic side of the plasma membrane and some properties. Biochem Med Metab Biol. 1988;40(2):186–96.
Aerts-Toegaert C, Heirman C, Tuyaerts S, Corthals J, Aerts JL, Bonehill A, et al. CD83 expression on dendritic cells and T cells: correlation with effective immune responses. Eur J Immunol. 2007;37(3):686–95.
Jin P, Han TH, Ren J, Saunders S, Wang E, Marincola FM, et al. Molecular signatures of maturing dendritic cells: implications for testing the quality of dendritic cell therapies. J Transl Med. 2010;8:4.
Hodi FS, Ballinger M, Lyons B, Soria JC, Nishino M, Tabernero J, et al. Immune-modified response evaluation criteria in solid tumors (imRECIST): refining guidelines to assess the clinical benefit of Cancer immunotherapy. J Clin Oncol. 2018;36(9):850–8.
Chiou VL, Burotto M. Pseudoprogression and immune-related response in solid tumors. J Clin Oncol. 2015;33(31):3541–3.
Funding
This study was supported entirely by Kiromic, Inc.
Author information
Authors and Affiliations
Contributions
LM ran experiments, analyzed the data and wrote the manuscript, MC-I designed the study and analyzed the data, RB analyzed the data, FM analyzed the data, JF designed the study, and SD analyzed the data and contributed to writing the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The safety, immunogenicity, and clinical activity of BSK01™ were evaluated in a Phase I clinical trial (ClinicalTrials.gov Identifier: NCT02705703). Patients were enrolled upon signing informed consent per IRB specifications (Salus central IRB, https://www.salusirb.com).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Figure S1.
Representative immune-phenotype results from pulsed, mDC from subject #1.
Additional file 2:
Figure S2. Representative immune-phenotype results from pulsed, mDC from subject #2.
Additional file 3:
Figure S3. Representative immune-phenotype results from pulsed, mDC from subject #3.
Additional file 4:
Figure S4. Comparison between first-generation DC vaccines and BSK01™
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Mirandola, L., Chiriva-Internati, M., Bresalier, R. et al. Preliminary report of a novel formulation of clinical-grade, fully matured, tumor-associated peptide-loaded dendritic cells for cancer immunotherapy. transl med commun 4, 17 (2019). https://doi.org/10.1186/s41231-019-0049-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41231-019-0049-0