
Abstract
Tamoxifen (TAM) has been prescribed worldwide to patients with and women at high-risk of breast cancer. However, long-term use of TAM increases the incidence of endometrial cancer. The carcinogenic mechanisms of TAM have been extensively investigated. TAM is hydroxylated and sulfonated at α-carbon to form α-hydroxytamoxifen-O-sulfonate. This metabolite readily reacts with genomic DNA, particularly with 2′-deoxyguanosine, leading to DNA replication error. TAM also exerts estrogenic activity at endometrial tissue to induce endometrial hyperplasia. Therefore, our efforts focused on the development of novel and safer anti-estrogens to diminish carcinogenic potential of TAM based on chemical modifications. In this review, we describe a crucial idea of our drug design and introduce our compounds SS1020 and SS5020, possessing high effectiveness, and no genotoxic and estrogenic activities.
Similar content being viewed by others

Background
International Agency for Research on Cancer (IARC) reported in 2018 that new cases of all cancers in women was 8.6 million, among which 24.2% was of breast cancer [1]. Tamoxifen (TAM) has been prescribed for breast cancer prevention and treatment worldwide [2]. In Japan, TAM was launched in 1981 for treatment of estrogen receptor (ER)-positive breast cancer. Besides the beneficial effect of TAM, several epidemiological studies reported that TAM increased endometrial cancer incidence by 2–3 times as compared with placebo group [3,4,5]. Indeed, TAM-DNA adducts were detected in endometrial samples from TAM-treated patients [6,7,8]. Therefore, our research group has been developing safer anti-breast cancer agents since 2003 [9, 10]. Before introducing our compounds, we describe the mechanisms of carcinogenic effect induced by TAM and ideas for developing next-generation anti-breast cancer drugs.
Carcinogenic mechanisms of tamoxifen and design of safer anti-breast cancer agents
Carcinogenic mechanisms of TAM have already been proposed to be its initiation and promotion effects. For the development of safer anti-estrogens, these two carcinogenic effects must be counteracted.
Metabolic pathway of TAM leading to DNA modification was shown in Fig. 1. TAM proceeds hydroxylation at α-position, and then undergoes sulfonation by sulfotransferases to form α-hydroxytamoxifen-O-sulfonate, which is transformed to carbocation and readily reacts with 2′-deoxyguanosine (dG) in genomic DNA [11,12,13]. In contrast, toremifene (TOR), a chlorinated analog of TAM, also undergoes hydroxylation at α-position, whereas its α-hydroxylated metabolite is not a suitable substrate of sulfotransferases because of steric hindrance of bulky chloride atom [14, 15]. Therefore, DNA adducts were not detected in rat liver and human leukocyte samples [16,17,18]. The lack of genotoxicity in TOR is one of the critical findings for developing safer alternatives.

Genotoxic mechanisms of tamoxifen, not toremifene, via metabolic activation. TAM (a) proceeds four steps leading to genotoxicity which are 1) α-hydroxylation by cytochrome p450, 2) sulfonation by sulfotransferase (SULT), and 3) formation of carbocation to react with DNA, particularly with 2′-deoxyguanosine (dG) [11]. In contrast, TOR (b) is also hydroxylated at α-position. However, this metabolite is not a good substrate for sulfotransferases because of steric hindrance of bulky chloride atom [14]. As a result, toremifene doesn’t produce stable DNA adducts
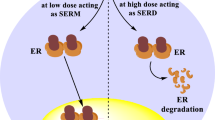
Regarding promotion activity, TAM is known as a selective estrogen receptor modulator (SERM), which exerts tissue-selective agonistic/antagonistic effects. TAM shows antagonistic effects on breast tissue, thereby suppresses the development of ER-positive breast cancer. However, TAM exerts agonistic effects on endometrial tissue, leading to endometrial cell proliferation. Unfortunately, non-genotoxic TOR also possesses agonistic activity in endometrial tissue in ovariectomized rats [9, 10]. Coactivator is a crucial determinant of ligand-dependent agonistic activity. X-ray crystal structure analysis revealed that 17β-estradiol (E2) induces appropriate relocation of the helix (H) 12 in ER-LBD (ligand-binding domain) [19], and allows ER to interact with specific coactivators, thereby leads to initiating transactivation (Fig. 2 [20]). In contrast, TAM induces H12 relocation different from that of E2, and suppresses recruitment of several coactivators, except SRC-1 (steroid receptor coactivator-1) [21]. SRC-1 expressing in endometrial cells can bind to TAM-bound ER complex; therefore TAM, probably TOR as well, shows an agonistic effect on endometrial tissue [22]. As shown in Fig. 3, there are many drug candidates for breast cancer treatment, among which GW5638 (GW) shows a unique anti-breast cancer property. GW is reported as effective against TAM-resistant breast cancer model with less estrogenic activity [23,24,25]. In hERα-LBD, acrylate side-chain of GW interacts with aspartate 351 residue in H12, leading to increased exposure of hydrophobic surface [26]. Since intracellular protein-degradation is increased depending on a surface hydrophobicity [27], acrylate side-chain would play an essential role in the decrease of intracellular ER protein by which GW shows differential anti-breast cancer spectrum as compared with TAM and TOR.

Overall structures of ligand-bound human estrogen receptor α ligand-binding domain complex. X-Ray crystal structures of human estrogen receptor α-ligand binding domain bound to E2 (left; PDB ID 1ERE [19]), 4-OH TAM (center; PDB ID 3ERT [21]) and GW (right; PDB ID 1R5K [26]) were illustrated using NGL software [20] on RCSB PDB website (http://www.rcsb.org/). These structures indicate the difference of helix (H) 12-relocation between three ligands. GW5638-induced H12 positioning increases surface hydrophobicity of ERα LBD, leading to ER instability

Chemical structures of SS1010, SS1020, SS5020, and related anti-estrogens
Safer anti-breast cancer agents: SS1020 and SS5020
To overcome the adverse effects of TAM, we designed SS1020 and SS5020 as safer alternatives (Fig. 4). Our compounds contain several essential structures as below. Chloride atom of SS1020 might diminish genotoxicity of TAM as shown in TOR. Also, 4-hydroxyl group of SS1020 promises to increase ER binding affinity as reported for 4-OH TAM [28]. Finally, the acrylate side-chain of SS1020 expects to diminish the endometrial activity of TAM with decreasing ER stability. Furthermore our 32P-postlabeling analysis showed that raloxifene (RAL) did not produce any DNA adduct in rat liver [17], the mimic of RAL structure is another choice for drug design. However, RAL, having two hydroxyl moieties, can be conjugated rapidly through phase II metabolism and excreted, making it difficult to achieve adequate bioavailability by oral administration [29]. Therefore, we synthesized SS5020 as a RAL analog with a slight structural modification to improve bioavailability [10].

Basic ideas of structural design for potent and non-genotoxic anti-estrogens (SS1020 and SS5020)
To confirm estrogenic activity of anti-estrogens, we carried out uterotrophic assay using ovariectomized rats in which rats were treated subcutaneously with test compounds for 3 d, and rat uteri were excised and weighted at 24 h after the final administration [9, 10]. TAM significantly increased uterine wet weight as shown in Fig. 5. 4-OH TAM, TOR, and ospemifene (OSP) also showed uterotrophic activity in this assay. RAL, SP500263 (SP) and SS1010 exerted moderate effects. As expected, SS1020 and SS5020 did not show any significant activity. Although the exact ER binding model of SS1020 and SS5020 remains to be unknown, our compounds might exhibit, at least in part, similar binding model, H12 relocation, and subsequent coactivator recruitment as seen in GW and SP. Therefore, we selected SS1020 and SS5020 for further determination of DNA adduct formation in rats (Fig. 6). Rats were orally treated with 20 mg of TAM or equimolar of test chemicals for 7 d, and rat liver DNA was analyzed using 32P-postlabeling/polyacrylamide gel electrophoresis assay [30]. In this assay, rat liver DNA was digested with nuclease P1 and micrococcal nuclease, and then DNA adduct was labeled with 32P using T4-polynucleotide kinase treatment in the presence of γ32P-ATP. Labeled DNA digests were separated on 30% polyacrylamide gel, and DNA adducts were observed as a single radioactive band. TAM treatment produced DNA adducts, especially dG-N2-TAM (fr-2), whereas no DNA adduct was detected in rat liver treated with TOR, SS1020 and SS5020. These results indicate that SS1020 and SS5020 have neither estrogenic activity nor genotoxicity in rats.

Uterotrophic effects of antiestrogens (left panel, SS1020 and its related compounds; right panel, SS5020 and its related compounds) in ovariectomized SD rats. Ovariectomized rats were subcutaneously treated with each test compound (0.3 μmol/rat/d of 17β-estradiol, or equivalent molar of test compound), and uterine wet weight was measured. TAM, 4-OH TAM, TOR, and OSP clearly increased uterine wet weight. RAL, GW, SP, and SS1010 exerted moderate uterotrophic effects. No detectable effects were observed in SS1020 [9] and SS5020 [10]

32P-postlabeling/PAGE analysis of SS1020, SS5020 and related anti-estrogens. Female SD rats were orally treated with test compound (20 mg/kg/d of TAM, or equivalent molar of test compound), and liver DNA was used for DNA adduct analysis. TAM treatment clearly formed dG-TAM adducts, whereas SS1020, SS5020 or TOR treatment didn’t observed any detectable DNA adducts [9, 10]
Anti-breast cancer potential of SS1020 and SS5020 was tested using two animal models which are dimethylbenz(a)anthracene-induced rat mammary tumor model and human breast cancer MCF-7 xenograft mouse model [9, 10]. In these animal experiments, SS1020 and SS5020 exhibit potent anti-breast cancer activity as compared with TAM, RAL, and GW. Although the mode of action should be further determined, SS1020 and SS5020 are considered to be safer alternatives for breast cancer therapy and prevention.
Conclusions
In the last several decades, TAM has been prescribed for ER-positive breast cancer patients and women at high-risk for breast cancer. Epidemiological data indicated that TAM increased the incidence of endometrial cancer. This fact prompted us to develop a safer alternative, which our group succeeded to develop novel anti-breast cancer agents lacking genotoxicity and estrogenic activity. This project would provide an alternative option for women who hesitate to use TAM because of the concern about secondary endometrial cancer. However, most important, our project would pave the way for toxicologists to apply fundamental genotoxicity data for developing a novel drug design.
Abbreviations
- dG:
-
2′-Deoxyguanosine
- E2:
-
17β-Estradiol
- ER:
-
Estrogen receptor
- GW:
-
GW5638
- OSP:
-
Ospemifene
- RAL:
-
Raloxifene
- SERM:
-
Selective estrogen receptor modulator
- SP:
-
SP500263
- SRC-1:
-
Steroid receptor coactivator-1
- TAM:
-
Tamoxifen
- TOR:
-
Toremifene
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. https://doi.org/10.3322/caac.21492.
Jordan VC, Fourteenth Gaddum Memorial Lecture. A current view of tamoxifen for the treatment and prevention of breast cancer. Br J Pharmacol. 1993;110(2):507–17.
Bissett D, Davis JA, George WD. Gynaecological monitoring during tamoxifen therapy. Lancet. 1994;344(8932):1244.
Fisher B, Costantino JP, Redmond CK, Fisher ER, Wickerham DL, Cronin WM. Endometrial cancer in tamoxifen-treated breast cancer patients: findings from the National Surgical Adjuvant Breast and bowel project (NSABP) B-14. J Natl Cancer Inst. 1994;86(7):527–37.
Sismondi P, Biglia N, Volpi E, Giai M, de Grandis T. Tamoxifen and endometrial cancer. Ann N Y Acad Sci. 1994;734:310–21.
Shibutani S, Suzuki N, Terashima I, Sugarman SM, Grollman AP, Pearl ML. Tamoxifen-DNA adducts detected in the endometrium of women treated with tamoxifen. Chem Res Toxicol. 1999;12(7):646–53. https://doi.org/10.1021/tx990033w.
Shibutani S, Ravindernath A, Suzuki N, Terashima I, Sugarman SM, Grollman AP, et al. Identification of tamoxifen-DNA adducts in the endometrium of women treated with tamoxifen. Carcinogenesis. 2000;21(8):1461–7.
Hernandez-Ramon EE, Sandoval NA, John K, Cline JM, Wood CE, Woodward RA, et al. Tamoxifen-DNA adduct formation in monkey and human reproductive organs. Carcinogenesis. 2014;35(5):1172–6. https://doi.org/10.1093/carcin/bgu029.
Laxmi YR, Liu X, Suzuki N, Kim SY, Okamoto K, Kim HJ, et al. Anti-breast cancer potential of SS1020, a novel antiestrogen lacking estrogenic and genotoxic actions. Int J Cancer. 2010;127(7):1718–26. https://doi.org/10.1002/ijc.25167.
Suzuki N, Liu X, Laxmi YR, Okamoto K, Kim HJ, Zhang G, et al. Anti-breast cancer potential of SS5020, a novel benzopyran antiestrogen. Int J Cancer. 2011;128(4):974–82. https://doi.org/10.1002/ijc.25659.
Dasaradhi L, Shibutani S. Identification of tamoxifen-DNA adducts formed by alpha-sulfate tamoxifen and alpha-acetoxytamoxifen. Chem Res Toxicol. 1997;10(2):189–96. https://doi.org/10.1021/tx960114h.
Shibutani S, Dasaradhi L, Terashima I, Banoglu E, Duffel MW. Alpha-hydroxytamoxifen is a substrate of hydroxysteroid (alcohol) sulfotransferase, resulting in tamoxifen DNA adducts. Cancer Res. 1998;58(4):647–53.
Kim SY, Suzuki N, Laxmi YR, Shibutani S. Genotoxic mechanism of tamoxifen in developing endometrial cancer. Drug Metab Rev. 2004;36(2):199–218. https://doi.org/10.1081/DMR-120033997.
Shibutani S, Ravindernath A, Terashima I, Suzuki N, Laxmi YR, Kanno Y, et al. Mechanism of lower genotoxicity of toremifene compared with tamoxifen. Cancer Res. 2001;61(10):3925–31.
Kim SY, Suzuki N, Santosh Laxmi YR, Rieger R, Shibutani S. Alpha-hydroxylation of tamoxifen and toremifene by human and rat cytochrome P450 3A subfamily enzymes. Chem Res Toxicol. 2003;16(9):1138–44. https://doi.org/10.1021/tx0300131.
Rajaniemi H, Mantyla E, Hemminki K. DNA adduct formation by tamoxifen and structurally-related compounds in rat liver. Chem Biol Interact. 1998;113(2):145–59.
Kim SY, Suzuki N, Laxmi YR, Umemoto A, Matsuda T, Shibutani S. Antiestrogens and the formation of DNA damage in rats: a comparison. Chem Res Toxicol. 2006;19(6):852–8. https://doi.org/10.1021/tx060052n.
Umemoto A, Lin CX, Ueyama Y, Komaki K, Santosh Laxmi YR, Shibutani S. Absence of DNA adduct in the leukocytes from breast cancer patients treated with toremifene. Chem Res Toxicol. 2006;19(3):421–5. https://doi.org/10.1021/tx0503045.
Brzozowski AM, Pike AC, Dauter Z, Hubbard RE, Bonn T, Engstrom O, et al. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature. 1997;389(6652):753–8. https://doi.org/10.1038/39645.
Rose AS, Bradley AR, Valasatava Y, Duarte JM, Prlic A, Rose PW. NGL viewer: web-based molecular graphics for large complexes. Bioinformatics. 2018;34(21):3755–8. https://doi.org/10.1093/bioinformatics/bty419.
Shiau AK, Barstad D, Loria PM, Cheng L, Kushner PJ, Agard DA, et al. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell. 1998;95(7):927–37.
Shang Y, Brown M. Molecular determinants for the tissue specificity of SERMs. Science. 2002;295(5564):2465–8. https://doi.org/10.1126/science.1068537.
Willson TM, Henke BR, Momtahen TM, Charifson PS, Batchelor KW, Lubahn DB, et al. 3-[4-(1,2-Diphenylbut-1-enyl)phenyl]acrylic acid: a non-steroidal estrogen with functional selectivity for bone over uterus in rats. J Med Chem. 1994;37(11):1550–2.
Bentrem D, Dardes R, Liu H, MacGregor-Schafer J, Zapf J, Jordan V. Molecular mechanism of action at estrogen receptor alpha of a new clinically relevant antiestrogen (GW7604) related to tamoxifen. Endocrinology. 2001;142(2):838–46. https://doi.org/10.1210/endo.142.2.7932.
Dardes RC, O'Regan RM, Gajdos C, Robinson SP, Bentrem D, De Los Reyes A, et al. Effects of a new clinically relevant antiestrogen (GW5638) related to tamoxifen on breast and endometrial cancer growth in vivo. Clin Cancer Res. 2002;8(6):1995–2001.
Wu YL, Yang X, Ren Z, McDonnell DP, Norris JD, Willson TM, et al. Structural basis for an unexpected mode of SERM-mediated ER antagonism. Mol Cell. 2005;18(4):413–24. https://doi.org/10.1016/j.molcel.2005.04.014.
Bohley P. Surface hydrophobicity and intracellular degradation of proteins. Biol Chem. 1996;377(7–8):425–35.
Borgna JL, Rochefort H. High-affinity binding to the estrogen receptor of [3H]4-hydroxytamoxifen, an active antiestrogen metabolite. Mol Cell Endocrinol. 1980;20(1):71–85.
Okamoto Y, Liu X, Suzuki N, Okamoto K, Sekimoto M, Laxmi YR, et al. Increased antitumor potential of the raloxifene prodrug, raloxifene diphosphate. Int J Cancer. 2008;122(9):2142–7. https://doi.org/10.1002/ijc.23362.
Terashima I, Suzuki N, Shibutani S. 32P-Postlabeling/polyacrylamide gel electrophoresis analysis: application to the detection of DNA adducts. Chem Res Toxicol. 2002;15(3):305–11.
Acknowledgements
This research project was conducted at Shibutani’s laboratory in State University of New York at Stony Brook. These experiments could not accomplish without supports from the past laboratory members and co-workers, Kanako Okamoto, Naomi Suzuki, Dr. Xiaoping Liu, and Dr. Y. R. Santosh Laxmi. The authors dedicate this review article for them.
Funding
National Institute of Environmental Health Sciences; Grant numbers: ES09418, ES012408; Grant sponsors: Walk-for-Beauty Foundation Research Award in Breast Cancer, the School of Medicine, SUNY at Stony Brook.
Availability of data and materials
Not applicable.
Author information
Authors and Affiliations
Contributions
Both authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All the authors have read the manuscript and have approved this submission and future publication.
Competing interests
The authors declare no conflicts of interest associated with this manuscript.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Okamoto, Y., Shibutani, S. Development of novel and safer anti-breast cancer agents, SS1020 and SS5020, based on a fundamental carcinogenic research. Genes and Environ 41, 9 (2019). https://doi.org/10.1186/s41021-019-0124-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41021-019-0124-9