
Abstract
Background
Pathogenic germline variants in MLH1, MSH2 and MSH6 genes account for the majority of Lynch syndrome (LS). In this first report from Pakistan, we investigated the prevalence of pathogenic MLH1/MSH2/MSH6 variants in colorectal cancer (CRC) patients.
Methods
Consecutive cases (n = 212) were recruited at the Shaukat Khanum Memorial Cancer Hospital and Research Centre (SKMCH&RC), between November 2007 to March 2011. Patients with a family history of > 3 or 2 HNPCC-associated cancers were classified as HNPCC (n = 9) or suspected-HNPCC (n = 20), respectively (group 1; n = 29). Cases with no family history were designated as non-HNPCC (group 2; n = 183). MLH1/MSH2/MSH6 genes were comprehensively screened in group 1. Pathogenic/likely pathogenic variants identified in group 1 were subsequently evaluated in group 2.
Results
Eight distinct pathogenic/likely pathogenic MLH1/MSH2 variants were found in group 1 (10/29; 34.5%), belonging to HNPCC (5/9; 55.6%) and suspected-HNPCC (5/20; 25%) families and in group 2 (2/183; 1.1%) belonging to non-HNPCC. Overall, three recurrent variants (MSH2 c.943-1G > C, MLH1 c.1358dup and c.2041G > A) accounted for 58.3% (7/12) of all families harboring pathogenic/likely pathogenic MLH1/MSH2 variants. Pathogenic MSH6 variants were not detected.
Conclusion
Pathogenic/likely pathogenic MLH1/MSH2 variants account for a substantial proportion of CRC patients with HNPCC/suspected-HNPCC in Pakistan. Our findings suggest that HNPCC/suspected-HNPCC families should be tested for these recurrent variants prior to comprehensive gene screening in this population.
Similar content being viewed by others

Background
Colorectal cancer (CRC) is the fifth most common malignancy in Pakistan and endometrial cancer (EC) is the third most common gynecologic malignancy in Pakistani women [1]. The age-standardized (world) annual rates of CRC and EC are 4.0 and 3.6 per 100,000 in Pakistan, respectively. Affected individuals generally present at a young age. The majority of CRC and EC are not linked with inherited cancer syndromes. Up to 30% of CRC are hereditary and these may be divided into polyposis and non-polyposis syndromes. The term hereditary non-polyposis colorectal cancer (HNPCC) refers to patients and families who fulfill the Amsterdam criteria and differentiates familial aggregation of CRC from the polyposis phenotype. Up to 50% of HNPCC families have the Lynch syndrome (LS), with a DNA mismatch repair (MMR) defect, while the rest comprise those with a Lynch-like syndrome and a familial colorectal cancer type X (FCCTX) with no DNA MMR defects [2]. LS refers to families with a pathogenic germline variant in one of the DNA MMR genes (MLH1, MSH2, MSH6, and PMS2) or the EPCAM gene 3′ end deletions [3]. The most common pathogenic MMR gene variants (up to 90%) in LS are reported in MLH1 and MSH2 [4, 5], less commonly in MSH6 (up to 10%) and uncommonly in PMS2 [6]. Deletions in EPCAM gene (1–3%) in LS are rarely reported [7]. Individuals with LS have a lifetime risk of CRC, EC, and ovarian cancer ranging from 50 to 80%, 31.5–62%, and 6.7–13.5%, respectively. These individuals also face increased lifetime risks of developing cancer of the small bowel, stomach, upper urologic tract, biliary tract, pancreas and brain [8,9,10,11,12]. Identification of individuals harboring pathogenic MMR gene variants is clinically important and has a significant impact on surveillance and management [13].
Various clinical criteria such as the Amsterdam II criteria [14, 15] or the Bethesda guidelines exist for identifying patients at high risk of HNPCC. These criteria are based on a strong family history of at least three HNPCC-associated cancers, age at diagnosis and tumor histology. However, these stringent criteria have reported under-diagnosis of LS [16, 17]. Less stringent criteria of suspected-HNPCC, based on a family history of only two HNPCC-linked cancers, have also been found useful in identifying pathogenic variants in MMR genes [18,19,20].
The prevalence and spectrum of pathogenic MMR gene variants show considerable variation by ethnicity and by geographic origin worldwide [21,22,23]. However, little is known about the contribution of MMR gene variants to CRC in Pakistan. In the current study, we comprehensively investigated the contribution of pathogenic germline variants in MLH1, MSH2 and MSH6 genes to 212 Pakistani cases with HNPCC/suspected-HNPCC or non-HNPCC.
Methods
Study subjects
Consecutive cases were identified at the Shaukat Khanum Memorial Cancer Hospital and Research Centre (SKMCH&RC) in Lahore, Pakistan, from November 2007 to March 2011. These study cases were stratified into two groups: HNPCC/suspected-HNPCC group (n = 29) and non-HNPCC group (n = 183). Stringent criteria were applied for inclusion in the HNPCC subgroup. These included: (i) at least three relatives affected by histologically verified CRC or EC, small bowel or urinary tract; at least one of whom was a first degree relative of the other two, (ii) at least two of the above individuals were first degree relatives from two different generations, (iii) at least one of the above persons had cancer diagnosed at age under 50 years, (iv) familial adenomatous polyposis (FAP) had been excluded [14, 15]. Somewhat less stringent criteria used for the suspected-HNPCC subgroup included: (i) diagnosis of at least one CRC, EC, small bowel or urinary tract malignancy amongst first degree relatives of a CRC patient (or in him/herself), (ii) at least one of the above cancers diagnosed under age 50, (iii) FAP had been excluded [18]. The remaining 183 enrolled CRC cases did not fulfill the diagnostic criteria of HNPCC/suspected-HNPCC and were assigned to the non-HNPCC group. Clinical and histopathological data of all index patients were collected from medical records and pathology reports. A detailed description of the 212 index cases is shown in Table 1.
The control population included 100 healthy individuals of Pakistani origin, having no family history of CRC. These were care-givers or family members of hospital registered patients or those visiting the hospital for medical reasons other than cancer. All study participants were furnished with and signed an informed written consent. The study was approved by the Institutional Review Board (IRB) of the SKMCH&RC (IRB approval number SKMCH-CRC-001).
Molecular analysis
Genomic DNA was extracted as previously described [24]. The entire coding region and exon-intron junctions of the MLH1, MSH2 and MSH6 genes (GenBank accession numbers NM_000249.3; NM_000251.2; NM_000179.2, respectively) were screened in 29 index patients of HNPCC/suspected-HNPCC group using denaturing high-performance liquid chromatography (DHPLC) analysis. The DHPLC analysis was carried out with the WAVE system (Transgenomics, Omaha, NE, US). PCR-primer pairs and DHPLC running conditions for MLH1/MSH2 genes were according to Kurzawski and colleagues [4] and for MSH6 gene was according to Kolodner et al. with some modifications [25] and are available upon request. When available, a positive control for each exon with a known variant was included in the DHPLC analyses.
Each sample showing variants detected by DHPLC analyses was sequenced using BigDye Terminator v.3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, US), as described elsewhere [26]. Bidirectional genomic DNA sequencing was performed on an independent sample to verify the presence of a sequence variant.
Pathogenic/likely pathogenic variants identified in the HNPCC/suspected-HNPCC group were subsequently screened in the non-HNPCC group by DHPLC. Novel pathogenic variants and in silico predicted likely pathogenic variants were further analyzed in 100 healthy individuals.
Classification of MMR gene variants
The MMR gene variants were stratified according to the following 5 tier classification, as described elsewhere: class 5 (pathogenic), class 4 (likely pathogenic), class 3 (uncertain significance), class 2 (likely benign) and class 1 (benign) [27]. The variants were designated as novel or previously reported variants by searching the following six databases: Exome Aggregation Consortium (ExAC), http://exac.broadinstitute.org/; Exome Sequence Project (ESP), http://evs.gs.washington.edu/EVS/; Human Gene Mutation Database (HGMD), http://www.hgmd.cf.ac.uk/ac/index.php; Leiden Open Variation Database (LOVD), https://databases.lovd.nl/shared/genes/; International Society for Gastrointestinal Hereditary Tumours (InSiGHT), https://insight-database.org/; Mismatch Repair Genes Variant Database (MMRGVD), http://www.med.mun.ca/mmrvariants/ or Universal Mutation Database (UMD), http://www.umd.be/ (by October 2016). The MMR gene variants identified in two or more unrelated patients were considered as recurrent variants.
In silico analyses
The novel missense variants identified in MLH1/MSH2 and previously reported class 3 variants of uncertain significance (VUS) in MMR genes were analyzed for their potential effect on protein function using the default settings of web tools Align-GVGD (http://agvgd.hci.utah.edu/agvgd_input.php), PolyPhen2 (http://genetics.bwh.harvard.edu/pph2/), SIFT (https://sift.bii.a-star.edu.sg/), Mut Pred (http://mutpred.mutdb.org/), SNPs&GO (http://snps.biofold.org/snps-and-go/snps-and-go.html), PhD SNP (http://snps.biofold.org/phd-snp/phd-snp.html), and SNAP (https://www.rostlab.org/services/snap/). Furthermore, all novel and previously reported intronic VUS in MMR genes were analyzed for their potential effect on splicing using the splice prediction algorithms SpliceSiteFinder-like (http://www.umd.be/searchSpliceSite.html), MaxEntScan (http://genes.mit.edu/burgelab/maxent/Xmaxentscan_scoreseq.html), NNSPLICE (http://www.fruitfly.org/seq_tools/splice.html), GeneSplicer (http://www.ccb.jhu.edu/software/genesplicer/) and HumanSpliceFinder (http://www.umd.be/HSF3/) via the Alamut software interface (Interactive Biosoftware) in default settings.
Statistical analysis
The comparison of the distribution of clinical and histopathological characteristics between HNPCC/suspected-HNPCC group vs. non-HNPCC group and carriers of pathogenic/likely pathogenic MLH1/MSH2 variant vs. non-carriers was performed using Fisher’s exact test for categorical variables and the Wilcoxon rank-sum test for quantitative variables. All statistical tests were two-sided. Results were deemed statistically significant if the P value was 0.05 or less. All statistical computations were done using StatXact 4 for Windows (Cytel Inc., Cambridge, US), SAS version 9.3 and R, version 2.1.
Results
Characteristics of the study participants
In total, 212 unrelated Pakistani index patients were included in the current study. Of these, 86.3% were diagnosed with CRC with no family history (non-HNPCC group = 183) and 13.7% reported a family history of cancer within the spectrum of HNPCC (HNPCC/suspected-HNPCC group = 29; 9 fulfilled the HNPCC criteria and 20 met the suspected-HNPCC criteria). Characteristics of the index CRC cases are shown in Table 1. Of the index cases, 210 patients including 146 males and 64 females had a diagnosis of CRC. Two patients belonged to the suspected-HNPCC subgroup: one with breast-endometrial cancer and the other with ovarian cancer. A majority of patients were of Punjabi (38.7%) or Pathan (34.4%) ethnic origin. The mean age at onset of disease was 42.7 years (range 20–61) and 43.1 years (range 14–77) for cases belonging to HNPCC/suspected-HNPCC group and non-HNPCC group, respectively (P = 0.95, Wilcoxon rank-sum test). The HNPCC/suspected-HNPCC group in comparison to non-HNPCC group more often presented with proximal tumor site (14/24, 58.3% vs. 24/181, 13.2%; P < 0.0001) and greater tumor size (> 5 cm) (13/24, 54.2% vs. 21/71, 29.6%; P = 0.047). There were no differences in histological type, mucinous component, macroscopic appearance, histologic grade, lymphovascular or venous invasion, tumor stage and lymph node involvement between both groups.
Pathogenic germline variants: HNPCC/suspected-HNPCC group
The index patients of HNPCC/suspected-HNPCC group (n = 29) were entirely screened for germline MLH1, MSH2 and MSH6 variants using DHPLC followed by DNA sequence analyses. Seven distinct pathogenic/likely pathogenic MLH1/MSH2 variants were identified in 10 cases (10/29; 34.5%) (Table 2). No pathogenic MSH6 variant was found. Of the identified carriers of pathogenic/likely pathogenic variants, five carriers (50%) met the HNPCC criteria and five carriers (50%) met the suspected-HNPCC criteria (Table 3).
MLH1 variants
Five pathogenic variants (including four distinct variants) were detected in MLH1 (5/29; 17.2%). Among these were two frame shift variants (including a recurrent variant), one nonsense variant and one missense variant (Table 4).
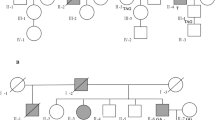
A recurrent frame shift variant in exon 12, c.1358dup (p.T455Dfs*24), was identified in two unrelated patients of Punjabi ethnicity. One patient presented with carcinoma of the sigmoid colon at 44 years of age (III:3, Fig. 1a). The other patient was diagnosed with carcinoma of the transverse colon at age 61 (III:18, Fig. 1b). Both reported a family history of HNPCC.

Pedigrees of HNPCC (a-d and f), suspected-HNPCC (e and g-i) and non-HNPCC (j, k) families with pathogenic/likely pathogenic MLH1 or MSH2 variants. a-k: Include families C203, H707, C202, C162, C92, C143, H1075, C49, C85, C122, and C164, respectively. Circles are females, squares are males, and a diagonal slash indicates a deceased individual. Symbols with filled left upper quadrant: unilateral breast cancer. Symbols with filled right lower quadrant: cancer other than breast, the name of that cancer is mentioned. Identification numbers of individuals are below the symbols. The index patient is indicated by an arrow. A, age; BC, breast cancer; CRC, colorectal cancer; D, death. The numbers following these abbreviations indicate age at enrollment, cancer diagnosis or death. M+, positive for pathogenic/likely pathogenic variant
Another frame shift variant in exon 1, c.67delG (p.E23Kfs*13), was detected in a 48-year-old patient (II:1, Fig. 1c) of Pathan ethnicity, who presented with carcinoma of the cecum and reported a family history of HNPCC.
A nonsense variant in exon 15, c.1672G > T (p.E558*), was identified in a 32-year-old patient (IV:2, Fig. 1d) of Kashmiri background, diagnosed with carcinoma of the transverse colon who also reported a family history of HNPCC.
One missense variant in exon 18, c.2041G > A (p.A681T), was identified in a 41-year-old patient (II:1, Fig. 1e) of Punjabi ethnicity with carcinoma of the transverse colon who reported a family history of suspected-HNPCC. This variant has been previously classified as a pathogenic variant [4, 30].
MSH2 variants
Five pathogenic/likely pathogenic variants (including three distinct variants) were identified in MSH2 (5/29; 17.2%). Among these were one recurrent splice site variant and two nonsense variants (Table 4).
A recurrent likely pathogenic splice site variant, c.943-1G > C, was found in three unrelated patients of Pathan ethnicity: one with rectosigmoid carcinoma at 32 years of age (III:1, Fig. 1f) and a family history of HNPCC. The remaining two patients harboring this variant presented with carcinoma of the ascending colon (III:2, Fig. 1g) and sigmoid colon (II:1, Fig. 1h) at age 43 and 60, respectively and both reported a family history of suspected-HNPCC.
A pathogenic nonsense variant in exon 12, c.1861C > T (p. R621*), was identified in a 45-year-old patient (II:1, Fig. 1i) of Punjabi ethnicity, who was diagnosed with carcinoma of the rectum and also reported a family history of suspected-HNPCC.
Another pathogenic nonsense variant in exon 16, c.2656G > T (p.E886*), was identified in a 67-year-old patient of Pathan ethnicity, who was diagnosed with endometrial and breast cancer at age 48 and 67, respectively. This patient had a family history of suspected-HNPCC and has been reported recently [28].
Pathogenic germline variants: non-HNPCC group
Screening of the index patients in the non-HNPCC group for the presence of the pathogenic/likely pathogenic MLH1/MSH2 variants identified in the HNPCC/suspected-HNPCC group revealed two additional pathogenic MLH1/MSH2 variants. The MLH1 missense variant, c.2041G > A (p.A681T) was detected in a 41-year-old patient (II:1, Fig. 1j) of Urdu speaking background, who was diagnosed with carcinoma of the rectum. His sister (II:2, Fig. 1j) was diagnosed with a brain tumor (Table 4). The MSH2in-frame deletion (c.1786_1788delAAT) was identified in a 39-year-old CRC patient (II:1, Fig. 1k) of Punjabi ethnicity with a family history of breast cancer.
Other MMR gene variants: novel or previously reported
In addition to the pathogenic/likely pathogenic variants, 35 distinct MMR variants including nine novel and 26 previously reported variants were detected. Among these were eight missense variants, six silent variants, and 21 intronic variants (Table 2).
The novel variants were analyzed for their potential functional effect by in silico analyses (Table 5). A novel MLH1splice-site variant, (c.116 + 3A > T), is predicted to be the likely pathogenic as suggested by four of the five splice-site prediction algorithms integrated into the Alamut software implying that this is disease-causative. This variant was identified in a 30-year-old patient of Punjabi origin, diagnosed with carcinoma of the sigmoid colon with no family history (Table 4). This variant was not found in 100 healthy controls, further supporting its pathogenicity.
A novel MSH2 missense variant, c.2120G > A (p.C707Y), is also predicted to be a likely pathogenic as suggested by five of the seven in silico prediction tools (Table 5). This variant was identified in three unrelated patients with CRC diagnosed at or below age 54: one patient of Pathan ethnicity reported a family history of HNPCC and two Punjabi patients of the non-HNPCC group (Table 4). Moreover, this variant was found in two out of 100 healthy controls including one with a family history of carcinoma of the pharynx and Ewing’s sarcoma. Characteristics of families harboring pathogenic/likely pathogenic MLH1/MSH2 variants are shown in Table 4. The remaining seven novel MMR gene variants were also analyzed for their potential functional effect by in silico analyses and classified as benign.
Among the 26 previously reported MMR gene variants, 25 were benign or likely benign (Table 2). One MLH1 missense variant, c.1919C > T (p.P640L), is predicted to be likely pathogenic as suggested by all seven in silico prediction tools used (Table 5). We identified this variant in eight unrelated CRC patients of Pathan ethnicity: six from the HNPCC/suspected-HNPCC group and two from the non-HNPCC group.
Patient and tumor characteristics by variant status
The index CRC patients with pathogenic/likely pathogenic MLH1/MSH2 variants (n = 11) and without pathogenic variants (n = 199) had a same median age of diagnosis, 43 years (range 32–61) and 43 years (range 14–77) of age, respectively (P = 0.74, Wilcoxon rank-sum test). The patients with pathogenic/likely pathogenic variants were more likely to present with proximal tumors (6/11, 54.5% vs. 26/194, 13.4%; P = 0.004) and greater tumor size (> 5 cm) (6/8, 75% vs. 28/87, 32.2%; P = 0.02) than non-carriers. No differences were detected between the carriers and non-carriers with regard to histologic type, mucinous component, macroscopic appearance, grade of malignancy, lymphovascular invasion, venous invasion, tumor stage, regional lymph node involvement and ethnic groups (data not shown).
Discussion
In this first comprehensive study from Pakistan, we investigated the contribution of MLH1, MSH2, and MSH6 pathogenic germline variants to 212 patients belonging to HNPCC/suspected-HNPCC group or non-HNPCC group. Initially, index patients from the HNPCC/suspected-HNPCC group (including HNPCC = 9 and suspected-HNPCC = 20; group 1) were screened for the entire coding sequence of these genes. The pathogenic/likely pathogenic variants identified in this group were then analyzed in the non-HNPCC group (n = 183; group 2). Eight different pathogenic/likely pathogenic variants in MLH1/MSH2 were identified, with an overall frequency of 34.5% (10/29) in group 1 and 1.1% (2/183) in group 2. No pathogenic variants were detected in the MSH6 gene. Among the group 1, five pathogenic MLH1/MSH2 variants were detected in each subgroup of HNPCC and suspected-HNPCC, with frequencies of 55.6% (5/9) and 25% (5/20), respectively. The stringent criteria of HNPCC are two times more sensitive for detection of a pathogenic variant than the less stringent criteria of suspected-HNPCC. Our findings are in agreement with an international collaborative study reporting pathogenic variant detection rates of 50% (109/217) and 26% (32/123) for HNPCC and suspected-HNPCC criteria, respectively [20]. In our study, one in two patients identified with pathogenic variant did not meet the criteria of HNPCC, suggesting the need to use the criteria of suspected-HNPCC in Pakistani population.
Of the identified distinct pathogenic/likely pathogenic MLH1/MSH2 variants (n = 8) in both groups, the MSH2 variant, c.2656G > T, is likely to be specific to the Pakistani population as it has not been reported in other populations. The other seven variants have been reported in Asia, Europe, and North America [3, 30,31,32,33,34,35,36,37]. These findings suggest that the spectrum of MLH1/MSH2 variants in Pakistan does not differ from other populations.
In the current study three distinct recurrent pathogenic/likely pathogenic variants in MLH1 (n = 2) and MSH2 (n = 1) were identified. The likely pathogenic MSH2 variant, c.943-1G > C, was identified in three unrelated HNPCC/suspected-HNPCC families of Pathan ethnicity. It was also frequently reported in HNPCC families from Germany [33]. The pathogenic MLH1 variant, c.1358dup, was found in two unrelated HNPCC families of Punjabi origin. This variant was recently found in HNPCC families from Australia [36]. The pathogenic MLH1 variant, c.2041G > A, was detected in two unrelated suspected-HNPCC or non-HNPCC families of Punjabi and Urdu-speaking background, respectively. This variant was first reported in Poland as a potential founder variant [4, 31], has been reported as a recurrent variant in Scotland [30] and has also been described once each in Germany [33], and Colombia [3]. These recurrent variants accounted for 58.3% (7/12) of all MLH1/MSH2 carriers from Pakistan. This further suggests a step-wise and cost-effective strategy of screening these recurrent variants, prior to the exhaustive analyses of MMR genes in our population. However, haplotype analysis of these recurrent variants is required to classify these as true Pakistani founder variants.
In addition to eight pathogenic/likely pathogenic variants found in twelve families, 35 MMR gene variants were detected: nine novel and 26 previously reported sequence variants. Of the novel sequence variants, two were suggested as in silico predicted likely pathogenic variants. The novel MLH1splice-site variant, c.116 + 3A > T, is predicted to be likely pathogenic as suggested by four of the five splice-site prediction algorithms. This variant was identified in a CRC patient of the non-HNPCC group and was not detected in 100 healthy controls. Further evidence of the impact of c.116 + 3A > T variant on aberrant mRNA splicing could not be provided because of the unavailability of an RNA sample from this patient. The novel MSH2 missense variant, p.C707Y, is predicted to be likely pathogenic on the basis of the effect on protein function predicted by five of the seven in silico prediction tools. This variant was identified in three unrelated patients, one belonged to HNPCC group and other two were from the non-HNPCC group. It is located in the highly conserved ATPase domain (amino acid residues 620 to 855), may disrupt interaction of MSH2 with other proteins in repair pathway and result in MMR defect [38]. This variant was detected in two out of 100 healthy controls with a family history of carcinoma of the pharynx or Ewing’s sarcoma. Functional analyses of both in silico predicted likely pathogenic novel variants (MLH1 c.116 + 3A > T and MSH2 p.C707Y) are warranted to further establish the association of these variants with the disease. One previously reported MLH1 missense variant, p.P640L, is a likely pathogenic variant as predicted by seven in silico prediction tools used. This variant was identified in eight unrelated CRC patients of Pathan origin: six belonged to the HNPCC/suspected-HNPCC group while the other two were from the non-HNPCC group. This variant is located in a highly conserved C-terminal interaction domain (amino acid residues 492 to 756) and may ablate interaction of MLH1 with PMS2 and result in the MMR defect. Previously, Hardt and colleagues performed two functional assays and characterized p.P640L as a pathogenic variant [29]. Overall, these findings suggest that MLH1 p.P640L is deemed to be a pathogenic variant.
In the current study, pathogenic/likely pathogenic MLH1/MSH2 variants were identified in 34.5% (10/29) of Pakistani HNPCC/suspected-HNPCC patients, which is in agreement with other Asian studies from Korea (54/188; 28.7%), China (7/23; 30.4%), and Singapore (17/59; 28.8%) [39,40,41], Poland (78/226; 34.5%) [32], US (26/71; 36.6%) [42], and Brazil (44/116; 38%) [5]. A higher frequency of pathogenic variants was observed in HNPCC families from Taiwan (82/135; 60.7%) [43]. This could be due to screening of families who only met Amsterdam II or HNPCC criteria, whereas in this study we have also screened families who met the less stringent criteria of suspected-HNPCC. No pathogenic variant in MSH6 was detected in the present study, in agreement with studies from China [44], and Singapore [40], suggesting a minimal contribution of MSH6 variants in Asia. The predominance of pathogenic MLH1/MSH2 variants and absence of MSH6 variant in Pakistani population are in line with other ethnic mutation database [45]. These findings suggest that the contribution of pathogenic MMR gene variants to HNPCC/suspected-HNPCC families varies in Asians as well as in other populations.
Several criteria have been reported for the identification of potential candidates for the detection of pathogenic MMR gene variant. The most stringent and commonly applied Amsterdam II criteria [14, 15] is based on a family history of at least three relatives with histologically verified CRC or cancers linked with HNPCC. In our study, five out of nine patients belonging to families fulfilling this criterion were found to harbor a pathogenic MLH1/MSH2 variant (5/9; 55.6%). The revised Bethesda guidelines recognize high-risk patients by the assessment of microsatellite instability and/or immunohistochemical testing of their tumors. However, this approach was not utilized due to limitations of normal/tumor tissue of study subjects. Nevertheless, the Amsterdam II criteria and Bethesda guidelines are shown to miss up to 72 and 27% of cases with HNPCC, respectively [17]. A recently suggested less stringent criteria of suspected-HNPCC are based on a family history of only two HNPCC-associated cancers [18,19,20]. In our study, five out of 20 patients belonging to families fulfilling this criterion were found to harbor a pathogenic MLH1/MSH2 variant (5/20; 25%). Of the identified twelve carriers of pathogenic/likely pathogenic variant, five carriers met the HNPCC criteria and five met the suspected-HNPCC criteria and only two carriers were found in the non-HNPCC group. Our data support the notion that the suspected-HNPCC criteria may be useful for the identification of Pakistani families. The suspected-HNPCC criteria have also been utilized in other studies from Turkey, Poland, Italy and Latvia [31, 32, 37, 46].
In the current study, the frequency of pathogenic MMR gene variants observed in HNPCC/suspected-HNPCC group may be an underestimate as the sensitivity of DHPLC can be below 100% and screening for large genomic rearrangements or EPCAM gene 3′ end deletions was not performed. Furthermore, PMS2 mutation screening was not performed. It is possible that we could have missed PMS2 variants. However, pathogenic PMS2 variants have only rarely been reported and accounted for less than 5% of all identified pathogenic MMR gene variants [7]. Finally, the contribution of additional undiscovered gene(s) in early onset CRC patients with a family history of LS-associated cancer who tested negative for any pathogenic MMR gene variants cannot be excluded. Thus, further studies in these patients are warranted.
Ethnic variations in frequencies of pathogenic MLH1/MSH2 variant carriers have been reported in selected HNPCC families from Europe and US [21,22,23]. Similar ethnic variations in carrier frequencies of pathogenic/likely pathogenic MLH1/MSH2 variants have been noted in our study. Of the identified variants, the majority of the families carrying MLH1 variants (3/6; 50%) belonged to the Punjabi ethnicity. Majority of the families harboring pathogenic/likely pathogenic MSH2 variants (4/5; 80%) had a Pathan background. These findings suggest that families with Punjabi or Pathan background should be first screened for the MLH1 or MSH2 gene, respectively. However, no firm conclusion could be made due to a small number of pathogenic MLH1/MSH2 variant carriers. Furthermore, this study is not population-based and therefore might have some ascertainment bias.
Previous studies in Caucasians have predominantly reported the proximal tumor location in CRC patients harboring pathogenic MMR gene variants [47]. Similarly, in our study, CRC patients with pathogenic/likely pathogenic MLH1/MSH2 variants more commonly presented with proximal tumor location compared to non-carriers. Similar observations have been noted in other Asian studies from Singapore [40], and Japan [48]. However, no such association was reported in studies from Korea [39] and China [49]. The differences in phenotypic manifestation may be due to ethnic variations or involvement of other genetic and/or non-genetic risk factors.
Conclusion
In summary, this is the first comprehensive study conducted in Pakistani CRC patients to assess the prevalence and spectrum of MLH1, MSH2, and MSH6 pathogenic germline variants. Pathogenic/likely pathogenic MLH1/MSH2 variants account for a substantial proportion (10/29; 34.5%) of CRC patients with HNPCC/suspected-HNPCC in Pakistan, whereas no pathogenic MSH6 variants were seen. Three recurrent MLH1/MSH2 variants accounted for 58.3% (7/12) of all families carrying pathogenic/likely pathogenic variants. We recommend that HNPCC families, even those fulfilling the less stringent criteria of suspected-HNPCC, should first be tested for the recurrent pathogenic/likely pathogenic MLH1/MSH2 variants prior to whole gene screening in Pakistani patients.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Abbreviations
- CRC:
-
Colorectal cancer
- DHPLC:
-
Denaturing high-performance liquid chromatography
- EC:
-
Endometrial cancer
- EPCAM :
-
Epithelial cell adhesion molecule
- FAP:
-
Familial adenomatous polyposis
- FCCTX:
-
Familial colorectal cancer type X
- HNPCC:
-
Hereditary non-polyposis colorectal cancer
- IRB:
-
Institutional Review Board
- LS:
-
Lynch syndrome
- MLH1 :
-
MutL Homolog 1
- MMR :
-
Mismatch repair
- MSH2 :
-
MutS Homolog 2
- MSH6 :
-
MutS Homolog 6
- PMS2 :
-
PMS1 Homolog 2
- SKMCH&RC:
-
Shaukat Khanum Memorial Cancer Hospital and Research Centre
References
Ferlay J, Soerjomataram I, Ervik M, Dikshit R, Eser S, Mathers C, et al. GLOBOCAN 2012. http://globocan.iarc.fr. Accessed 20 Jan 2018.
Dominguez-Valentin M, Therkildsen C, Da Silva S, Nilbert M. Familial colorectal cancer type X: genetic profiles and phenotypic features. Mod Pathol. 2015;28:30–6.
Espenschied CR, LaDuca H, Li S, McFarland R, Gau C-L, Hampel H. Multigene panel testing provides a new perspective on Lynch syndrome. J Clin Oncol. 2017;35:2568–75.
Kurzawski G, Safranow K, Suchy J, Chlubek D, Scott RJ, Lubiński J. Mutation analysis of MLH1 and MSH2 genes performed by denaturing high-performance liquid chromatography. J Biochem Biophys Methods. 2002;51:89–100.
da Silva FC, de Oliveira Ferreira JR, Torrezan GT, Figueiredo MCP, Santos ÉMM, Nakagawa WT, et al. Clinical and molecular characterization of Brazilian patients suspected to have Lynch syndrome. PLoS One. 2015;10:e0139753.
Silva FCC, Valentin MD, Ferreira FO, Carraro DM, Rossi BM. Mismatch repair genes in Lynch syndrome: a review. Sao Paulo Med J. 2009;127:46–51.
Tutlewska K, Lubinski J, Kurzawski G. Germline deletions in the EPCAM gene as a cause of Lynch syndrome–literature review. Hered Cancer Clin Pract. 2013;11:9.
Aarnio M, Mecklin JP, Aaltonen LA, Nyström-Lahti M, Järvinen HJ. Life-time risk of different cancers in hereditary non-polyposis colorectal cancer (HNPCC) syndrome. Int J Cancer. 1995;64:430–3.
Brosens LA, Offerhaus GJA, Giardiello FM. Hereditary colorectal cancer: genetics and screening. Surg Clin. 2015;95:1067–80.
Hampel H, Frankel WL, Martin E, Arnold M, Khanduja K, Kuebler P, et al. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer). N Engl J Med. 2005;352:1851–60.
Quehenberger F, Vasen H, Van Houwelingen H. Risk of colorectal and endometrial cancer for carriers of mutations of the hMLH1 and hMSH2 gene: correction for ascertainment. J Med Genet. 2005;42:491–6.
Watson P, Vasen HF, Mecklin JP, Bernstein I, Aarnio M, Järvinen HJ, et al. The risk of extra-colonic, extra-endometrial cancer in the Lynch syndrome. Int J Cancer. 2008;123:444–9.
Pearlman R, Frankel WL, Swanson B, Zhao W, Yilmaz A, Miller K, et al. Prevalence and spectrum of germline cancer susceptibility gene mutations among patients with early-onset colorectal cancer. JAMA Oncol. 2017;3:464–71.
Vasen H, Mecklin J, Khan P, Lynch H. The International Collaborative Group on Hereditary Non-Polyposis Colorectal Cancer (ICG-HNPCC). Dis Colon Rectum. 1991;34:424.
Vasen HF, Watson P, Mecklin JP, Lynch HT. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the international collaborative group on HNPCC. Gastroenterology. 1999;116:1453–6.
Shia J. Evolving approach and clinical significance of detecting DNA mismatch repair deficiency in colorectal carcinoma. Semin Diagn Pathol. 2015;32:352–61.
Palomaki GE, McClain MR, Melillo S, Hampel HL, Thibodeau SN. EGAPP supplementary evidence review: DNA testing strategies aimed at reducing morbidity and mortality from Lynch syndrome. Genet Med. 2009;11:42.
Kladny J, Lubiñski J. Lynch syndrome (HNPCC). Hered Cancer Clin Pract. 2008;6:99–102.
Park J-G, Vasen HF, Park KJ, Peltomaki P, De Leon MP, Rodriguez-Bigas MA, et al. Suspected hereditary nonpolyposis colorectal cancer. Dis Colon Rectum. 1999;42:710–5.
Park J-G, Vasen HF, Park Y, Park K, Peltomaki P, De Leon MP, et al. Suspected HNPCC and Amsterdam criteria II: evaluation of mutation detection rate, an international collaborative study. Int J Color Dis. 2002;17:109–14.
Caldes T, Godino J, De La Hoya M, Garcia Carbonero I, Perez Segura P, Eng C, et al. Prevalence of germline mutations of MLH1 and MSH2 in hereditary nonpolyposis colorectal cancer families from Spain. Int J Cancer. 2002;98:774–9.
Nyström-Lahti M, Wu Y, Moisio A-L, Hofstra RM, Osinga J, Mecklin J-P, et al. DNA mismatch repair gene mutations in 55 kindreds with verified or putative hereditary non-polyposis colorectal cancer. Hum Mol Genet. 1996;5:763–9.
Tannergård P, Lipford JR, Kolodner R, Frödin JE, Nordenskjöld M, Lindblom A. Mutation screening in the hMLH1 gene in Swedish hereditary nonpolyposis colon cancer families. Cancer Res. 1995;55:6092–6.
Rashid MU, Muzaffar M, Khan FA, Kabisch M, Muhammad N, Faiz S, et al. Association between the BsmI polymorphism in the vitamin D receptor gene and breast cancer risk: results from a Pakistani case-control study. PLoS One. 2015;10:e0141562.
Kolodner RD, Tytell JD, Schmeits JL, Kane MF, Gupta RD, Weger J, et al. Germ-line msh6 mutations in colorectal cancer families. Cancer Res. 1999;59:5068–74.
Rashid MU, Muhammad N, Faisal S, Amin A, Hamann U. Deleterious RAD51C germline mutations rarely predispose to breast and ovarian cancer in Pakistan. Breast Cancer Res Treat. 2014;145:775–84.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Rashid MU, Naeemi H, Muhammad N, Loya A, Yusuf MA, Lubiński J, et al. A novel deleterious c. 2656G> T MSH2 germline mutation in a Pakistani family with a phenotypic overlap of hereditary breast and ovarian cancer and Lynch syndrome. Hered Cancer Clin Pract. 2016;14:14.
Hardt K, Heick SB, Betz B, Goecke T, Yazdanparast H, Kuppers R, et al. Missense variants in hMLH1 identified in patients from the German HNPCC consortium and functional studies. Familial Cancer. 2011;10:273–84.
Barnetson RA, Tenesa A, Farrington SM, Nicholl ID, Cetnarskyj R, Porteous ME, et al. Identification and survival of carriers of mutations in DNA mismatch-repair genes in colon cancer. N Engl J Med. 2006;354:2751–63.
Dymerska D, Kurzawski G, Suchy J, Roomere H, Toome K, Metspalu A, et al. Lynch syndrome mutations shared by the Baltic States and Poland. Clin Genet. 2014;86:190–3.
Kurzawski G, Suchy J, Lener M, Kłujszo-Grabowska E, Kładny J, Safranow K, et al. Germline MSH2 and MLH1 mutational spectrum including large rearrangements in HNPCC families from Poland (update study). Clin Genet. 2006;69:40–7.
Mangold E, Pagenstecher C, Friedl W, Mathiak M, Buettner R, Engel C, et al. Spectrum and frequencies of mutations in MSH2 and MLH1 identified in 1,721 German families suspected of hereditary nonpolyposis colorectal cancer. Int J Cancer. 2005;116:692–702.
Nilbert M, Wikman FP, Hansen TV, Krarup HB, Örntoft TF, Nielsen FC, et al. Major contribution from recurrent alterations and MSH6 mutations in the Danish Lynch syndrome population. Familial Cancer. 2009;8:75–83.
Sheng J-Q, Zhang H, Ji M, Fu L, Mu H, Zhang M-Z, et al. Genetic diagnosis strategy of hereditary non-polyposis colorectal cancer. World J Gastroenterol: WJG. 2009;15:983.
Sjursen W, McPhillips M, Scott RJ, Talseth-Palmer BA. Lynch syndrome mutation spectrum in New South Wales, Australia, including 55 novel mutations. Mol Genet Genomic Med. 2016;4:223–31.
Pedroni M, Roncari B, Maffei S, Losi L, Scarselli A, Di Gregorio C, et al. A mononucleotide markers panel to identify hMLH1/hMSH2 germline mutations. Dis Markers. 2007;23:179–87.
Ollila S, Dermadi Bebek D, Jiricny J, Nyström M. Mechanisms of pathogenicity in human MSH2 missense mutants. Hum Mutat. 2008;29:1355–63.
Lee SY, Kim DW, Shin YK, Ihn MH, Lee SM, Oh HK, et al. Validation of prediction models for mismatch repair gene mutations in Koreans. Cancer Res Treat. 2016;48:668–75.
Liu Y, Chew MH, Goh XW, Tan SY, Loi CTT, Tan YM, et al. Systematic study on genetic and epimutational profile of a cohort of Amsterdam criteria-defined Lynch syndrome in Singapore. PLoS One. 2014;9:e94170.
Zhang J-X, Fu L, de Voer RM, Hahn M-M, Jin P, Lv C-X, et al. Candidate colorectal cancer predisposing gene variants in Chinese early-onset and familial cases. World J Gastroenterol: WJG. 2015;21:4136.
Mueller J, Gazzoli I, Bandipalliam P, Garber JE, Syngal S, Kolodner RD. Comprehensive molecular analysis of mismatch repair gene defects in suspected Lynch syndrome (hereditary nonpolyposis colorectal cancer) cases. Cancer Res. 2009;69:7053–61.
Kamiza AB, Hsieh L-L, Tang R, Chien H-T, Lai C-H, Chiu L-L, et al. Risk factors associated with colorectal cancer in a subset of patients with mutations in MLH1 and MSH2 in Taiwan fulfilling the Amsterdam II criteria for Lynch syndrome. PLoS One. 2015;10:e0130018.
Yan S-Y, Zhou X-Y, Du X, Zhang T-M, Lu Y-M, Cai S-J, et al. Three novel missense germline mutations in different exons of MSH6 gene in Chinese hereditary non-polyposis colorectal cancer families. World J Gastroenterol: WJG. 2007;13:5021.
Peltomäki P, Vasen H. Mutations associated with HNPCC predisposition—update of ICG-HNPCC/INSiGHT mutation database. Dis Markers. 2004;20:269–76.
Tunca B, Pedroni M, Cecener G, Egeli U, Borsi E, Zorluoglu A, et al. Analysis of mismatch repair gene mutations in Turkish HNPCC patients. Familial Cancer. 2010;9:365–76.
Cruz-Correa M, Diaz-Algorri Y, Pérez-Mayoral J, Suleiman-Suleiman W, del Mar G-PM, Bertrán C, et al. Clinical characterization and mutation spectrum in Caribbean Hispanic families with Lynch syndrome. Familial Cancer. 2015;14:415–25.
Suzuki O, Eguchi H, Chika N, Sakimoto T, Ishibashi K, Kumamoto K, et al. Prevalence and clinicopathologic/molecular characteristics of mismatch repair-deficient colorectal cancer in the under-50-year-old Japanese population. Surg Today. 2017;47:1135–46.
Hu F, Li D, Wang Y, Yao X, Zhang W, Liang J, et al. Novel DNA variants and mutation frequencies of hMLH1 and hMSH2 genes in colorectal cancer in the Northeast China population. PLoS One. 2013;8:e60233.
Acknowledgements
We are thankful to the index patients and healthy individuals for their participation in this study and to Saima Faisal for the recruitment of study subjects. We are also grateful to Grzegorz Kurzawski and Janina Suchy from the Department of Genetics and Pathology, Pomeranian Medical University, Poland for providing the sequences of MSH6 primers.
Funding
This work was supported by the Shaukat Khanum Memorial Cancer Hospital and Research Centre (SKMCH&RC), Lahore, Pakistan.
Author information
Authors and Affiliations
Contributions
MUR contributed to conception and design of the study, patient recruitment for colorectal cancer study and data acquisition. In addition, he was involved in data analysis, interpretation and in drafting and revising the manuscript. HN and NM performed the molecular analyses, contributed to data analysis, interpretation and manuscript writing. AL and MAY were involved in patient recruitment, clinical and pathological data acquisition and in critically reviewing the manuscript. JL and AJ contributed to design of the study, data analysis, and interpretation and in revising the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was approved by the Institutional Review Board (IRB) of the SKMCH&RC (IRB approval number SKMCH-CRC-001). All study participants were furnished with and signed an informed written consent.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Rashid, M.U., Naeemi, H., Muhammad, N. et al. Prevalence and spectrum of MLH1, MSH2, and MSH6 pathogenic germline variants in Pakistani colorectal cancer patients. Hered Cancer Clin Pract 17, 29 (2019). https://doi.org/10.1186/s13053-019-0128-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13053-019-0128-2