
Abstract
This study investigates the structural, electronic, and magnetic properties of pristine, hydrogenated, and oxygenated Cr2O3 monolayer honeycomb-kagome (HK) surface using first-principles calculations based on density functional theory (DFT). Molecular dynamics and phonon dispersion calculations demonstrate the thermal and dynamic stability of these systems, implying their feasibility for laboratory synthesis. The results revealed that the adsorption of hydrogen and oxygen atoms on the Cr2O3 monolayer surface exhibited negative adsorption energies, indicating that these structures are energetically favorable. Specifically, the adsorption of oxygen atoms transforms the initial half-metallic nature of the monolayer into semiconducting behavior. Moreover, the introduction of hydrogen and oxygen atoms considerably enhances the Curie temperature of the surface. These findings offer a practical approach for enhancing the electronic and magnetic properties of Cr2O3 monolayer HK surface, thereby facilitating their prospective application in spintronics.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Two-dimensional (2D) nanomaterials, including graphene, boron nitride, and transition metal disulfide compounds, have been extensively studied due to their excellent physical properties and promising applications in the next generation of nanoelectronic devices [1,2,3]. Scientists have been dedicated to creating and enhancing a diverse array of 2D materials, each with its unique physical properties [4,5,6,7,8]. In order to realize the practical application of 2D materials in the field of electronics and spintronics, it is crucial to have a large adjustable band gap and high Curie temperature [9, 10]. Researchers have made numerous attempts and efforts to engineer the band gap and induce magnetism in 2D materials, such as through the utilization of asymmetric grating [11], doping [12], and surface functionalization (including hydrogenation, fluorination, oxygenation, etc.) [13,14,15,16,17]. Indeed, chemical functionalization is a widely adopted and effective method to tailor and modify the electronic and magnetic properties of 2D materials [18,19,20]. For example, in graphene, functionalization with hydrogen, fluorine, or other elements can effectively open a band gap, transforming it from a semimetal to a semiconductor [21,22,23,24]. The hydrogenation process has also induced ferromagnetism in the graphene surface [25,26,27,28].
The successful experimental production of ultrathin Cr2O3 nanosheets through rapid thermal annealing has provided evidence for the viability of synthesizing 2D Cr2O3 [29]. Building on these findings, Hashmi et al. were motivated to study the stability, electronic, and magnetic properties of the Cr2O3 monolayer using first-principle calculations [30]. In their systematic investigation, Hashmi et al. revealed that the honeycomb-kagome lattice of the Cr2O3 monolayer is thermodynamically stable at room temperature. This unique structure exhibits a kagome band with an unusually large gap in one spin channel, making it a highly interesting system for further exploration. Additionally, they found that the Cr2O3 monolayer displays stable intrinsic Ising ferromagnetism, as evidenced by the magnetocrystalline anisotropy energy (MAE). The calculations of the magnetic anisotropy further unveiled a remarkable perpendicular MAE of approximately 0.9 meV, which exceeds the values reported for experimentally realized 2D layers [31,32,33,34]. Moreover, the simulations for the Curie temperature (Tc) indicated that the Cr2O3 monolayer exhibits a Tc estimated to be 185 K, which highlights its potential for magnetic applications even at relatively high temperatures. In addition to its intriguing magnetic properties, Hashmi et al. explored the spin transport properties of the Cr2O3 monolayer using the nonequilibrium Green's function method. Their investigation demonstrated that the Cr2O3 monolayer exhibits excellent spin filtering behavior, further enhancing its potential for spintronics applications.
This article is based on first-principles calculations and aims to investigate a method to improve the electronic, and magnetic properties of the Cr2O3 monolayer honeycomb-kagome surface. Specifically, we study the effect of hydrogen and oxygen atom adsorption on the electronic and magnetic properties of the Cr2O3 monolayer surface. We demonstrate that the electronic properties can vary in a wide range. In addition, the Curie temperature in the Cr2O3 monolayer can be significantly improved by the adsorption of hydrogen and oxygen atoms, which can be attributed to the interaction between direct exchange and superexchange interactions. These findings provide a promising path for tuning electronic properties and optimizing Curie temperature in Cr2O3 monolayers, bringing this material closer to potential applications in nanoelectronic and spintronic devices.
2 Method of calculation
In this study, the first-principles calculations were conducted using the Quantum Espresso (QE) electronic structure code [35, 36], which is a simulation package based on the density-functional theory (DFT) approach [37, 38]. The Kohn–Sham (KS) formulation [37, 38] within the plane-wave (PW) basis set was resolved to efficiently describe the electronic structure of the material. The calculation adopted the scalar-relativistic projector-augmented wave (PAW) method [39], utilizing pseudopotentials to treat the Cr-3d and O-2p electrons as valence orbitals. To find the most stable configuration of the materials, we performed complete optimization of the unit cell volume, shape, and atomic positions. The optimization process was carried out using the Broyden-Fletcher-Goldfarb-Shanno (BFGS) algorithm, which efficiently converges to the optimal geometry [40]. The kinetic energy cut-off for the valence orbitals was set to 60 Ry, determining the accuracy and efficiency of the wave function expansion. Furthermore, the Brillouin Zone (BZ) sampling used a 5 × 5 × 1 k-point grid. In addition, a smearing parameter of 0.01 Ry was considered for the electron population function, implementing the Methfessel-Paxton smearing technique to improve the convergence of certain electronic properties [41]. The cell relaxation process continued until the force on each atom reached a threshold of 1 × 10−6 eV Å−1 and the total energy was 1 × 10−5 eV Å−1. To investigate the electronic properties in detail, band structure, and projected density of states (PDOS) calculations were performed. Both self-consistent field (SCF) and non-self-consistent (NSCF) calculations were carried out using different Monkhorst–Pack mesh sizes for Brillouin Zone sampling 9 × 9 × 1 and 33 × 33 × 1, respectively.
To simulate an infinite 2D plane, we employed a periodic cell in both the x- and y-directions for our 2D models. Additionally, to prevent any interaction between adjacent layers, a vacuum spacing of 25 Å was introduced in the z-direction. This avoids any potential false interactions and ensures the accuracy of our calculations.
For the 3d electron systems, the presence of strong Coulomb interaction between the localized 3d electrons of transition metals can significantly affect their properties. To account for this interaction, the Hubbard correction term is often used. In this calculation, the effective Hubbard parameters U = 2.0 eV with J = 0.7 eV were employed to describe the 3d electrons of the Cr atom, as employed in a previous study [30]. The Heyd–Scuseria–Ernzerhof (HSE) hybrid exchange–correlation functional calculations were performed to obtain an accurate electronic band structure.
3 Results and discussion
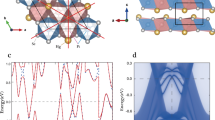
The study begins by conducting the optimization of the geometric structures of a pristine honeycomb-kagome Cr2O3 monolayer. In the original unit cell, there are two Cr atoms and three O atoms in one plane. The Cr atoms are bonded to three neighboring O atoms through covalent bonding, resulting in a Cr–O interatomic distance of approximately 1.79 Å. The hexagonal lattice parameters of the single layer have been optimized, with both the ɑ and ƅ parameters equal to 6.187 Å. These optimized values are found to be in good agreement with previous studies [30]. For completeness, Fig. 1 displays different views of the 3 × 3 × 1 supercell atomic structures of the pristine honeycomb-kagome Cr2O3, providing a visual representation of the arrangement of atoms in the material.

Atomic structure of the 3×3 Cr2O3 supercell monolayer. The area marked by black dashed lines indicates the unit cell
Figure 2 displays the electronic band structure along the high-symmetry points ΓKMΓ and the corresponding total density of state (DOS) of a pristine Cr2O3 monolayer. As shown, the monolayer Cr2O3 is a half-metallic 2D material with a large spin gap of ~ 4.1 eV at Γ-point and this also agrees with the previous result [30]. The observation of typical kagome band characteristics in the electronic band structure is indeed intriguing. Below the Fermi level, the band structure exhibits a linearly dispersive band that forms a Dirac point at the high symmetry K-point in the Brillouin zone. Around this Dirac point, two flat bands are seen to quadratically contact with the Dirac bands at the Γ-point, which are located both above and below the Fermi level. This specific arrangement of bands with Dirac points and flat bands at specific high symmetry points in the Brillouin zone is a hallmark feature of the Kagome band structure.

The electronic band structures and corresponding DOS of a pristine Cr2O3 monolayer. The spin-up bands are represented by the blue line and the spin-down bands are represented by the red line
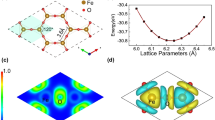
The investigation of the hydrogenation and oxygenation properties of the Cr2O3 monolayer is the main focus of our research. To begin our study, we examined the adsorption of hydrogen and oxygen on the Cr2O3 monolayer. In Fig. 3, we depict four distinct adsorption sites, namely top-on Cr (TCr), top-on O (TO), hollow (H), and bridge (B). These adsorption sites serve as crucial locations for the interaction between the Cr2O3 monolayer and hydrogen or oxygen atoms. By studying the behavior of hydrogen and oxygen adsorption at these sites, we can gain important insights into the reactivity and catalytic properties of the Cr2O3 monolayer.

The four different adsorption sites of a Cr2O3 monolayer surface
Therefore, we performed full surface relaxation for each adsorption site and calculated the adsorption energy (Ea) for each site using the relevant equation provided in the study.
where \(E_{{Cr_{2} O_{3} + n\left( {H/O} \right)}}\) is the total energy of the relaxed H/O-Cr2O3 system, \(E_{{Cr_{2} O_{3} }}\) is the total energy of pristine Cr2O3 layer, \(E_{{\left( {H/O} \right)}}\) is the total energy of an isolated H/O atom, and \(n\) is the total number of adatom H/O in the system. Figure 4 shows the adsorption energy (Ea) for an H/O adatom on the Cr2O3 surface for each site. Among the four possible adsorption sites for hydrogen atom, the top-on O (TO) adsorption became the most stable configuration. While for the oxygen atom, the top-on Cr (TCr) adsorption became the most stable configuration. Thus, we will focus on this adsorption configuration. Figure 5a–b shows the optimized atomic structure of a single H/O impurity adsorbed system on top of the O/Cr atom, respectively.

Adsorption energy in the four different adsorption sites of a Cr2O3 monolayer surface

Optimized atomic structure of 2 × 2 Cr2O3 supercell with a 1H-Cr2O3, and b 1O-Cr2O3 systems
3.1 Structure characterization
The adsorbed H/O atom is covalently bonded to one O/Cr atom and is located right above this atom. The O/Cr atom in the adsorption site extrudes out of the surface plane to form the σ H–O/O–Cr bond of distance d equal to 0.97 and 1.59 Å, respectively. We found that the bond lengths between the adsorptive O atom and its nearest neighbors were slightly increased to 2.00 Å while the adsorptive Cr atom and its nearest neighbors distance was 1.76 Å.
3.2 Dynamical and thermal stability
Structural stability is a primary requirement for two-dimensional materials in experimental preparation and applications. Therefore, the dynamic and thermal stability of the three different adsorption sites of a Cr2O3 monolayer surface were evaluated through calculations. Firstly, the phonon spectrum of the three different systems (1H-Cr2O3, 1O-Cr2O3, and 2O-Cr2O3) were calculated, as shown in Fig. 6. Throughout the entire Brillouin zone, no imaginary phonon modes were found, indicating its excellent dynamic stability. Consequently, these structures exhibit dynamic stability and can exist as independent two-dimensional crystals.

Vibrational phonon spectra of the a 1H-Cr2O3, b 1O-Cr2O3, and c 2O-Cr2O3 systems
Furthermore, Fig. 7 shows the fluctuations in the total energy of the three different systems during AIMD simulation at 300 K, as well as the crystal structure after an 8 ps simulation. The MD method was used for temperature control during the calculations, with a time step of 1 fs in the canonical ensemble. From Fig. 7, it can be observed that throughout the entire simulation, both the energy and system temperature fluctuations are small, and all atoms vibrate near their equilibrium positions, maintaining the two-dimensional periodic structure. These calculations indicate that all the different systems exhibit good thermal stability and are predicted to remain stable at room temperature. The corresponding atomic structures after the AIMD simulation are presented in Fig. 7c, f, and i.

AIMD simulations of the temperature (left panels), total energy (middle panels), and atomic structures after an AIMD simulation (right panels) period of 8 ps for (a, b, c) 1H-Cr2O3, (d, e, f) 1O-Cr2O3, and (g, h, i) 2O-Cr2O3 systems
3.3 Electronic structure
Figure 8a shows the electronic band structure of the hydrogenated Cr2O3 monolayer. As shown, the monolayer hydrogenated Cr2O3 is a half-metallic material with a large spin gap of ~ 4.18 eV at Γ-point. The exploration of electronic properties extends to the oxygenated Cr2O3 monolayer, as shown in Fig. 8b. In stark contrast to its hydrogenated counterpart, the band structure of the oxygenated monolayer takes on the form of a semiconductor. Notably, a narrow direct bandgap of 0.39 eV is distinctly present at the Γ-point. This alteration in electronic behavior upon oxygenation is a testament to the pivotal role played by oxygen in modulating the monolayer electronic properties.

The electronic band structures of the a 1H-Cr2O3, b 1O-Cr2O3, and c 2O-Cr2O3 systems using PBE + U and d 1H-Cr2O3, e 1O-Cr2O3, and f 2O-Cr2O3 systems using HSE
Furthermore, as we delve deeper into the influence of oxygenation, Fig. 8c illustrates the electronic band structure of the oxygenated Cr2O3 monolayer, featuring the presence of two oxygen atoms. Notably, the bandgap is observed to expand to 0.65 eV, discernible at the K-point. This observation underscores the dynamic interplay between oxygen content and the material bandgap, offering intriguing prospects for the tailoring of its electronic properties.
In the HSE calculations, the band gap values for the 1O-Cr2O3 and 2O-Cr2O3 systems are 0.43 eV and 0.72 eV, respectively, and they closely align with the values obtained using the PBE + U method.
In the analysis of the projected density of states (PDOS), we gain valuable insights into the orbital contributions at the Fermi level (EF) within the Cr2O3 monolayer. The result reveals that at EF, the principal contributions originate from the d-orbitals of the Cr atoms and the p-orbitals of the O atoms. Notably, the d-orbitals of the Cr atom significantly outweigh the contribution of the p-orbitals of the O atoms, as shown in Fig. 9a. This dominance of Cr d-orbitals at the EF is a key factor in maintaining the half-metallic behavior observed in the electronic band structure. Interestingly, when exploring the influence of hydrogen and oxygen adatoms, we find that hydrogen adatoms do not contribute significantly through their s-orbital, and there is no noteworthy change in the electronic band structure, which continues to exhibit half-metallic behavior, as shown in Fig. 9b.

Partial density of states (PDOS) of a Cr2O3, b 1H-Cr2O3, c 1O-Cr2O3, and d 2O-Cr2O3 systems
In contrast, oxygen adatoms attached to Cr atoms introduce a distinct behavior. The electronic band structure changes from its initial half-metallic state to a semiconducting nature, featuring a small direct band gap of 0.39 eV. This change can be understood by considering the contribution of the p-orbitals originating from the oxygen adatoms at the EF, as shown in Fig. 9c. With the introduction of two oxygen adatoms on the Cr2O3 surface, this band gap further increases to 0.65 eV. This is due to the elevated dominance of the p-orbitals from the two oxygen adatoms, which contribute significantly to both the p-orbitals of oxygen atoms and the d-orbitals of Cr atoms at the EF, as shown in Fig. 9d.
These observations highlight the significant role of oxygen adatoms in tailoring the electronic properties of the Cr2O3 monolayer, demonstrating the potential for precise band gap engineering in this material.
3.4 Magnetic properties
This section aims to study the magnetic properties of the pristine, hydrogenated, and oxygenated Cr2O3 monolayer by investigating the magnetic state, and Curie temperature. To calculate the magnetic state, we perform spin-polarized DFT calculations for a 2 × 2 supercell of ferromagnetic (FM) and various antiferromagnetic (AFM) phases, such as AFM Néel (AFM-N), AFM-zigzag (AFM-ZZ), and AFM-stripy (AFM-SR), respectively, for Cr elements, as shown in Fig. 10. The exchange energy, Eex, is calculated using the formula Eex = (EAFM − EFM). The results reveal that the FM configuration is the most energetically favorable magnetic state and the next higher energy one is AFM-N for all systems. The results of the calculation are presented in Table 1, which lists the exchange energy (Eex) for each system.

Schematics of different magnetic configurations
The Curie temperature is estimated using the VAMPIRE software package [42, 43] and a magnetization curve obtained from Metropolis Monte Carlo (MC) simulations [26]. The Hamiltonian describes the magnetic energy of the system and is a sum of all possible pairs of magnetic moments. It was described using the Heisenberg model with the equation:
The first term represents the exchange interaction, where J is the magnetic exchange coupling constant and \(\hat{m}_{i}\) and \(\hat{m}_{j}\) are the magnetic moments of atoms i and j, respectively. The dot product between \(\hat{m}_{i}\) and \(\hat{m}_{j}\) represents the interaction between the two magnetic moments. The magnetic exchange coupling constant J was obtained using the formula \(J = \frac{{E_{ex} }}{{Nm^{2} }}\), where Eex is the exchange energy, N is the number of magnetic atoms per unit cell, and m is the magnitude of the magnetic moment. The simulation was performed using 50 × 50 supercells, which were considered sufficiently large to minimize the impact of periodic boundary conditions. In this simulation, the temperature-dependent magnetization curves were calculated using 60,000 equilibration steps and 60,000 averaging steps with critical damping (λ = 1) and a temperature step interval of 0.2 K. Subsequently, the obtained magnetization curves were fitted by using the Curie-Bloch equation in the classical limit, expressed as \(m\left( T \right) = \left[ {1 - \frac{T}{{T_{c} }}} \right]^{\beta }\) (represented by the solid red line in Fig. 11) where T is the temperature and Tc represents the Curie temperature. The calculated critical exponent was determined to be β = 0.48.

Magnetization curve for a Cr2O3, b 1H-Cr2O3, c 1O-Cr2O3, and d 2O-Cr2O3 systems
The magnetic moment variations of pristine, hydrogenated, and oxygenated Cr2O3 monolayer as a function of temperature were calculated using MC simulations. The results of average magnetic moments as a function of temperature are shown in Fig. 11. As shown in the results, the magnetic moment curves decrease as the temperature increases, dropping to near zero at a certain temperature, referred to as the Curie temperature. The estimated Tc for the different monolayers is listed in Table 1, with values of 170, 410, 277, and 380 K for pristine Cr2O3, 1H-Cr2O3, 1O-Cr2O3, and 2O-Cr2O3 systems, respectively. To confirm the accuracy of the MC calculations, we calculated the Tc of the CrI3 monolayer, which was found to be 46 K. This value is in excellent agreement with the experimental CrI3 monolayer Tc value of 45 K, thereby verifying the accuracy of our MC calculations.
4 Conclusions
In summary, our study provides insights into the structural, electronic, and magnetic properties of pristine, hydrogenated, and oxygenated Cr2O3 monolayer honeycomb-kagome (HK) surfaces. Our findings indicate that both hydrogenated and oxygenated Cr2O3 monolayers exhibit stability, as confirmed by the calculated phonon dispersion spectrum and AIMD simulations. The optimized structures are energetically favorable structures due to the negative adsorption energy. The results found that hydrogen adatoms preferentially adsorb on top of O atoms, while the oxygen adatoms preferentially adsorb on top of Cr atoms presenting much higher adsorption energy. The electronic properties can also be tuned by the oxygenation of the Cr2O3 monolayer, transforming from the half-metallic to the semiconducting nature. The band gap varies from 0 to 0.65 eV and can be achieved by the adsorption of two O atoms on the Cr2O3 monolayer surface. With the oxygenation of the Cr2O3 monolayer, the O atoms are strongly hybridized with the Cr atoms in the Cr2O3 surface due to which a band gap can be achieved. Interestingly, we have predicted that the intrinsic ferromagnetism and corresponding Curie temperature can be largely enhanced by the adsorption of H and O atoms. These theoretical findings present an innovative strategy to elevate the electronic and magnetic performance of Cr2O3 layers, with the potential for broader applications across the realm of advanced 2D materials.
Data Availability
No Data associated in the manuscript.
References
L.-P. Tang, L.-M. Tang, D. Wang, H.-X. Deng, K.-Q. Chen, Metal and ligand effects on the stability and electronic properties of crystalline two-dimensional metal-benzenehexathiolate coordination compounds. J. Phys. Condens. Matter 30, 465301 (2018). https://doi.org/10.1088/1361-648X/aae618
G. Xie, D. Ding, G. Zhang, Phonon coherence and its effect on thermal conductivity of nanostructures. Adv. Phys. X. 3, 1480417 (2018). https://doi.org/10.1080/23746149.2018.1480417
D. Wang, L.-M. Tang, X.-X. Jiang, J.-Y. Tan, M.-D. He, X.-J. Wang, K.-Q. Chen, High bipolar conductivity and robust in-plane spontaneous electric polarization in selenene. Adv. Electron. Mater. 5, 1800475 (2019). https://doi.org/10.1002/aelm.201800475
C. Liu, G. Zhao, T. Hu, Y. Chen, S. Cao, L. Bellaiche, W. Ren, Ferromagnetism, Jahn-Teller effect, and orbital order in the two-dimensional monolayer perovskite ${\mathrm{Rb}}_{2}{\mathrm{CuCl}}_{4}$. Phys. Rev. B 104, L241105 (2021). https://doi.org/10.1103/PhysRevB.104.L241105
B. Wu, Y. Song, W. Ji, P. Wang, S. Zhang, C. Zhang, Quantum anomalous Hall effect in an antiferromagnetic monolayer of MoO. Phys. Rev. B 107, 214419 (2023). https://doi.org/10.1103/PhysRevB.107.214419
H. Sun, S.-S. Li, W. Ji, C.-W. Zhang, Valley-dependent topological phase transition and quantum anomalous valley Hall effect in single-layer RuClBr. Phys. Rev. B 105, 195112 (2022). https://doi.org/10.1103/PhysRevB.105.195112
S. Zhang, C. Zhang, S. Zhang, W. Ji, P. Li, P. Wang, S. Li, S. Yan, Intrinsic Dirac half-metal and quantum anomalous Hall phase in a hexagonal metal-oxide lattice. Phys. Rev. B 96, 205433 (2017). https://doi.org/10.1103/PhysRevB.96.205433
K. Jia, X.-J. Dong, S.-S. Li, W.-X. Ji, C.-W. Zhang, Spontaneous valley polarization and valley-nonequilibrium quantum anomalous Hall effect in Janus monolayer ScBr I. Nanoscale 15, 8395–8405 (2023). https://doi.org/10.1039/D2NR07221A
F.H.L. Koppens, T. Mueller, P. Avouris, A.C. Ferrari, M.S. Vitiello, M. Polini, Photodetectors based on graphene, other two-dimensional materials and hybrid systems. Nat. Nanotechnol. 9, 780–793 (2014). https://doi.org/10.1038/nnano.2014.215
F. Schwierz, J. Pezoldt, R. Granzner, Two-dimensional materials and their prospects in transistor electronics. Nanoscale 7, 8261–8283 (2015). https://doi.org/10.1039/C5NR01052G
Y. Zhang, T.-T. Tang, C. Girit, Z. Hao, M.C. Martin, A. Zettl, M.F. Crommie, Y.R. Shen, F. Wang, Direct observation of a widely tunable bandgap in bilayer graphene. Nature 459, 820–823 (2009). https://doi.org/10.1038/nature08105
W. Zhang, C.-T. Lin, K.-K. Liu, T. Tite, C.-Y. Su, C.-H. Chang, Y.-H. Lee, C.-W. Chu, K.-H. Wei, J.-L. Kuo, L.-J. Li, Opening an electrical band gap of bilayer graphene with molecular doping. ACS Nano 5, 7517–7524 (2011). https://doi.org/10.1021/nn202463g
M.Z.S. Flores, P.A.S. Autreto, S.B. Legoas, D.S. Galvao, Graphene to graphane: a theoretical study. Nanotechnology 20, 465704 (2009). https://doi.org/10.1088/0957-4484/20/46/465704
J.O. Sofo, A.S. Chaudhari, G.D. Barber, Graphane: a two-dimensional hydrocarbon. Phys. Rev. B 75, 153401 (2007). https://doi.org/10.1103/PhysRevB.75.153401
M. Moaied, Y.S. Lim, J. Hong, Hydrogenated black phosphorus single layer. Phys. E Low-Dimens. Syst. Nanostructures. 104, 333–339 (2018). https://doi.org/10.1016/j.physe.2018.07.013
M.U. Farooq, I. Khan, M. Moaied, J. Hong, Hydrogen functionalization induced two-dimensional ferromagnetic semiconductor in Mn di-halide systems. Phys. Chem. Chem. Phys. 19, 29516–29524 (2017). https://doi.org/10.1039/C7CP05732F
M. Moaied, S. Soliman, Two-dimensional semiconductors of Cr–X3–H3 (X = O, S, Se, and Te) structures with large magnetic anisotropy and high Curie temperature. Phys. Scr. 97, 125813 (2022). https://doi.org/10.1088/1402-4896/ac9d71
X. Ma, W. Mi, Surface functionalization tailored electronic structure and magnetic properties of two-dimensional CrC2 monolayers. J. Phys. Chem. C 124, 3095–3106 (2020). https://doi.org/10.1021/acs.jpcc.9b10598
S.N. Reed-Lingenfelter, M. Wang, N.L. Williams, J.J. Cha, Surface functionalization for magnetic property tuning of nonmagnetic 2D materials. Adv. Mater. Interfaces 9, 2100463 (2022). https://doi.org/10.1002/admi.202100463
L. Daukiya, J. Seibel, S. De Feyter, Chemical modification of 2D materials using molecules and assemblies of molecules. Adv. Phys. X. 4, 1625723 (2019). https://doi.org/10.1080/23746149.2019.1625723
H. Gao, L. Wang, J. Zhao, F. Ding, J. Lu, Band gap tuning of hydrogenated graphene: H coverage and configuration dependence J. Phys. Chem. C 115, 3236–3242 (2011). https://doi.org/10.1021/jp1094454
V. Georgakilas, M. Otyepka, A.B. Bourlinos, V. Chandra, N. Kim, K.C. Kemp, P. Hobza, R. Zboril, K.S. Kim, Functionalization of graphene: Covalent and non-covalent approaches, derivatives and applications. Chem. Rev. 112, 6156–6214 (2012). https://doi.org/10.1021/cr3000412
J.H. Jørgensen, A.G. Čabo, R. Balog, L. Kyhl, M.N. Groves, A.M. Cassidy, A. Bruix, M. Bianchi, M. Dendzik, M.A. Arman, L. Lammich, J.I. Pascual, J. Knudsen, B. Hammer, P. Hofmann, L. Hornekaer, Symmetry-driven band gap engineering in hydrogen functionalized graphene. ACS Nano 10, 10798–10807 (2016). https://doi.org/10.1021/acsnano.6b04671
F. Withers, S. Russo, M. Dubois, M.F. Craciun, Tuning the electronic transport properties of graphene through functionalisation with fluorine. Nanoscale Res. Lett. 6, 526 (2011). https://doi.org/10.1186/1556-276X-6-526
H. González-Herrero, J.M. Gómez-Rodríguez, P. Mallet, M. Moaied, J.J. Palacios, C. Salgado, M.M. Ugeda, J.-Y. Veuillen, F. Yndurain, I. Brihuega, Atomic-scale control of graphene magnetism by using hydrogen atoms. Science 352, 437–441 (2016). https://doi.org/10.1126/science.aad8038
M. Moaied, J.V. Alvarez, J.J. Palacios, Hydrogenation-induced ferromagnetism on graphite surfaces. Phys. Rev. B 90, 115441 (2014). https://doi.org/10.1103/PhysRevB.90.115441
M. Moaied, J.A. Moreno, M.J. Caturla, F. Ynduráin, J.J. Palacios, Theoretical study of the dynamics of atomic hydrogen adsorbed on graphene multilayers. Phys. Rev. B 91, 155419 (2015). https://doi.org/10.1103/PhysRevB.91.155419
M. Moaied, J. Hong, Tuning the magnetic properties of hydrogenated bilayer graphene and graphene/h-BN heterostructures by compressive pressures. Carbon 131, 266–274 (2018). https://doi.org/10.1016/j.carbon.2018.01.102
C. Zhao, H. Zhang, W. Si, H. Wu, Mass production of two-dimensional oxides by rapid heating of hydrous chlorides. Nat. Commun. 7, 12543 (2016). https://doi.org/10.1038/ncomms12543
A. Hashmi, K. Nakanishi, M.U. Farooq, T. Ono, Ising ferromagnetism and robust half-metallicity in two-dimensional honeycomb-kagome Cr2O3 layer. Npj 2D Mater. Appl. 4, 1–8 (2020). https://doi.org/10.1038/s41699-020-00174-0
B. Huang, G. Clark, E. Navarro-Moratalla, D.R. Klein, R. Cheng, K.L. Seyler, D. Zhong, E. Schmidgall, M.A. McGuire, D.H. Cobden, W. Yao, D. Xiao, P. Jarillo-Herrero, X. Xu, Layer-dependent ferromagnetism in a van der Waals crystal down to the monolayer limit. Nature 546, 270–273 (2017). https://doi.org/10.1038/nature22391
C. Gong, L. Li, Z. Li, H. Ji, A. Stern, Y. Xia, T. Cao, W. Bao, C. Wang, Y. Wang, Z.Q. Qiu, R.J. Cava, S.G. Louie, J. Xia, X. Zhang, Discovery of intrinsic ferromagnetism in two-dimensional van der Waals crystals. Nature 546, 265–269 (2017). https://doi.org/10.1038/nature22060
H.L. Zhuang, Y. Xie, P.R.C. Kent, P. Ganesh, Computational discovery of ferromagnetic semiconducting single-layer ${\mathrm{CrSnTe}}_{3}$. Phys. Rev. B 92, 035407 (2015). https://doi.org/10.1103/PhysRevB.92.035407
M. Moaied, J. Lee, J. Hong, A 2D ferromagnetic semiconductor in monolayer Cr-trihalide and its Janus structures. Phys. Chem. Chem. Phys. 20, 21755–21763 (2018). https://doi.org/10.1039/C8CP03489C
P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G.L. Chiarotti, M. Cococcioni, I. Dabo, A.D. Corso, S. de Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A.P. Seitsonen, A. Smogunov, P. Umari, R.M. Wentzcovitch, QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 21, 395502 (2009). https://doi.org/10.1088/0953-8984/21/39/395502
P. Giannozzi, O. Andreussi, T. Brumme, O. Bunau, M. Buongiorno Nardelli, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, M. Cococcioni, N. Colonna, I. Carnimeo, A. Dal Corso, S. de Gironcoli, P. Delugas, R.A. DiStasio, A. Ferretti, A. Floris, G. Fratesi, G. Fugallo, R. Gebauer, U. Gerstmann, F. Giustino, T. Gorni, J. Jia, M. Kawamura, H.-Y. Ko, A. Kokalj, E. Küçükbenli, M. Lazzeri, M. Marsili, N. Marzari, F. Mauri, N.L. Nguyen, H.-V. Nguyen, A. Otero-de-la-Roza, L. Paulatto, S. Poncé, D. Rocca, R. Sabatini, B. Santra, M. Schlipf, A.P. Seitsonen, A. Smogunov, I. Timrov, T. Thonhauser, P. Umari, N. Vast, X. Wu, S. Baroni, Advanced capabilities for materials modelling with Quantum ESPRESSO, J. Phys. Condens. Matter Inst. Phys. J. 29, 465901 (2017). https://doi.org/10.1088/1361-648X/aa8f79.
P. Hohenberg, W. Kohn, Inhomogeneous electron gas. Phys. Rev. 136, B864–B871 (1964). https://doi.org/10.1103/PhysRev.136.B864
W. Kohn, L.J. Sham, Self-consistent equations including exchange and correlation effects. Phys. Rev. 140, A1133–A1138 (1965). https://doi.org/10.1103/PhysRev.140.A1133
P.E. Blöchl, Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994). https://doi.org/10.1103/PhysRevB.50.17953
D.C. Liu, J. Nocedal, On the limited memory BFGS method for large scale optimization. Math. Program. 45, 503–528 (1989). https://doi.org/10.1007/BF01589116
D. Vanderbilt, Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 41, 7892–7895 (1990). https://doi.org/10.1103/PhysRevB.41.7892
R.F.L. Evans, W.J. Fan, P. Chureemart, T.A. Ostler, M.O.A. Ellis, R.W. Chantrell, Atomistic spin model simulations of magnetic nanomaterials. J. Phys. Condens. Matter 26, 103202 (2014). https://doi.org/10.1088/0953-8984/26/10/103202
R.F.L. Evans, Vampire, (2016). http://vampire.york.ac.uk/.
Acknowledgements
The authors acknowledge the computational support provided by the Supercomputing Facility of the Bibliotheca Alexandrina, Egypt.
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
There are no conflicts to declare.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mohamed, N., Moaied, M. Enhancement of electronic and magnetic properties in Cr2O3 monolayer honeycomb-kagome by hydrogenation and oxygenation. Eur. Phys. J. Plus 138, 1075 (2023). https://doi.org/10.1140/epjp/s13360-023-04728-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1140/epjp/s13360-023-04728-1