
Abstract
This paper reports on the characterization of the stereo-dynamic controlling three different chemi-ionization reactions, recent objective of our study, since they participate to the balance of phenomena occurring in plasma, interstellar medium, planetary atmospheres, flames and lasers. The optical potential, obtained by a phenomenological method and defined in the whole space of the relative configurations of reagents, has been formulated in an accurate and internally consistent way for three different systems. Some cuts of the multidimensional potential, that asymptotically correlate with a specific fine level of the open shell atom and/or with a defined orientation of the molecular reagent, have been exploited in the present study to emphasize crucial features of the collision dynamics along selected entrance channels of the reactions. Consistently, basic quantities determining the topology of the reaction stereo-dynamics have been properly defined, emphasizing in the three cases relevant changes in the microscopic reaction evolution. Much attention focused on the selectivity of the orbital angular momentum, affecting each collision event at any chosen collision energy. It controls the relative weight of two different reaction mechanisms. The direct reaction mechanism is driven by short-range chemical forces, promoting, by direct electron transfer between reagents, a prototypical elementary oxidation reaction. The indirect mechanism, controlled by the combination of long-range chemical and physical forces, can be triggered by a virtual photon exchanged between reagents, promoting a sort of photo-ionization process. Obtained results and emphasized differences appear to be of general interest for many other elementary processes, more difficult to characterize at this level of detail.
Graphic abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Assessment and control of all quantities determining the stereo-dynamics of elementary processes, including single-step chemical reactions, represent an important challenge for the advanced research in fundamental and applied fields, since they provide the ground of the scientific development in many areas of the Chemistry and of the Matter Structure (see for instance Refs. [1,2,3,4,5,6]). At present, particular attention is devoted to basic phenomena occurring both at very low temperature (see for instance Refs. [7,8,9,10,11,12]), of interest for the quantum behavior of matter and for the balance of elementary processes occurring in interstellar media, and at room and higher temperature, of relevance for the chemistry and physics of planetary atmospheres and plasmas [13,14,15]. The final target of this challenge is the full characterization of all crucial effects (basic quantities), controlling, under a variety of conditions of interest, the stereo-dynamical evolution of each elementary process. In particular, determination, control and role of capture effects, probed by reagents and due to anisotropic long-range attractions, appear to be of great and general interest [16,17,18].
For most of elementary processes, the proper characterization of most of these quantities is very hard or even impossible to obtain in a straightforward way, since controlled by intermolecular interactions whose strength can fall even in the scale of few meV, a fraction of 1 kJ/mol (difficult to obtain with ab initio methods), and is masked by many other effects, operative especially under several bulk conditions. Accordingly, the attention must be addressed to the detailed investigation of some prototype systems, for which a large lot of experimental and theoretical information, crucial to this purpose, is available. The investigation of such systems, for which the interaction is known in detail, allows to obtain the characterization of the topology mentioned above and to extract basic general guidelines, permitting an extended assessment and exploitation of static and dynamical properties of matter under a variety of conditions.
The present study is stimulated by the recent investigation of some prototypical chemi-ionization reactions (CHEMI), an important class of bimolecular processes discovered several years ago by Penning [19], for which a lot of information here available allows a proper mapping of their stereo-dynamics [20,21,22,23,24,25]. CHEMI participate to the balance of phenomena occurring in the interstellar medium, planetary atmospheres and plasmas. Under specific conditions, CHEMI can also be identified as high exothermic elementary oxidation reactions. The latter often represent some intermediate elementary passages in chemical reactions occurring with a complex (multi-steps) mechanism.
Indeed, the general scheme of CHEMI is [26,27,28,29,30,31]
Usually, X* is an electronically excited noble gas atom Ng*, and M is an atomic/molecular partner. Ng* is formed in a high energetic electronic level, having metastable character, by collisions with energetic particles, such as electrons or cosmic rays, and it is exhibiting a lifetime sufficiently long to permit several collisions with M. In addition to emitted electrons, other reaction products are the associated ion NgM+, the parent (also called Penning) ion M+ and its fragmentation species. Because of the high energetic content of reagents, most efficient CHEMI are those promoted by metastable He*(3S1,1S0) and Ne*(3P2,0) atoms which reach with all molecules and most atoms available in nature.
In the following sections, it will be emphasized that promoted CHEMI can occur with two completely different reaction mechanisms, whose relative role depends on important aspects of the reaction topology. For the purpose of the present investigation, it is important to note that the Ne*(3PJ) reagent shows an outer electron in the 3s1 atomic orbital, while its internal 2p5 ionic core is iso-electronic of a high electron affinity fluorine atom. Moreover, the fine levels J = 2,0 of Ne*(3P) correlate, along the intermolecular electric field axis, to states showing a different alignment degree of the half-filled 2p orbital of its ionic core [23, 32].
As nuclear processes, also CHEMI are affected by an optical potential W [26,27,28,29,30,31,32], whose real part V controls the collision dynamics of reagents in the entrance channels, while the imaginary part Γ, also known as resonance width, triggers the passage from entrance to exit channels, that is, the state-to-state formation of ionic products from neutral reagents. Therefore, Γ defines the opacity of the system: its state-to-state selectivity and strength are often elusive even to most advanced theoretical methods.
In this study, we exploited three different CHEMI for which we have recently formulated W, in an internally consistent way both in its real and imaginary parts, triggering their state-to-state microscopic evolution. In particular, the phenomenological method, successfully applied in the last years to describe the dynamics of several elementary processes, has been here exploited the investigate basic features of the reactivity.
For the present purposes, we selected CHEMI involving Ne* + NH3, N2 and Ar as reagents for which the intermolecular forces involved and related collision dynamics have been tested, with high detail, on several experimental findings obtained in our and in other laboratories [20, 21, 23,24,25]. In particular, the combined analysis of absolute value and energy dependence of total and partial ionization cross sections, branching ratios and Penning ionization spectra of emitted electrons have been exploited. These observables probe complementary features of W.
Therefore, the proper characterization of the topology of the reaction stereo-dynamics, affecting such prototypical CHEMI, becomes here possible, exploiting specific cuts of their multidimensional W. It provides general guidelines permitting the control of many other elementary processes, including elementary oxidation and their inverse (reduction) reactions.
The basis of the phenomenological method is summarized in Sect. 2, fundamental quantities affecting the topology of the reaction stereo-dynamics are emphasized in the next Sect. 3, while Sect. 4 presents the detailed study of the three prototype CHEMI with all general information obtained. The discussion with some general conclusions is given in Sect. 5.
2 The phenomenological method
The leading interaction components, identified by the theory of the intermolecular forces and operative in several systems involving closed shell and open shell atoms, ions and molecules [33], have been accurately characterized by performing scattering experiments with the molecular beam technique. The analysis of the experimental observables suggested the phenomenological method, which adopts semi-empirical and empirical formulas [see ref. 34 and references therein] to provide the force fields in the whole space of the relative configurations of formed adducts. Such formulas represent the leading interaction components in terms of fundamental physical properties of the interacting partners. In some cases, the predictions of such method have been tested on results of ab initio calculations, permitting often improvements of the method itself.
Important applications of the phenomenological method were carried out in collaboration with the Bari group [35,36,37,38,39,40], and very recently, it has been adopted by the NASA, to investigate transport properties in environments involving systems of remarkable applied interest [41].
The present study exploits an accurate an internally consistent formulation of both parts of the optical potential, W, whose real component V controls the collision dynamics in the entrance channels, while the imaginary part Γ is determining the probability of state-to-state passage to the exit channels. Adopted W has been previously obtained [20, 23,24,25], and their formulation has been suggested by the complex phenomenology of open shell “P” atoms, investigated by advanced experimental and theoretical methods, by the behavior of ion-neutral systems, coupled by charge transfer, and by the spectroscopic properties of excimers. The obtained formulation has been tested on experimental findings of CHEMI, investigated in our and other laboratories by coupling scattering and spectroscopic techniques [21,22,23,24,25]. To emphasize innovative aspects of the reaction stereo-dynamics, we have found convenient to refer to particular geometries of precursor (or pre-reactive) state that opens specific reaction channels. However, in the analysis and tests on the experimental findings, the full space of the relative geometries of reagents has been considered. In the case of NH3 CHEMI, the considered geometry is the most relevant one promoting, within a selected angular cone [21], the formation of NH3+ ionic product in the ground (X).
3 Basic quantities affecting the reaction stereo-dynamics
The focus of the present study is on:
-
Capture efficiency, promoted by the long-range attraction, in determining the reaction precursor state formation and its modulation by the different approach geometry of reagents.
-
Role of the collision energy \({E}_{\mathrm{coll}}\);
-
Role of the orbital angular momentum defined by the quantum number ℓ that, in a classical picture of the collision dynamics, corresponds to the impact parameter b.
-
Combined effect of \({E}_{\mathrm{coll}}\) and ℓ.
-
Role of the centrifugal barrier, defined by ℓ, on the capture efficiency.
-
Range of the intermolecular distance R mainly probed during collision events of reagents occurring from sub-thermal up to hyper-thermal conditions.
-
Different reaction mechanisms and their selective dependence on all effects, quantities and parameters, indicated above.
We adopt a semi-classical treatment of the collision events [30, 31], occurring from thermal up to hyper-thermal conditions that properly account for all dynamical effects accompanying both approaches and remove of reagents and the formation of products. We are also conscious that quantum mechanical corrections, due to resonances as orbiting effects, observable with lighter systems, become relevant at very low \({E}_{\mathrm{coll}}\) (that is, under sub-thermal conditions), but all leading aspects of the reaction stereo-dynamics here obtained should be conservative (i.e., they are still accounted for, although at a semi-quantitative level) in suggesting the topology of interest for many other processes.
4 Detailed investigation of prototypical reactions
In this section, a detailed investigation of the stereo-dynamics of three different CHEMI is presented: It is stimulated by the results of recent our studies carried out in an internally consistent way for the three reactions [20, 21, 23,24,25]. Both real and imaginary parts of the optical potential have been formulated (see Sect. 2) by using a phenomenological approach [32, 34,35,36,37,38,39,40,41,42], that is, adopting empirical and semi-empirical formulas to represent strength and range and angular dependence of the leading interaction components involved. The two parts are found to be interdependent [23,24,25, 32, 43], and, as stressed above, the predictions of the phenomenological method have been tested on the experimental results obtained in our and other laboratories under single collision conditions with the molecular beam technique [20, 21, 23,24,25]. The critical comparison between predictions and experimental findings has also been exploited to improve the formulation of phenomenological method itself.
4.1 Microscopic CHEMI mechanisms
It has been recently proposed [23,24,25, 32, 43] that CHEMI occur through two complementary microscopic mechanisms: The direct mechanism (DM) triggered by short-range intermolecular forces of chemical nature; the indirect mechanism (IM) stimulated by the critical balance of intermolecular forces components operating at intermediate and long range and having both chemical and physical nature. Chemical forces manifest under a pronounced overlap between valence orbital of reagents and in most cases increase their role in scattering events occurring at high \({E}_{coll},\) probing short intermolecular distances. Under such conditions, a closer approach of reagents is favored, and CHEMI are confined in the behavior of high exothermic elementary oxidation reactions, since involving direct electron exchanges between valence orbital of reagents. Physical forces combine with weak chemical components operative at intermediate and large intermolecular distances, as those usually probed by scattering events occurring at low \({E}_{\mathrm{coll}}\). The critical balance of such forces promotes a different reaction mechanism, basically driven by energy transfer effects, as those associated to virtual photon exchanges between reagents. Under such conditions, CHEMI can be classified as photo-ionization processes. As stressed above, the target of the present study is to discover, for such favorable cases of CHEMI, further selectivity of general interest, that is, affecting the collision dynamics under a variety of conditions.
4.2 The crucial role of the interaction potential
According to expectations, some features of the interaction potential play a fundamental role in the evolution of elementary processes.
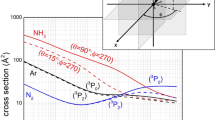
For Ne*(3PJ) + NH3, the radial dependence of the intermolecular interaction V in the entrance channels [20], associated to a specific cut of multidimensional potential energy surfaces (PESs) defining the real part of the optical potential, not including the explicit dependence on the spin orbit level J = 2,0 of Ne*(3P) reagent, is plotted in Fig. 1. In this case, the selected V controls the formation of the precursor state in its most stable C3v configuration, promoting the direct passage to ionic products involving NH3+ in its 2X ground electronic state. The same figure is also plotted the V components of the optical potentials referred to a specific entrance channel of the Ne*(3P0) + N2 (collinear approach) [25] and of Ne*(3P0) + Ar reactions [23]. They are promoted by particular alignment of the half-filled 2p orbital of Ne* ionic core (see below) and lead again to the formation of the ionic product in the ground electronic state. The comparison of the V components is important to emphasize the different capture efficiencies of the reagents in the selected channels of the three Ne* + Ar, N2 and NH3 CHEMI reactions.

The real part of the optical potential describing the interaction potential energy associated to specific cuts of PESs controlling the entrance channels of the three Ne* + Ar, N2 and NH3 CHEMI
The selective role of the centrifugal barrier depends on orbital angular momentum of the collision complex, defined by the quantum number ℓ. In particular, ℓ determines the centrifugal component \({V}_{c}\) of the interaction that accompanies any scattering event. The R dependence of the total potential \({V}_{t}\), defined as \({V}_{t}=V+{V}_{c},\) is plotted in Fig. 2. Note that \({V}_{c}\) is calculated using specific quantized ℓ values, while the V values are the same of Fig. 1.

The total interaction energy sum of the real V and of the centrifugal Vc component, defined as \({V}_{C}\)=\(\frac{{\hbar }^{2}}{2\mu }\)·\(\frac{\ell(\ell+1)}{{R}^{2}},\) where μ is the reduced mass of each system, evaluated at defined ℓ values. θ and ϕ are the polar orientation angles of molecular reagents. The different roles of the centrifugal barrier for the three systems are emphasized at a defined collision energy, Ecoll = 10 meV
In Fig. 2, the selective role of the centrifugal barrier, associated to the different \({V}_{t}\) for the three systems, is properly emphasized at \({E}_{\mathrm{coll}}\)=10 meV. Moreover, the imaginary Γ components, referred to the same three reaction channels [23,24,25], are plotted in the upper panel, Fig. 3.

Comparison between the different dependences on intermolecular distance R of Γ components determining the direct and indirect mechanisms. The overall ionization width for each investigated system is approximately given by the sum of the two correlated Γ-functions plotted [24]. Associated branching ratios for the three Ne* + Ar, N2 and NH3 CHEMI are also plotted in the lower panel as a function of R
In order to obtain a more consistent comparison, the relative role of \({\Gamma }_{\mathrm{DM}}\)/\({(\Gamma }_{\mathrm{IM}}+{\Gamma }_{\mathrm{DM}})\) and \({\Gamma }_{\mathrm{IM}}\)/\({(\Gamma }_{\mathrm{IM}}+{\Gamma }_{\mathrm{DM}})\) components (defined as branching ratio), triggering direct (DM) and indirect (IM) reaction mechanisms, is reported as a function of the intermolecular distance R in the lower part of the same Fig. 3.
4.3 The dependence of the collision dynamics on basic quantities
From the methodology adopted to represent W and to evaluate the collision dynamics, it has been possible to extract the dependence of some of the important quantities mentioned above on the fate of collision events occurring at selected Ecoll as a function of ℓ values along the selected entrance channels. In particular, it has been characterized:
-
The distance of closer approach \({R}_{c}\), where CHEMI have the highest probability of occurring, since here the system spends the highest time, corresponding to that required to invert the relative motion direction. The proper identification of Rc leads to defines, on quantitative ground, the relative role of DM and IM at each ℓ.
-
The probability of ionization \(P\left(b\right)\) at the related impact parameter b. Note that b depends linearly on ℓ, as quantified by the semiclassical relation
$$b \simeq \frac{\ell }{k}$$where \(k\) is the way number associated to each scattering event.
-
The partial cross section contribution defined as \(\pi {b}^{2}P\left(b\right)\);
The results obtained for the three systems at \({E}_{\mathrm{coll}}\)= 0.1, 1.0, 10, 100 and 1000 meV are plotted, respectively, in Figs. 4, 5, 6, 7 and 8 for an important comparison. The five values of \({E}_{\mathrm{coll}}\) have been chosen in order to cover the range from sub-thermal up to hyper-thermal collision energies. Plots so obtained permit to emphasize important differences between the three systems under various conditions of interest.

Comparison for the three Ne* + Ar, N2 and NH3 CHEMI between the distance Rc of closed approach, the reaction probability P(b) and the ionization cross section contribution πb2P(b) evaluated as a function of the orbital angular momentum quantum number ℓ at fixed \({E}_{\mathrm{coll}}\)=0.1 meV. Note that for such \({E}_{\mathrm{coll}}\) value, the semiclassical relation \(b \simeq \frac{\ell }{k}\) provides \(b\left( {\text{\AA}} \right) \simeq \frac{\ell }{0.6633}\) for ammonia, \(b\left( {\text{\AA}} \right) \simeq \frac{\ell }{0.7471}\) for N2 and \(b\left( {\text{\AA}} \right) \simeq \frac{\ell }{0.7987}\) for Ar

As shown in Fig. 4, at \(E_{{{\text{coll}}}}\)=1 meV. For such \(E_{coll}\) value, the semiclassical relation \(b \simeq \frac{\ell }{k}\) provides \(b\left( {\text{\AA}} \right) \simeq \frac{\ell }{2.097}\) for ammonia, \(b\left( {\text{\AA}} \right) \simeq \frac{\ell }{2.369}\) for N2 and \(b\left( {\text{\AA}} \right) \simeq \frac{\ell }{2.526}\) for Ar

As shown in Fig. 4, at \(E_{{{\text{coll}}}}\)=10 meV. For such \(E_{{{\text{coll}}}}\) value, the semiclassical relation \(b \simeq \frac{\ell }{k}\) provides \(b\left( {\text{\AA}} \right) \simeq \frac{\ell }{6.633}\) for ammonia, \(b\left( {\text{\AA}} \right) \simeq \frac{\ell }{7.471}\) for N2 and \(b\left( {\text{\AA}} \right) \simeq\) \(\frac{\ell }{7.987}\) for Ar

As shown in Fig. 4, at \(E_{{{\text{coll}}}}\)=100 meV. For such \(E_{{{\text{coll}}}}\) value, the semiclassical relation \(b \simeq \frac{\ell }{k}\) provides \(b\left( {\text{\AA}} \right) \simeq \frac{\ell }{20.98}\) for ammonia, \(b\left( {\text{\AA}} \right) \simeq \frac{\ell }{23.63}\) for N2 and \(b\left( {\text{\AA}} \right) \simeq\) \(\frac{\ell }{25.26 }\) for Ar

As shown in Fig. 4, at \(E_{{{\text{coll}}}}\) =1000 meV. For such \(E_{coll}\) value, the semiclassical relation \(b \simeq \frac{\ell }{k}\) provides \(b\left( {\text{\AA}} \right) \simeq \frac{\ell }{66.33}\) for ammonia, \(b\left( {\text{\AA}} \right) \simeq \frac{\ell }{7.471}\) for N2 and \(b\left( {\text{\AA}} \right) \simeq \frac{\ell }{7.987}\) for Ar
The first aspect to be stressed is that, because of the market capture by the sufficiently strong long-range attraction, determined by the combination of attractive electrostatic, induction and dispersion components, CHEMI of NH3 occurs with a direct mechanism even at very low collision energy. In particular, the ℓ and Rc quantities, for which the reaction effectively occurs, are all confined, respectively, at small values and at short range. When the centrifugal barrier becomes effective to limit the capture, the collision dynamics for a specific ℓ provides a Rc shift toward very large values, where the reaction probability vanishes since the reagents are too distant. Therefore, for such system, affected by a pronounced attraction (capture effect) by long-range forces, the centrifugal barrier represents a selector of the reaction mechanism, permitting the occurrence of the reaction at low and intermediate \({E}_{\mathrm{coll}}\) exclusively via DM. The role of IM becomes appreciable only at the highest \({E}_{\mathrm{coll}}\) values, that is, when the selection of the centrifugal barrier is overcame for a large lot of ℓ values. Under such conditions, the reaction can occur in an extended range of Rc, covering the gradual transition from DM to IM as ℓ increases.
The second relevant aspect is that CHEMI of Ar behaves in an opposite way because of a less pronounced role of the long-range attraction (see Fig. 1), here mostly determined by dispersion forces. In particular, from Figs. 4–8, it emerges that at low \({E}_{coll}\), CHEMI promoted by Ne* + Ar reagents, exclusively occurs, as expected, via IM since the trapping effect of weak attraction forces is overcome by the size repulsion combined with the repulsion of the centrifugal barrier. Consequently, the reagents remain always confined at large R. The transition to DM gradually occurs with the increase in \({E}_{\mathrm{coll}}\) that permits a smooth passage to shorter R probing. This transition becomes evident for \({E}_{\mathrm{coll}}\)≥ 100 meV. The Ne* (3P0) + N2 reaction (see Figs. 4–8) behaves in an intermediate way. Here, the approach of reagents is affected by two potential wells of limited depth (4–5 meV, as shown in Figs. 1 and 2): The first one occurs at about R = 5.5 Å, and it is mostly determined by the dispersion attraction; the second one manifests at about R = 3.3 Å and arises from the floppy cloud polarization of external 3 s electron of Ne* atom that originates the interaction between the disclosed atomic internal ionic core and the molecular electric quadrupole. IM exclusively drives the reaction for \({E}_{\mathrm{coll}}\) lower than 1 meV, and, because of the centrifugal barrier, the system probes exclusively the potential well at larger separation distance. DM promptly emerges at collision energy larger than 1 meV and becomes dominant already at \({E}_{\mathrm{coll}}\)=10 meV, that is, when the access to the second potential well, located at shorter R, is allowed. As for the other two CHEMI, also for this reaction, the role of IM tends to become less relevant further increasing \({E}_{\mathrm{coll}}\).
5 Discussion and conclusions
In the present study, the detailed knowledge of the optical potential has been exploited to characterize the different dynamics along selected reaction channels of CHEMI with its dependence on the collision energy. In particular, the choice of specific cuts of the multidimensional potential, that is, without the averaging effects due to the combined role of more configurations of the precursor state, has been useful to better characterize reaction selectivity. The three cases investigated clearly show that all basic details of the topology of reaction stereo-dynamics are controlled, especially at low \({E}_{\mathrm{coll}}\) values, by the critical balance of intermolecular forces having both chemical and physical origins. In general, this critical balance determines all features of the precursor state of the processes that for the barrier-less CHEMI investigated in this study coincides with the reaction transition state [23,24,25, 32, 43].
It must be stressed again that related intermolecular interactions (and in particular, the Γ components, see Fig. 3) show a strength that often amounts to few meV (a fraction of kJ/mol), and this represents a challenge to the theoretical methods used for their predictions. Another crucial point is that such interactions must be known both in an extended R range and in the whole space of the relative configurations of involved partners, and they must be given in analytical form in order to formulate related force fields (FF). The detailed knowledge of the latter represents a necessary condition to carry out any type of molecular dynamics simulations. In our investigation, an important help in the formulation of FF has been provided by the phenomenological method [32, 34,35,36,37,38,39,40,41,42], whose predictions have been tested on the experimental findings available, often permitting an improvement of the potential formulation [20, 21, 23,24,25].
The adoption of the semi-classical treatment of the collision events [30, 31], occurring from thermal up to hyper-thermal conditions, accounts for all dynamical effects accompanying the precursor state formation and properly includes the role of back dissociation and the probability of its evolution toward the final products. However, quantum mechanical corrections are expected to be relevant at very low \({E}_{coll}\) (that is, under sub-thermal conditions), but all leading aspects of the reaction stereo-dynamics here obtained should be conservative.
The present study demonstrates that the role of capture effects by long-range forces can be completely different, especially at low \({E}_{\mathrm{coll}}\), even for processes having the same origin. In particular, such role critically depends on the balance of long-range attraction contributions and of the centrifugal barrier that represents a sort of selector of angular momentum components promoting reactions. Moreover, it is of great relevance to assess the consequences by the opening of different reaction channels at short separation distances [44]. Note that the long-range interactions have basically physical origin, being determined by dispersion, induction/polarization and electrostatic contributions, while the components emerging at intermediate and short separation distances arise from chemical contributions, as those due to electron sharing and electron/charge transfer between valence orbital of reagents. The chemical components, introducing important selectivity determined by energy and symmetry of the precursor state formed at short R, become operative only if the reagents can overcome the centrifugal barrier located at large intermediate separation. It has been also demonstrated [23, 25, 43, 45] that while the Ne*(3P0) reagent tends to align the half-filled 2p orbital parallel to the intermolecular axis, favoring the reactivity promoted by short-range chemical forces, Ne*(3P2) behaves in an opposite way, that is, its reactivity at short range is hindered, because of the perpendicular 2p orbital alignment. Moreover, in both cases, the alignment degree, strongly dependent on the separation distance, that is on the effectiveness of the intermolecular electric field mainly probed by the reagents, represents a key quantity in the control of the relative role of the two reaction mechanisms [23, 25, 43]. In other words, the stereo-dynamics of elementary processes directly depends on all features (energy, symmetry, angular momentum couplings and structure) of the precursor (pre-reactive) state formed by collision events of reagents under the various conditions. More in detail, at low \({E}_{coll}\), the variation of centrifugal barrier selects the formation of the precursor state, affected by interaction components of chemical nature, from that binding through weak intermolecular forces having pure physical nature. Therefore, a fundamental aspect emphasized by the present study is that under low collision energy conditions, the centrifugal barrier, related to the orbital angular momentum of each collision complex, becomes the selector of ℓ values promoting the two different reaction mechanisms of CHEMI, that is, it separates the DM, triggered by chemical forces, from the IM, driven by the combination of weak intermolecular forces both of chemical and physical origins.
The results here obtained refer to phenomena occurring under conditions of two body collisions, as those operating in high-resolution experiments performed with the molecular beam technique [23, 25, 43]. They cast light on further important details of the topology of CHEMI stereo-dynamics [45]. All such details are of interest also for many other processes for which they are more difficult to obtain directly being masked by many other effects, as the role of many-body collision events and the different structures of the most stable precursor state respect to the reaction transition state showing the lowest energy barrier [45]. Moreover, the efficiency of atomic alignment and of the molecular orbital orientation, induced in a natural way by field gradients associated to anisotropic intermolecular interactions, still under investigation since not completely quantified, is another important point.
All the selectivity emphasized by the present investigation play a crucial role especially under conditions characterized by low density, as the interstellar environments, where only two body conditions are effective, and bimolecular collisions occur at very low \({E}_{\mathrm{coll}}\). In particular, low-temperature stereo-dynamics effects are basic to rationalize Arrhenius and anti-Arrhenius behavior in the dependence of the reactivity from the temperature. Moreover, some propensities in the stereo-dynamics operate also under more drastic conditions as those operating in flames and plasmas.
Finally, it is interesting to note that CHEMI can be considered as the inverse of the electron attachment that participates to the balance of processes occurring in several environments of great interest, as low- and high-energy plasmas including even nuclear fusion [46].
Data Availability Statement
This manuscript has no associated data or the data will not be deposited. [Authors’ comment: All data generated or analysed during this study are included in this published article].
References
R.B. Bernstein, D.R. Herschbach, R.D. Levine, J. Phys. Chem. 91(21), 5365–5377 (1987)
R.N. Zare, Angular Momentum: Understanding Spatial Aspects in Chemistry and Physics (Wiley-Interscience, New York, 1998), pp.1–335
R.N. Zare, Science 279, 1875–1879 (1998)
B. Jiang, M. Yang, D. Xie, H. Guo, Chem. Soc. Rev. 45, 3621–3640 (2016)
F. Wang, J.-S. Lin, K. Liu, Science 331, 900–903 (2011)
H. Chadwick, R.D. Beck, Annu. Rev. Phys. Chem. 68, 39–61 (2017)
C.A. Arango, M. Shapiro, P. Brumer, Phys. Rev. Lett. 97, 193202 (2006)
A.B. Henson, S. Gersten, Y. Shagam, J. Narevicius, E. Narevicius, Science 338, 234–238 (2012)
W.E. Perreault, N. Mukherjee, R.N. Zare, Science 358, 356–359 (2017)
O. Dulieu, A. Osterwalder, Cold Chemistry. Molecular Scattering and Reactivity Near Absolute Zero (Royal Society of Chemistry, Cambridge, 2018), pp.1–670
G.D.S. Gordon, J.J. Omiste, J. Zou, S. Tanteri, P. Brumer, A. Osterwalder, Nat. Chem. 10, 1190–1195 (2018)
P. Paliwal, N. Deb, D.M. Reich, A. van der Avoird, C.P. Koch, E. Narevicius, Nat. Chem. 13, 94–98 (2021)
T.M. Sugden, Annu. Rev. Phys. Chem. 13(1), 369–390 (1962)
S. Falcinelli, F. Pirani, F. Vecchiocattivi, Atmosphere 6(3), 299–317 (2015)
D. Ascenzi, A. Cernuto, N. Balucani, P. Tosi, C. Ceccarelli, L.M. Martini, F. Pirani, Astron. Astrophys. 625, A72 (2019)
Y.-P. Chang, K. Długołęcki, J. Küpper, D. Rösch, D. Wild, S. Willitsch, Science 342, 98–101 (2013)
D. Rösch, S. Willitsch, Y.-P. Chang, J. Küpper, J. Chem. Phys. 140, 124202 (2014)
A. Kilaj, J. Wang, P. Straňák, M. Schwilk, U. Rivero, L. Xu, O.A. von Lilienfeld, J. Küpper, S. Willitsch, Nat. Com. 12, 6047 (2021)
F.M. Penning, Naturwissenschaften 15, 818 (1927)
S. Falcinelli, A. Bartocci, S. Cavalli, F. Pirani, F. Vecchiocattivi, Chem. Eur. J. 22, 764–771 (2016)
S. Falcinelli, M. Rosi, S. Cavalli, F. Pirani, F. Vecchiocattivi, Chem. Eur. J. 22, 12518–12526 (2016)
S. Falcinelli, F. Vecchiocattivi, F. Pirani, J. Chem. Phys. 150, 044305 (2019)
S. Falcinelli, J.M. Farrar, F. Vecchiocattivi, F. Pirani, Acc. Chem. Res. 53(10), 2248–2260 (2020)
S. Falcinelli, F. Vecchiocattivi, J.M. Farrar, F. Pirani, J. Phys. Chem. A 125, 3307–3315 (2021)
S. Falcinelli, F. Vecchiocattivi, F. Pirani, Sci. Rep. 11, 19105 (2021)
H. Nakamura, J. Chem. Phys. Jpn. 26, 1473–1479 (1969)
W.H. Miller, J. Chem. Phys. 52, 3563–3572 (1970)
A. Niehaus, Ber. Bunsenges Phys. Chem. 77, 632–640 (1983)
H. Morgner, A. Niehaus, J. Phys. B At. Mol. Phys. 12, 1805–1820 (1979)
P.E. Siska, Rev. Mod. Phys. 65, 337–412 (1993)
B. Brunetti, F. Vecchiocattivi, in Current Topic on Ion Chemistry and Physics, ed. C.Y. Ng, T. Baer, I. Powis (John Wiley & Sons Ltd, New York,1993), pp. 359–445
S. Falcinelli, F. Vecchiocattivi, F. Pirani, Commun. Chem. 3(1), 64 (2020)
G.C. Maitland, M. Rigby, E.B. Smith, W.A. Wakeham, in Intermolecular Forces: Their Origin and Determination (Clarendon Press, Oxford, 1987)
F. Pirani, G.S. Maciel, D. Cappelletti, V. Aquilanti, Int. Rev. Phys. Chem. 25, 165–199 (2006)
M. Capitelli, D. Cappelletti, G. Colonna, C. Gorse, A. Laricchiuta, G. Liuti, S. Longo, F. Pirani, Chem. Phys. 338, 62–68 (2007)
A. Laricchiuta, G. Colonna, D. Bruno, R. Celiberto, C. Gorse, F. Pirani, M. Capitelli, Chem. Phys. Lett. 445, 133–139 (2007)
A. Laricchiuta, D. Bruno, M. Capitelli, C. Catalfamo, R. Celiberto, G. Colonna, P. Diomede, D. Giordano, C. Gorse, S. Longo, D. Pagano, F. Pirani, Eur. J. Phys. D 2009(54), 607–612 (2009)
A. Laricchiuta, F. Pirani, G. Colonna, D. Bruno, C. Gorse, R. Celiberto, M. Capitelli, J. Phys. Chem. A 113, 15250–15256 (2009)
D. Bruno, C. Catalfamo, M. Capitelli, G. Colonna, O. De Pascale, P. Diomede, C. Gorse, A. Laricchiuta, S. Longo, D. Giordano, F. Pirani, Phys. Plasmas 17, 112315 (2010)
G. Colonna, A. D’Angola, L.D. Pietanza, M. Capitelli, F. Pirani, E. Stevanato, A. Laricchiuta, Plasma Sour. Sci. Techonl. 27, 015007 (2018)
G. Bellas Chatzigeorgis, J.B. Haskins, J.B. Scoggins, Phys. Fluids 34, 087106 (2022)
A. Bartocci, L. Belpassi, D. Cappelletti, S. Falcinelli, F. Grandinetti, F. Tarantelli, F. Pirani, J. Chem. Phys. 142(18), 184304 (2015)
S. Falcinelli, F. Vecchiocattivi, F. Pirani, Phys. Rev. Lett. 121, 163403 (2018)
A. Tsikritea, A. Jake, J.A. Diprose, T.P. Softley, B.R. Heazlewood, J. Chem. Phys. 157, 060901 (2022)
S. Falcinelli, F. Vecchiocattivi, F. Pirani, Commun. Chem. 6(1), 30 (2023)
N. Bacal, Nucl. Fusion 46, S250–S259 (2006)
Funding
Open access funding provided by Università degli Studi di Perugia within the CRUI-CARE Agreement.
Author information
Authors and Affiliations
Contributions
All authors contributed equally to the paper.
Corresponding author
Additional information
Guest editors: Annarita Laricchiuta, Iouli E. Gordon, Christian Hill, Gianpiero Colonna, Sylwia Ptasinska.
T.I. : Atomic and Molecular Data and Their Applications: ICAMDATA 2022.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Falcinelli, S., Parriani, M., Vecchiocattivi, F. et al. The selective role of the orbital angular momentum on the reaction stereo-dynamics. Eur. Phys. J. D 77, 65 (2023). https://doi.org/10.1140/epjd/s10053-023-00643-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1140/epjd/s10053-023-00643-3