
Abstract
New thiazole and thiadiazole derivatives bound to the acetanilide moiety were synthesized and evaluated for their cytotoxic activity. The precursor N-(4-acetamidophenyl)-N'-phenylthiourea (2) was cyclocondensed with ethyl bromoacetate to afford a mixture of the two isomers, 2-(4-acetamidophenylimino)-3-phenylthiazolidin-4-one (3a, 23%) and 3-(4-acetamidophenyl)-2-phenyliminothiazolidin-4-one (3b, 71%). The Knoevenagel reaction of 3b with various aromatic aldehydes afforded 5-arylidene-2-phenyliminothiazolidin-4-one derivatives 5a–5e. Intramolecular cyclization of thiourea scaffold 2 with chloroacetone and/or phenacyl chloride gave the conforming thiazole derivatives 6a and 6b. A new series of thiadiazole derivatives 9a–9c and 11a–11c was synthesized by the reaction of N-(4-acetamidophenyl)-N'-phenylthiourea (2) with selected derivatives of hydrazonoyl halide in ethanol and triethylamine. The structures of the synthesized thiazole and thiadiazole compounds were elucidated by their compatible spectral data. The cytotoxic activity of the synthesized thiazole and thiadiazole derivatives was screened against four human cancer cell lines and showed promising results. Thiazolidin-4-one compound 5d showed the strongest cytotoxic effects on hepatocellular carcinoma (IC50 = 8.80 ± 0.31 μg/mL), mammary gland breast cancer (IC50 = 7.22 ± 0.65 μg/mL) and colorectal carcinoma (IC50 = 9.35 ± 0.61 μg/mL) cell lines.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
INTRODUCTION
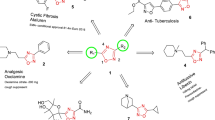
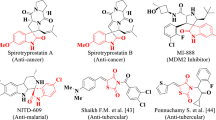
Thiourea derivatives are important synthons in the synthesis of biologically active heterocyclic compounds that have a wide range of biological applications [1]. In particular, the most significant thiourea derivatives demonstrated antiviral [2], cytotoxic potential [3], antibacterial and antifungal [4] and anticancer activities [5]. 4-Thiazolidinone derivatives have also attracted continuing interest due to their valuable biological activities [6-8] such as anti-inflammatory [9], anti-microbial [10], antidiabetic and antiviral [11, 12], anti-tuberculostatic [13], anti-malarial [14], COX-2 inhibitor activities [15], anti-HIV [16], anti-oxidant [17], Anti-urease Agents [18] and anti-cancer [19, 20]. A number of thiazolidinone-based compounds with various substituents surrounding the core nucleus are being investigated as potential anticancer agents (Fig. 1) [21, 22]. While, thiazole scaffold have medical claims such as bacteriostatic, antibiotics [23], anti-inflammatory [24], analgesics [25, 26], and anti-HIV [27]. Some of these thiazole-containing compounds have been transferred into clinical trials and cancer therapy—Dasatinib and Dabrafenib (Fig. 1) [28, 29].

Structure of some thiazole and thiadiazole derivatives showing anticancer activity.
In addition, 1,3,4-thiadiazoles exhibit a wide range of biological activities, including antimicrobial [30, 31], anticancer [32–34], antioxidant [35], antidepressant [36], antibacterial [37], anti-fungal [38], anticonvulsant [39], anti-inflammatory [40], against covid-19 [41], and antiproliferative activities [42], as well as the treatment of Alzheimer disease [43, 44]. 2-(4-Fluorophenylamino)-5-(2,4-dihydroxyphenyl)-1,3,4-thiadiazole (5) (Fig. 1) inhibited tumor cell proliferation derived from nervous system cancers (medulloblastoma/neuroblastoma, glioma, and rhabdosarcoma) and peripheral cancers (lung and colon carcinoma). The thiadiazole derivatives 5 and 6 (Fig. 1) are not toxic to normal cells [45, 46]. The present research article reported on the efficient synthesis of some new highly functionalized thiazole and 1,3,4-thiadiazole derivatives utilizing N-(4-acetamidophenyl)-N'-phenylthiourea as a key starting synthon, and an evaluation of their cytotoxicity against HepG2, MCF-7, HTC-116, and PC-3 cell lines.
RESULTS AND DISCUSSION
Reaction of 4-aminoacetanilide (1) with phenyl isothiocyanate furnished the key start N-(4-acetamidophenyl)-N'-phenylthiourea (2) [47]. The regioselective reaction of 1,3-diarylthiourea derivative 2 with ethyl bromoacetate proceeded in ethanol containing fused CH3COONa (0.5 g) to afford two isomeric thiazolidin-4-ones 3a and 3b (Scheme 1). The comparable electronic nature of the thiourea nitrogen atoms appears to dominate the outcome of the cyclization reaction and an almost 1:3 mixture of the two 2-iminothiazolidine-4-ones 3a and 3b is obtained, the ratio was determined from the 1H NMR spectrum. The cyclization’s regioselectivity is influenced not only by the reaction conditions and the δ-halocarbonyl derivative used, but also by the nature of the thiourea intermediate’s two substituents. The regioselective reaction of unsymmetrical thiourea with alpha-haloesters is dependent on the pKa’s of the amines. The regioselective product 2-imino-4-thiazolidinone is formed when an amine is attached to a thiourea with a lower pKa as part of the imino component and another amine with a higher pKa contributes to the other heterocyclic nitrogen.

Synthesis of 2-imino-4-thiazolidinone derivatives 3a and 3b.
When unsymmetrical thiourea 2 having a phenyl and a p-acetamidophenyl groups reacted with ethyl bromoacetate, a mixture of two isomers acetamidophenylimino 3a and phenylimino 3b (with a ratio of 1 : 3) has been obtained. The measured pKa of aniline and p-acetamidoaniline are 4.63 and 5.46, respectively. Thus, the amine with the lower pKa contributes to the imino component, while the other amine attached to the thiourea with the higher pKa contributes to the other heterocyclic nitrogen in the 2-imino-4-thiazolidinone skeleton (3b). Fortunately, the 3-(4-acetamidophenyl)-2-phenyliminothiazolidin-4-one (3b) was isolated from its isomeric 2-(4-acetamidophenylimino)-3-phenylthiazolidin-4-one (3a) by recrystallization of the regioselective mixture from ethyl alcohol. The proposed structure of the isolated product 3b was assessed by spectral and analytical data. The IR spectrum of 3b identified absorptions at 3295 and 1728 cm–1 due to the presence of N–H and thiazolidinone (C=O) functions, respectively. The 1H NMR spectrum displayed a singlet at δ 2.01 ppm for the methyl group and a singlet at δ 4.15 ppm for the methylene group of the thiazolidinone ring. The upfielded chemical shift of the AA′BB′ system of the isolated isomer 3b at δ 6.79 ppm refers to the ortho-aromatic protons of phenylimino-moiety. In addition to that, the singlet for one proton at δ 9.89 ppm indicated the acetamido group (CONH). The 13C NMR spectrum displayed the carbon signal of the methylene group (thiazolidine-4-one ring) at δ 37.18 ppm. Also, the carbon signals of carbonyl groups were observed at δ 169.05 (C=O, amide) and δ 172.88 ppm (C=O, thiazolidinone ring) respectively.
In an attempt to prepare the pyranothiazole derivatives 4, the thiazolidin-4-one derivative 3b was heated with 2-(4-methylbenzylidene)malononitrile in ethanol and piperidine. Unfortunately the reaction did not furnish our targeting pyranothiazole derivatives 4 while the 5-arylidenethiazolidin-4-one scaffolds 5 are isolated as a sole product in each case (Scheme 2). Compounds 5 were also obtained directly by refluxing the 2-phenyliminothiazolidine-4-one derivative 3b with various substituted benzaldehydes (4-toulaldehyde, 4-anisaldehyde, 2,5-dimethoxybenzaldehyde, 4-nitrobenzaldehyde and/or 3-(4-chlorophenyl)-4-formyl-1-phenyl-1H-pyrazole) in acetic acid containing sodium acetate (Scheme 2). The IR of thiazolidine-4-one 5b identified absorptions at 3308 cm–1 (N–H), 1714 and 1660 cm–1 (two carbonyl groups). Whereas, 1H NMR spectrum displayed singlet signals at δ 2.06 and 3.79 ppm for the protons of methyl (CH3) and methoxy (OCH3) groups. The two singlet signals for the methine (CH=C) and N–H protons are observed at δ 7.76 and 10.11 ppm. The 1H NMR of the synthesized compounds 5a–5e clearly shows the disappearance of thiazole methylene group and detection of CH=C. The 13C NMR spectrum demonstrated carbon signals at δ 24.39 (CH3) and 55.47 ppm (OCH3). The signal at δ 143.07 ppm identified the carbon of thiazole-C5. The signal of imine group (C=N) was resonated at δ 160.60 ppm. Furthermore, the carbon signals at δ 163.61 and 168.23 ppm indicated the carbonyl groups (C=O, amide), and (C=O, ring), respectively.

Synthesis of 5-arylidene-2-phenyliminothiazolidin-4-one derivatives 5a–5e.
Furthermore, the reaction of diaryl thiourea derivative 2 with α-halogenated reagents (chloroacetone and/or phenacyl chloride) in boiling ethanol and triethylamine furnished the conforming thiazole compounds 6a and 6b. The reaction starts via nucleophilic substitution of the chlorine atom from α-chloroketone to yield the alkylated intermediate A, which underwent intramolecular nucleophilic addition of N–H function to the carbonyl group. The produced intermediate B undergoes elimination of water molecule to the targeting thiazole derivative 6a and 6b (Scheme 3). The IR spectrum of 6a indicated absorptions at 3240 and 1669 cm–1 for the N–H and C=O functions, respectively. The 1H NMR spectrum showed singlet signals for two methyl protons at δ 1.89 and 2.09 ppm, a singlet for the proton of thiazole-C5 at δ 6.95 ppm, and a singlet for the proton of N–H function at δ 9.17 ppm. The aromatic protons are observed in the region from δ 7.07 to 7.43 ppm. The 13C NMR spectrum of 6a exhibited carbon signals at δ 16.90 (CH3) and 24.55 ppm (CH3). The carbon signal of thiazole-C5 was resonated at δ 95.82 ppm. Furthermore, the carbon signal of carbonyl group was resonated at δ 169.70 ppm (C=O, amide group).

Synthesis of thiazole derivatives 6a and 6b.
Heating the diaryl thiourea derivative 2 in ethanol and triethylamine with the 2-oxo-N-arylpropanehydrazonoyl chlorides 7a–7c produced the conforming 5-acetylthiadiazole derivatives 9a–9c rather than the thiazole-based skeleton 8a–8c (Scheme 4). The reaction was initiated by nucleophilic substitution of the chlorine atom from the hydrazonoyl chloride derivative 7 to yield the alkylated intermediate C1. This intermediate C1 failed to tautomerize into the intermediate C2, and the loss of a water molecule resulted in the formation of the assumed arylazothiazoles 8. However, the sulfide intermediate C1 favors the nucleophilic addition of the N–H function (from the hydrazone moiety) to the C=N group to constitute the intermediate D, which undergoes elimination of the aniline molecule and furnishes the corresponding 5-acetyl-1,3,4-thiadiazoles 9a–9c (Scheme 4). Thiadiazole compound 9b showed the characteristic IR absorptions at 3290, 1665, 1657 cm–1 due to N–H and two carbonyl (C=O) groups. The 1H NMR spectrum exhibited singlet signals for the protons of three methyl groups at δ 2.03, 2.38 and 2.45 ppm, a multiplet for eight aromatic protons in the region δ 6.70–7.53 ppm and a singlet for the proton of the N–H function at δ 10.12 ppm. The 13C NMR spectrum displayed carbon signals at δ 23.81 (CH3), 24.52 (CH3), and 26.12 ppm (CH3). The aromatic carbon atoms were indicated by the signals at δ 114.51, 114.83, 122.62, 124.77, 128.65, 129.34, 131.25, and 133.70 ppm. Furthermore, the carbon signals at δ 158.17 and 162.94 ppm indicated the signals of carbonyl groups (C=O, amide group) and (C=O, acetyl group), respectively.

Synthesis of 5-acetylthiadiazole derivatives 9a–9c.
Furthermore, the reactivity of the N-(4-acetamidophenyl)-N′-phenylthiourea (2) with various ethyl 2-chloro-2-(2-phenylhydrazono)acetates 10a–10c was also conducted. The reaction proceeds by refluxing the reactants in ethanol containing triethylamine to afford the respective 5-ethoxycarbonyl-1,3,4-thiadiazole derivatives 11a–11c (Scheme 5). 1,3,4-Thiadiazoles 11a–11c were formed through nucleophilic substitution of the chlorine atom (from hydrazonoyl chlorides 10a-c) to yield the alkylated intermediate E, which undergoes intramolecular cyclization of the N–H group (hydrazone moiety) on the carbon activated of the imino group (C=N). The formed intermediate F rapidly loses an aniline molecule, yielding the corresponding 1,3,4-thiadiazoles 11a–11c. The structures of thiadiazole products 11a–11c were established based on their analytical and spectral data. The thiadiazole compound 11a showed the cacteristic infrared absorptions at 3365 for the N–H stretching and at 1737 and 1657 cm–1 due to the two carbonyl (C=O) groups. The 1H NMR spectrum of 11a displayed triplet and quartet signals that resonated at δ 1.18 and 4.20 ppm due to the protons of the ethyl group, and a singlet for the protons of the methyl group (CH3CO) at δ 2.07 ppm. The aromatic protons are observed as multiplet signals at δ 6.82–7.65 ppm. The proton of the N–H group was identified as a singlet at δ 10.11 ppm. The 13C NMR spectrum of 11a showed carbon signals at δ 14.34 (CH3), 23.55 (CH3), and 61.50 ppm (OCH2). The aromatic carbon atoms are identified by the carbon signals at δ 114.93, 121.62, 123.85, 125.51, 129.06, 129.87, 133.44, and 139.29 ppm. The signals at δ 144.57 and 150.23 ppm indicated the carbon atoms of the thiadiazole ring. Moreover, the signals of carbonyl groups were observed at δ 162.80 (amide group) and 169.78 ppm (ester group).

Synthesis of 5-ethoxycarbonyl-thiadiazole derivatives 11a–11c.
In vitro antitumor activity. Cancer is recognized as the most serious disease in the world, with severity and lethality caused by inconsistency in cell growth and proliferation [48,49]. Chemotherapy is the most fundamental and important approach in cancer treatment, employing a wide range of natural and synthetic compounds to destroy cancer cells [50]. The inability of the current chemotherapeutic system to distinguish between normal and cancerous cells has been its main drawback [51]. As a result, the major challenge for medicinal chemists has been the development of safe and effective chemotherapeutic agents. The current study aims to look into the anticancer activity of chemically synthesized thiazolidine-4-one, thiazole, and 1,3,4-thiadiazole derivatives against four human cancer cell lines: HepG2 (hepatocellular carcinoma), MCF-7 (mammary gland breast cancer), HTC-116 (colorectal carcinoma) and PC-3 (human prostate cancer carcinoma). The effects of thiazole and thiadiazole compounds 3b, 5a–5e, 6a, 6b, 9a–9c, and 11a–11c on the viability of these cell lines were studied by applying the MTT assay [52, 53] using 5-fluorouracil (5-FU) as a reference drug. The results (Table 1) showed that the majority of the prepared compounds had moderate to strong cytotoxicity effects on the cancer cell lines tested. 5-(4-Nitrobenzylidene)-thiazolidin-4-one compound 5d displayed the highest cytotoxicity on two cancer cell lines HepG2 (IC50 8.80 ± 0.31 μg/mL) and MCF-7 (IC50 7.22 ± 0.65 μg/mL), their IC50 values are close to the standard anticancer drug 5-fluorouracil. In addition, it showed strong cytotoxic effects among the other two cancer cell lines HTC-116 (IC50 9.35 ± 0.61 μg/mL) and PC-3 (IC50 12.57 ± 1.09 μg/mL). Furthermore, 2-phenylimino-thiazolidin-4-one derivatives 5b, 5c, and 5e showed equipotent anticancer activity against all cell lines with IC50 values ranging from 19.07 to 11.80 μg/mL. In contrast, the investigated thiadiazole derivatives 9a–9c and 11a–11c showed considering activity against breast cancer cell lines as indicted by the MCF-7 profile (Table 1). Thiadiazole compounds 9c and 11c, in particular, demonstrated the most pronounced activity against the MCF-7 cell line when compared to 5-fluorouracil, with IC50 values of 15.64 and 17.32 μg/mL, respectively.
The structure-activity relationship gave a clear picture of the activity of these title compounds. The addition of a substituent on the benzylidene moiety that is linked to the thiazolidine-4-one ring affects anticancer acidity. The presence of an electron-donating methoxy group (OCH3) in the benzylidene fragment boosts the anticancer activities. Moreover, the activity is enhanced by the presence of an electron-withdrawing group (NO2) at the fourth position of the benzylidene moiety. In fact, the most interesting results were obtained for 9c and 11c, cacterized by the presence of a thiadiazole moiety and of a chlorine atom on the aromatic ring bonded at the p-position, of the aromatic ring, as evidenced by half-maximal inhibitory concentration (IC50) values (Table 1).
EXPERIMENTAL
Gallenkamp electric apparatus is used to measure the melting points. Infrared spectra (IR) were recorded on a Thermo Scientific Nicolet iS10 FTIR spectrometer. The 1H NMR and 13C NMR spectra were recorded on a Bruker AV 400 MHz using DMSO-d6 as a solvent. The mass analyses were obtained by a Kratos MS equipment (EI mode at 70 eV). C, H, and N analyses were determined on Perkin-Elmer 2400 analyzer.
Preparation of N-(4-acetamidophenyl)-N′-phenylthiourea (2). Phenyl isothiocyanate (1.20 g, 10 mmol) was added to a hot stirred suspension of 4-aminoacetanilide (1.50 g, 10 mmol) in hot ethanol (20 mL). The mixture was refluxed for 10–15 min. The precipitated product was filtered and dried to afford the thiourea derivative 2. Gray crystals, yield 84%, mp 212–213°C, lit. mp 213°C [47].
Synthesis of 3-(4-acetamidophenyl)-2-phenylimino-thiazolidine-4-one (3b). A suspension of diaryl thiourea compound 2 (1.71 g, 6 mmol), ethyl bromoacetate (0.99 g, 6 mmol) and 0.50 g fused sodium acetate in 20 mL ethanol was refluxed for 3 h. The solid that formed was collected, dried, and then subjected to recrystallization form 50 mL boiling ethanol to afford the targeting thiazolidin-4-one derivative 3b. White solid, yield 68% mp 139–140°C. IR spectrum, ν, cm–1: 3295 (N–H), 1728 (C=O), 1658 (C=O), 1625 (C=N). 1H NMR spectrum, δ, ppm: 2.01 s (3H, CH3), 4.15 s (2H, CH2), 6.79 d (2H, J = 8.40 Hz, HAr), 7.38 d (2H, J = 8.40 Hz, HAr), 7.42 (t, 1H, J = 7.60 Hz, HAr), 7.49 d (2H, J = 7.20 Hz, HAr), 7.51 d (2H, J = 8.0 Hz, HAr), 9.89 s (1H, CONH). 13C NMR spectrum, δ, ppm: 24.30, 34.18, 114.07 (2C), 114.57 (2C), 123.26, 125.71, 129.15 (2C), 129.86 (2C), 133.24, 147.61, 156.20, 169.05, 172.88. Found, %: C 62.88; H 4.60; N 12.97. C17H15N3O2S. Calculated, %: C 62.75; H 4.65; N 12.91. MS: m/z: 325 [M]+.
Synthesis of 3-(4-acetamidophenyl)-2-phenylimino-5-(4-substitutedbenzylidene)-thiazolidin-4-one derivatives 5a–5e. To a solution of thiazolidin-4-one derivative 3b (0.65 g, 2 mmol) in 20 mL glacial acetic acid, each of the substituted benzaldehyde derivative (2 mmol) and fused sodium acetate (0.5 g) were added. The mixture was refluxed for 4 h and then diluted with 20 mL ice-cold water. The precipitate that formed was picked up by filtration and then subjected to recrystallization by heating in ethyl alcohol.
3-(4-Acetamidophenyl)-5-(4-methylbenzylidene)-2-phenylimino-thiazolidin-4-one (5a). Yellow crystals, yield 74%, mp 278–280°C. IR spectrum, ν, cm–1: 3306 (N–H), 1715 (C=O), 1661 (C=O), 1632 (C=N). 1H NMR spectrum, δ, ppm: 2.05 s (3H, CH3), 2.34 s (3H, CH3), 6.92 d (2H, J = 8.40 Hz, HAr), 7.33 d (2H, J = 8.40 Hz, HAr), 7.47–7.59 m (9H, HAr), 7.77 s (1H, CH=C), 9.96 (s, 1H, CONH). 13C NMR spectrum, δ, ppm: 20.13, 24.35, 118.91 (2C), 119.87 (2C), 120.12, 121.36 (2C), 128.47 (2C), 129.08, 129.53 (2C), 129.95 (2C), 130.41, 130.63, 136.32, 139.52, 140.23, 142.97, 152.40, 163.74, 168.08. Found,%: C 70.12; H 4.98; N 9.90. C25H21N3O2S. Calculated, %: C 70.24; H 4.95; N 9.83. MS: m/z: 427 [M]+.
3-(4-Acetamidophenyl)-5-(4-methoxybenzylidene)-2-phenylimino-thiazolidin-4-one (5b). Yellow crystals, yield 70%, mp 284–285°C. IR spectrum, ν, cm–1: 3308 (N–H), 1714 (C=O), 1660 (C=O), 1632 (C=N). 1H NMR spectrum, δ, ppm: 2.06 s (3H, CH3), 3.79 s (3H, OCH3), 6.91 d (2H, J = 8.80 Hz, HAr), 7.08 d (2H, J = 8.80 Hz, HAr), 7.52–7.55 m (7H, HAr), 7.60 d (2H, J = 8.40 Hz, HAr), 7.76 s (1H, CH=C), 10.11 (s, 1H, CONH). 13C NMR spectrum, δ, ppm: 24.39, 55.47, 114.91 (2C), 117.94, 118.88 (2C), 120.01 (2C), 122.30 (2C), 125.73, 129.04 (2C), 129.79, 130.24, 132.15 (2C), 135.86, 138.66, 143.17, 152.58, 161.09, 163.61, 167.83. Found,%: C 67.86; H 4.82; N 9.55. C25H21N3O3S. Calculated, %: C 67.70; H 4.77; N 9.47. MS: m/z: 443 [M]+.
3-(4-Acetamidophenyl)-5-(2,5-dimethoxybenzylidene)-2-phenylimino-thiazolidin-4-one (5c). Yellow crystals, yield 81%, mp 269–270°C. IR spectrum, ν, cm–1: 3368 (N–H), 1711, 1685 (C=O), 1633 (C=N). 1H NMR spectrum, δ, ppm: 2.09 s (3H, CH3), 3.68 s (3H, OCH3), 3.79 s (3H, OCH3), 6.84–7.18 m (6H, HAr), 7.39 d (2H, J = 8.40 Hz, HAr), 7.44 d (2H, J = 8.80 Hz, HAr), 7.74 d (2H, J = 8.80 Hz, HAr), 7.92 s (1H, CH=C), 10.32 (s, 1H, CONH). 13C NMR spectrum, δ, ppm: 24.31, 55.48, 56.33, 112.84, 114.55, 115.91, 119.39 (2C), 120.07 (2C), 120.95, 121.76 (2C), 122.43, 123.67, 125.52, 128.48 (2C), 129.60, 135.31, 138.85, 142.73, 152.11, 158.52, 160.03, 168.58. Found,%: C 65.85; H 4.94; N 8.95. C26H23N3O4S. Calculated, %: C 65.94; H 4.90; N 8.87. MS: m/z: 473 [M]+.
3-(4-Acetamidophenyl)-5-(4-nitrobenzylidene)-2-phenylimino-thiazolidin-4-one (5d). Yellow crystals, yield 82%, mp 260–262°C. IR spectrum, ν, cm–1: 3322 (N–H), 1704 (C=O), 1639 (broad, C=O and C=N). 1H NMR spectrum, δ, ppm: 2.09 s (3H, CH3), 6.92 d (2H, J = 8.40 Hz, HAr), 6.98 d (2H, J = 8.00 Hz, HAr), 7.20– 7.80 m (7H, HAr), 7.93 s (1H, CH=C), 8.31 d (2H, J = 8.4 Hz, HAr), 10.29 (s, 1H, CONH). 13C NMR spectrum, δ, ppm: 24.31, 119.17 (2C), 120.09 (2C), 121.28 (2C), 123.45, 127.83 (2C), 129.01, 129.68 (2C), 130.99 (2C), 132.36, 134.44, 137.05, 139.53, 143.18, 149.87, 164.50, 166.71, 168.24. Found, %: C 62.71; H 3.90; N 12.32. C24H18N4O4S. Calculated, %: C 62.87; H 3.96; N 12.22. MS: m/z: 458 [M]+.
3-(4-Acetamidophenyl)-5-((3-(4-chlorophenyl)-1-phenyl-1H-pyrazol-4-yl)methylene)-2-phenyliminothiazolidin-4-one (5e). Yellow crystals, yield 75%, mp 265–267°C. IR spectrum, ν, cm–1: 3255 (N–H), 1715, 1646 (C=O). 1H NMR spectrum, δ, ppm: 2.05 s (3H, CH3), 6.93 d (2H, J = 8.80 Hz, HAr), 7.38–7.68 m (15H, 14 HAr and CH=C), 7.96 d (2H, J = 8.40 Hz, HAr), 8.70 s (1H, CH=C), 10.10 (s, 1H, CONH). 13C NMR spectrum, δ, ppm: 24.36, 116.89, 119.37 (2C), 120.06 (2C), 121.17 (2C), 121.88 (2C), 122.44, 127.73, 129.01 (2C), 129.56 (2C), 130.05, 130.14, 131.15 (2C), 131.65 (2C), 131.78, 133.71, 134.51, 139.22, 139.93, 150.57, 150.86, 164.80, 165.03, 168.56. Found, %: C 67.28; H 4.15; N 11.93. C33H24ClN5O2S. Calculated, %: C 67.17; H 4.10; N 11.87. MS: m/z: 589 [M]+.
Preparation of 3-(4-acetamidophenyl)-2-phenyliminothiazole 6a and 6b. In a 50 mL RBF, aryl thiourea derivative 2 (1.42 g, 5 mmol) and each of the α-chloroketone(5 mmol) (chloroacetone and/or phenacyl chloride) were refluxed for 3 h in 25 mL of ethanol (HPLC), and 0.2 mL triethylamine. Upon cooling to 20°C, the solid that formed was collected and recrystallized from ethanol to obtain the thiazole derivatives 6a and 6b.
N-(4-(4-Methyl-2-(phenylimino)thiazol-3(2H)-yl)phenyl)acetamide (6a). Pale Yellow crystals, yield 69%, mp 100–102°C. IR spectrum, ν, cm–1: 3240 (N–H), 1669 (C=O). 1H NMR spectrum, δ, ppm: 1.89 s (3H, CH3), 2.09 s (3H, CH3), 6.95 s (1H, C5Hthiazole), 7.07–7.18 m (5H, HAr), 7.37 d (2H, J = 8.40 Hz, HAr), 7.43 d (2H, J = 8.40 Hz, HAr), 9.32 s (1H, NH). 13C NMR spectrum, δ, ppm: 16.90, 24.55, 95.82, 115.30 (2C), 123.31 (2C), 124.09, 128.12 (2C), 128.95 (2C), 129.06, 131.54, 139.45, 144.64, 152.61, 169.70. Found, %: C 66.78; H 5.33; N 12.89. C18H17N3OS. Calculated, %: C 66.85; H 5.30; N 12.99. MS: m/z: 323 [M]+.
N-(4-(4-Phenyl-2-(phenylimino)thiazol-3(2H)-yl)phenyl)acetamide (6b). Yellow powder, yield 64%, mp 116–118°C. IR spectrum, ν, cm–1: 3292 (N–H), 1668 (C=O). 1H NMR spectrum, δ, ppm: 2.03 s (3H, CH3), 6.97 s (1H, C5Hthiazole), 7.09–7.25 m (10H, HAr), 7.39 d (2H, J = 8.80 Hz, HAr), 7.47 d (2H, J = 8.80 Hz, HAr), 9.89 s (1H, NH). 13C NMR spectrum, δ, ppm: 24.58, 98.67, 114.07 (2C), 114.92 (2C), 122.19 (2C), 123.35 (2C), 124.09, 128.18, 128.97, 129.05 (2C), 129.77 (2C), 130.32, 131.54, 139.07, 144.61, 159.26, 170.16. Found, %: C 71.72; H 4.93; N 10.81. C23H19N3OS. Calculated, %: C 71.66; H 4.97; N 10.90. MS: m/z: 385 [M]+.
Synthesis of 5-acetylthiadiazoles 9a–9c and 5-ethoxycrbonylthiadiazoles 11a–11c. A mixture of diaryl thiourea derivative 2 (0.85 g, 3 mmol) and each of the hydrazonoyl halides 7a–7c or 10a–10c (3 mmol) was refluxed in ethanol (25 mL) containing 0.2 mL of triethylamine for 4–6 h. The precipitate that formed was isolated by filtration and recrystallized from EtOH/DMF mixture to yield the targeting thiadiazole compounds 9a–9c and 11a–11c, respectively.
N-(4-((5-Acetyl-3-phenyl-1,3,4-thiadiazol-2(3H)-ylidene)amino)phenyl)acetamide (9a). Yellow powder, yield 77%, mp 248–250°C. IR spectrum, ν, cm–1: 3320 (N–H), 1670, 1654 (C=O). 1H NMR spectrum, δ, ppm: 2.09 s (3H, CH3), 2. 43 s (3H, CH3), 6.77 d (2H, J = 8.80 Hz, HAr), 7.12 d (2H, J = 8.40 Hz, HAr), 7.35–7.58 m (5H, HAr), 10.13 s (1H, CONH). 13C NMR spectrum, δ, ppm: 23.63, 25.12, 114.43, 114.92 (2C), 123.53 (2C), 125.30, 128.91 (2C), 129.65 (2C), 133.46, 134.72, 141.90, 145.34, 158.17, 162.92. Found, %: C 61.18; H 4.53; N 15.97. C18H16N4O2S. Calculated, %: C 61.35; H 4.58; N 15.90. MS: m/z: 352 [M]+.
N-(4-((5-Acetyl-3-(p-tolyl)-1,3,4-thiadiazol-2(3H)-ylidene)amino)phenyl)acetamide (9b). Yellow powder, yield 74%, mp 238–240°C. IR spectrum, ν, cm–1: 3290 (N–H), 1665, 1657 (C=O). 1H NMR spectrum, δ, ppm: 2.03 s (3H, CH3), 2.38 s (3H, CH3), 2.45 s (3H, CH3), 6.70 d (2H, J = 8.00 Hz, HAr), 6.91 d (2H, J = 7.60 Hz, HAr), 7.38–7.53 m (4H, HAr), 10.12 s (1H, CONH). 13C NMR spectrum, δ, ppm: 23.81, 24.52, 26.12, 114.51, 114.83 (2C), 122.62 (2C), 124.77, 128.65 (2C), 129.34 (2C), 131.25, 133.70, 143.93, 147.35, 158.87, 162.94. Found, %: C 62.41; H 5.03; N 15.38. C19H18N4O2S. Calculated, %: C 62.28; H 4.95; N 15.29. MS: m/z: 366 [M]+.
N-(4-((5-Acetyl-3-(4-chlorophenyl)-1,3,4-thiadiazol-2(3H)-ylidene)amino)phenyl)-acetamide (9c). Yellow powder, yield 69%, mp 259–270°C. IR spectrum, ν, cm–1: 3370 (N–H), 1671, 1648 (C=O). 1H NMR spectrum, δ, ppm: 2.07 s (3H, CH3), 2.46 s (3H, CH3), 6.79 d (2H, J = 8.80 Hz, HAr), 7.23 d (2H, J = 8.80 Hz, HAr), 7.55–7.78 m (4H, HAr), 10.15 (s, 1H, NH). 13C NMR spectrum, δ, ppm: 23.94, 26.33, 114.11, 115.30 (2C), 122.87 (2C), 126.35, 129.20 (2C), 129.91 (2C), 131.83, 135.94, 143.57, 146.28, 158.93, 162.71. Found, %: C 55.77; H 3.94; N 14.59. C18H15ClN4O2S. Calculated, %: C 55.88; H 3.91; N 14.48. MS: m/z: 386 [M]+.
Ethyl 5-((4-acetamidophenyl)imino)-4-phenyl-4,5-dihydro-1,3,4-thiadiazole-2-carboxylate (11a). Brown powder, yield 75%, mp 271–273°C. IR spectrum, ν, cm–1: 3365 (N–H), 1737, 1657 (C=O). 1H NMR spectrum, δ, ppm: 1.18 s (3H, CH3), 2.07 s (3H, CH3), 4.20 s (2H, OCH2), 6.82 d (2H, J = 8.80 Hz, HAr), 7.23 d (2H, J = 8.80 Hz, HAr), 7.41–7.65 m (5H, HAr), 10.11 s (1H, NH). 13C NMR spectrum, δ, ppm: 14.34, 23.55, 61.50, 114.93 (2C), 121.62 (2C), 123.85, 125.51, 129.06 (2C), 129.87 (2C), 133.44, 139.29, 144.57, 150.23, 162.80, 169.78. Found, %: C 59.53; H 4.78; N 14.75. C19H18N4O3S. Calculated, %: C 59.67; H 4.74; N 14.65. MS: m/z: 382 [M]+.
Ethyl 5-((4-acetamidophenyl)imino)-4-(p-tolyl)-4,5-dihydro-1,3,4-thiadiazole-2-carboxylate (11b). Brown powder, yield 74%, mp 266–268°C. IR spectrum, ν, cm–1: 3360 (N–H), 1735, 1647 (C=O). 1H NMR spectrum, δ, ppm: 1.27 s (3H, CH3), 2.09 s (3H, CH3), 2.39 s (3H, CH3), 4.18 s (2H, OCH2), 6.93 d (2H, J = 8.80 Hz, HAr), 7.38 d (2H, J = 8.90 Hz, HAr), 7.56–7.78 m (4H, HAr), 10.01 s (1H, NH). 13C NMR spectrum, δ, ppm: 16.05, 23.32, 24.71, 61.56, 114.58, 118.40 (2C), 119.71 (2C), 123.83, 128.55 (2C), 129.70 (2C), 131.91, 133.22, 140.53, 150.70, 165.81, 168.58. Found, %: C 60.76; H 5.03; N 14.25. C20H20N4O3S. Calculated, %: C 60.59; H 5.08; N 14.13. MS: m/z: 396 [M]+.
Ethyl 5-((4-acetamidophenyl)imino)-4-(4-chlorophenyl)-4,5-dihydro-1,3,4-thiadiazole-2-carboxylate (11c). Brown powder, yield 70%, mp 284–286°C. IR spectrum, ν, cm–1: 3369 (N–H), 1739, 1665 (C=O). 1H NMR spectrum, δ, ppm: 1.28 s (3H, CH3), 2.09 s (3H, CH3), 4.20 s (2H, OCH2), 6.93 d (2H, J = 8.80 Hz, HAr), 7.35 d (2H, J = 8.40 Hz, HAr), 7.61–7.75 m (4H, HAr), 10.15 s (1H, NH). 13C NMR spectrum, δ, ppm: 14.92, 24.35, 61.82, 114.30 (2C), 122.34 (2C), 123.66, 125.81, 129.10 (2C), 129.93 (2C), 133.24, 139.15, 144.64, 152.61, 162.77, 169.85. Found, %: C 54.56; H 4.18; N 13.35. C19H17ClN4O3S. Calculated, %: C 54.74; H 4.11; N 13.44. MS: m/z: 416 [M]+.
Cytotoxicity assay. Using the MTT assay, the above-mentioned cell line was used to determine the inhibitory effects of the thiazole and thiadiazole compounds on cell growth [52,53]. This colorimetric test relies on mitochondrial succinate dehydrogenase in practical cells to convert the yellow 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) to a purple formazan derivative. Hep2 was cultivated in RPMI-1640 medium containing 10% fetal bovine serum. At 37°C in a 5% CO2 incubator, anti-toxins included 100 units/mL penicillin and 100 µg/mL streptomycin. The cell line was seeded in a 96-well plate at a density of 1 × 104 cells/well [54] for 48 h at 37°C in a 5 percent CO2 environment. The cells were incubated for 24 h after being treated with different concentrations of compounds. Following 24 h of medication treatment, 20 µL of 5 mg/mL MTT solution was added and incubated for 4 h. To dissolve the purple formazan formed, 100 µL of dimethyl sulfoxide (DMSO) is added to each well. The colorimetric test is measured and recorded at 570 nm absorbance using a plate reader (EXL 800) from the United States. The relative cell viability was calculated as (A570 of treated samples/A570 of untreated samples) ×100.
CONCLUSIONS
Treatment of 2-iminothiazolidine-4-one compound 3b with some aromatic aldehydes affords 5-arylidene-2-phenyliminothiazolidin-4-one derivatives 5a–5e. Thiazole derivatives 6a and 6b were synthesized upon cyclizing N-(4-acetamidophenyl)-N'-phenylthiourea (2) with chloroacetone and/or phenacyl chloride. A new series of 5-substitiutedthiadiazole scaffolds, 9a–9c and 11a–11c were synthesized via intramolecular cyclization of thiourea derivative 2 with the appropriate hydrazonoyl halides 7a–c and 10a–c in ethanol containing an amount of triethylamine. The synthesized thiazole and thiadiazole compounds exhibited moderate to strong cytotoxic effects on four human cancer cell lines (HepG2, MCF-7, HTC-116, and PC-3). The best activity was obtained with compound 5d against hepatocellular carcinoma (IC50 8.80 ± 0.31 μg/mL), mammary gland breast cancer (IC50 7.22 ± 0.65 μg/mL) and colorectal carcinoma (IC50 9.35 ± 0.61 μg/mL) cell lines compared with the reference drug (5-fluorouracil).
REFERENCES
Khasawneh, M.A., AlKaabi, A., Samadi, A., Antony, P., Vijayan, R., Al-Keridis, L.A., Saadeh, H.A., and Abutah, N., Arab. J. Chem., 2022, vol. 15, no. 7, p. 103905. https://doi.org/10.1016/j.arabjc.2022.103905
Kondo, H., Koshizuka, T., Majima, R., Takahashi, K., Ishioka, K., Suzutani, K., and Inoue, N., Antivir. Res., 2021, vol. 196, p. 105207. https://doi.org/10.1016/j.antiviral.2021.105207
Ray, U., John, F., Pooppadi, S., George, J., Sma, S., and Raghavan, S.C., J. Heterocycl. Chem., 2021, vol. 58, no. 1, p. 40. https://doi.org/10.1002/jhet.4145
Abbas, S.Y., El-Sief, M.A.M.Sh., Basyouni, W.M., Fakhr, I.M.I., and El-Gammal, E.W., Eur. J. Med. Chem., 2013, vol. 64, p. 111. https://doi.org/10.1016/j.ejmech.2013.04.002
Al-Harbi, R.A., El-Sharief, M.A.S., and Abbas, S.Y., Bioorg. Chem., 2019, vol. 90, p. 103088. https://doi.org/10.1016/j.bioorg.2019.103088
Nirwan, S, Chahal, V., and Kakkar, R., J. Heterocycl. Chem., 2019, vol. 56, no. 4, p. 1239. https://doi.org/10.1002/jhet.3514
Abdellatif, K.R., Abdelgawad, M.A., Elshemy, H.A., and Alsayed, S.S., Bioorg. Chem., 2016, vol. 64, p. 1. https://doi.org/10.1016/j.bioorg.2015.11.001
Aqlan, F.M., Al-Bogami, A.S., Alqahtani, N.F., Wani, M.Y., and Khan, S.A., J. Mol. Struct., 2022, vol. 1250, no. 2, p. 131771. https://doi.org/10.1016/j.molstruc.2021.131771
Сhulovska, Z., Chaban, T., Drapak, I., Matiychuk, V., Chaban, I., and Nektegaev, I., , Biointerface Res. Appl. Chem., 2021, vol. 11, no. 1, p. 8009. https://doi.org/10.33263/BRIAC111.80098017
Mandal, M.K., Ghosh, S., Naesens, L., RajBhat, H., and Singh, U.P., Bioorg. Chem., 2021, vol. 114, p. 105153. https://doi.org/10.1016/j.bioorg.2021.105153
Gummidi, L., Kerru, N., Ebenezer, O., Awolade, P., Sanni, O., Islam, Md. S., and Singh, P., Bioorg. Chem., 2021, vol.115, p. 105210. https://doi.org/10.1016/j.bioorg.2021.105210
Vaarla, K., Vishwapathi, V., Vermeire, K., Vedula, R.R., and Kulkarni, C.V., J. Mol. Struct., 2022, vol. 1249, p. 131662. https://doi.org/10.1016/j.molstruc.2021.131662
Trotsko, N., Eur. J. Med. Chem., 2021, vol. 215, p. 113266. https://doi.org/10.1016/j.ejmech.2021.113266
Jain, S., Kumar, A., and Saini, D., Exp. Parasitol., 2018, vol. 185, p. 107. https://doi.org/10.1016/j.exppara.2018.01.015
Omar, Y.M., Abdel-Moty, S.G., and Abdu-Allah, H.H.M., Bioorg. Chem., 2020, vol. 97, p. 103657. https://doi.org/10.1016/j.bioorg.2020.103657
Bielenica, A., Szulczyk, D., Olejarz, W., Madeddu, S., Giliberti, G., Materek, I.B., Koziol, A.E., and Struga, M., Biomed. Pharmacother., 2017, vol. 94, p. 804. https://doi.org/10.1016/j.biopha.2017.07.152
Djukica, M., Fesatidoub, M., Xenikakisb, I., Geronikakib, A., Angelovac, V.T., Savicd, V., Pasica, M., Krilovica, B., Djukice, D., Gobeljica, B., Pavlicae, M., Djurica, A., Stanojevicf, I., Vojvodicf, D., and Sasog, L., Chem. Biol. Interact., 2018, vol. 286, p. 119. https://doi.org/10.1016/j.cbi.2018.03.013
Rahim, F., Zaman, K., Ullah, H., Taha, M., Wadood, A., Javed, M.T., Rehman, W., Ashraf, M., Uddin, R., Uddin, I., Asg, H., Khan A.A., and Khan, K.M., Bioorg. Chem., 2015, vol. 63, p. 123. https://doi.org/10.1016/j.bioorg.2015.10.005
Abumelha, H.M.A., and Saeed, A., J. Heterocycl. Chem., 2020, vol. 57, p. 1816. https://doi.org/10.1002/jhet.3906
Kryshchyshyn-Dylevych, A., Radko, L., Finiuk, N., Garazd, M., Kashchak, N., Posyniak, A., Niemczuk, K., Stoika, R., Lesyk, R., Bioorg. Med. Chem., 2021, vol. 50, no.15, p. 116453. https://doi.org/10.1016/j.bmc.2021.116453
Appalanaidu, K., Kotcherlakota, R., Dadmal, T.L., Bollu, V.S., Kumbe, R.M., and Patra, C.R., Bioorg. Med. Chem. Lett., 2016, vol. 26, p. 5361. https://doi.org/10.1016/j.bmcl.2016.08.013
Kumar, K.S.S., Hanumappa, A., Vetrivel, M., Hegde, M., Girish, Y.R., Byregowda, T.R., Rao, S., Raghavan, S.C., and Rangappa, K.S., Bioorg. Med. Chem. Lett., 2015, vol. 25, p. 3616. https://doi.org/10.1016/j.bmcl.2015.06.069
Mabkhot, Y.N., Khaled, J.M.A., Alharbi, N.S.H.A., Mohammed, F.A.N., Almekhlafi, F.A., Abutaha, N.M., Kheder, N., Asiri, Y.I., Bin Muhsinah, A., and Alsayari, A., Polycycl. Aromat. Compd., 2022. https://doi.org/10.1080/10406638.2021.1984952
Modrić, M., Božičević, M., Faraho, I., Bosnar, M., and Škorić, I., J. Mol. Struct., 2021, vol. 1239, p. 130526. https://doi.org/10.1016/j.molstruc.2021.130526
Kumara, G. and Singh, N.P., Bioorg. Chem., 2021, vol. 107, p. 104608. https://doi.org/10.1016/j.bioorg.2020.104608
Pember, S.O., Mejia, G.L., Price, T.J., and Pasteris, R.J., Bioorg. Med. Chem. Lett., 2016, vol. 26, p. 2965. https://doi.org/10.1016/j.bmcl.2016.02.061
Kasralikar, H.M., Jadhavar, S.C, Goswami, S.V., Kaminwar, N.S., and Bhusare, S.R., Bioorg. Chem., 2019, vol. 86, p. 437. https://doi.org/10.1016/j.bioorg.2019.02.006
Rzeski, W., Matysiak, J., and Kandefer-Szerszen, M., Bioorg. Med. Chem., 2007, vol. 15, p. 3201. https://doi.org/10.1016/j.bmc.2007.02.041
Puszkiel, A., Noé, G., Bellesoeur, A., Kramkimel, N., Paludetto, M.N., Thomas-Schoemann, A., Vidal, M., Goldwasser, F., Chatelut, E., and Benoit, B., Clin. Pmacokinet., 2019, vol. 58, no. 4, p. 451. https://doi.org/10.1007/s40262-018-0703-0
Rashdan, H.R.M., Abdelmonsef, A.H., AbouKrisha, M.M., and Yousef, T.A., Molecules, 2021, vol. 26, no. 23, p. 7119. https://doi.org/10.3390/molecules26237119
Shehadi, I.A., Abdelrahman, M.T., Abdelraof, M., and Rashdan, H.R.M., Molecules, 2022, vol. 27, no. 2, p. 342. https://doi.org/10.3390/molecules27020342
Janowska, S., Khylyuk, D., Bielawska, A., Mandziuk, S., and Wujec, M., Molecules, 2022, vol. 27, no. 6, p. 1814. https://doi.org/10.3390/molecules27061814
Altıntop, M.D., Sever, B., Özdemir, A., Ilgın, S., Atlı, Ö., Turan-Zitouni, G., and Kaplancıklı, Z. A., Anti-Cancer Agents Med. Chem., 2018, vol. 18, no. 11, p. 1606. https://doi.org/10.2174/1871520618666180509111351
Obakachi, V.A., Kushwah, B., Kushwaha, N.D., Mokoena, S., Ganai, A.M., Pathan, T.K., Zyl, W.E., and Karpoormath, R., J. Sulfur Chem., 2021, vol. 42, no. 6, p. 670. https://doi.org/10.1080/17415993.2021.1963441
Khadri, M.J.N., Begum, A.B., Sunil, M.K., and Khanum, S.A., Results Chem., 2020, vol. 2, p. 100045. https://doi.org/10.1016/j.rechem.2020.100045
Can, N.Ö., Can, Ö.D., Osmaniye, D., and Özkay, Ü.D., Molecules, 2018, vol. 23, no. 4, p. 716. https://doi.org/10.3390/molecules23040716
Rashdan, H.R.M., Shehadi, I.A., Abdelrahman, M.T., and Hemdan, B.A., Molecules, 2021, vol. 26, no. 16, p. 4817. https://doi.org/10.3390/molecules26164817
Sabt, A., Abdelrahman, M.T., Abdelraof, M., and Rashdan, H.R.M., ChemistrySelect, 2022, vol. 7, no. 17, p. e202200691. https://doi.org/10.1002/slct.202200691
Luszczki, J.J., Karpińsk, M., Matysiak, J., and Niewiadomy, A., Pharmacol. Rep., 2015, vol. 67, no. 3, p. 588. https://doi.org/10.1016/j.pharep.2014.12.008
Chaban, T., Matiychuk, V., Сhulovska, Z., Myrko, I., Drapak, I., Sogujko, R., Chaban, I, Ogurtsov, V., and Nektegaev, I., Biointerface Res. Appl. Chem., 2022, vol. 12, no.6, p. 7226. https://doi.org/10.33263/BRIAC126.72267238
Rashdan, H.R.M., Abdelmonsef, A.H., AbouKrisha, M.M., and Yousef, T.A., Biointerface Res. Appl. Chem., 2022, vol. 12, no. 6, p. 8258. https://doi.org/10.33263/BRIAC126.82588270
Toan, V.N., Thanh, N.D., and Tri, N.M., Arab. J. Chem., 2021, vol. 14, no. 4, p. 103053. https://doi.org/10.1016/j.arabjc.2021.103053
Taha, M., Rahim, F., Uddin, N., Khan, I.U., Iqbal, N., Anouar, E., Salahuddin, M., Farooq, R.K., Gollapalli, M., Khan, K.M., and Zafar A., Int. J. Biol. Macromol., 2021, vol. 188, p. 1025. https://doi.org/10.1016/j.ijbiomac.2021.08.065
Makhaeva, G.F., Kovaleva, N.V., Boltneva, N.P., Lushchekina, S.V., Rudakova, E.V., Stupina, T.S., Terentiev, A.A., Serkov, I.V., Proshin, A.N., Radchenko, E.V., Palyulin, V.A., Bachurin, S.O., and Ricdson, R.J., Bioorg. Chem., 2020, vol. 94, p. 103387. https://doi.org/10.1016/j.bioorg.2019.103387
Keating, G.M., Drugs, 2017, vol. 77, no. 1, p. 85. https://doi.org/10.1007/s40265-016-0677-x
Matysiak, J. and Opolski, A., Bioorg. Med. Chem., 2006, vol. 14, p. 4483. https://doi.org/10.1016/j.bmc.2006.02.027
Tiwari, S.S. and Swaroop, A., J. Indian Chem. Soc., 1961, vol. 38, p. 245.
Lagergren, P., Schandl, A., Aaronson, N.K., Adami, H.O., Lorenzo, F., Denis, L., Faithfull, S., Liu, L., Meunier, F., and Ulrich,C., Mol. Oncol., 2018, vol. 13, no. 3, p. 624. https://doi.org/10.1002/1878-0261.12428
Bhat, M., Poojary, B., Kalal, B.S., Swamy, P.M.G., Kabilan, S., Kumar, V., Shruthi, N., Anand, S.A.A., and Pai, V.R., Future Med. Chem. 2018, vol. 10, no. 9, p. 1017. https://doi.org/10.4155/fmc-2017-0191
Beretta, G.L., Cassinelli, G., Pennati, M., Zuco, V., and Gatti, L., Eur. J. Med. Chem., 2017, vol. 142, p. 271. https://doi.org/10.1016/j.ejmech.2017.07.062
Zwick, E., Bange, J. and Ullrich, A., Endocr. Relat. Cancer, 2001, vol. 8, p. 161.
Mosmann, T., J. Immunol. Methods. 1983, vol. 65, p. 55. https://doi.org/10.1016/0022-1759(83)90303-4
Denizot, F., and Lang, R., J. Immunol. Methods, 1986, vol. 89, p. 271. https://doi.org/10.1016/0022-1759(86)90368-6
Weichselbaum, R.R., Mauceri, H.J., Hanna, N.N., Beckett, M.A., Gorski, D.H., Staba, M.-J., Stellato, K.A., Bigelow, K., Heimann, R., Gately, S., Dhanabal, M., Soff, G.A., Sukhatme, V.P., and Kufe, D.W., Nature, 1998, vol. 394, p. 287. https://doi.org/10.1038/28412
ACKNOWLEDGMENTS
The authors extend their appreciation to the Deputyship for Research & Innovation, Ministry of Education in Saudi Arabia for funding this research work through the project number “IF_2020_NBU_234.”
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
No conflict of interest was declared by the authors.
Rights and permissions
About this article

Cite this article
El-Rayyes, A., Soliman, A.M. & Saeed, A. Synthesis and Anticancer Evaluation of New Thiazole and Thiadiazole Derivatives Bearing Acetanilide Moiety. Russ J Gen Chem 92, 2132–2144 (2022). https://doi.org/10.1134/S1070363222100267
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1070363222100267