
Abstract
The reaction of 2,6-di(tert-butyl)anthracene with potassium graphite and monocyclopentadienyllutetium dichloride tetrahydrofuranate in THF gave the anthracenide complex [(η5-C5H5)Lu(η2-2,6-tBu2C14H8)(THF)2] (I), which was studied by X-ray diffraction (CCDC no. 2215512). Complex I crystallizes in the orthorhombic space group P212121. The structural rigidity of the Lu(O)2Cp(anthracene) crystallographic node was demonstrated. The retention of the structure of complex I in solution was confirmed by NMR techniques.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
The choice of ligands plays a considerable role in the chemistry of coordination and organometallic compounds of rare earth elements (REE). According to the common views [1], the formation of kinetically stable REE complexes requires optimization of the electrostatic ligand–metal interactions, provided by negatively charged ligands, and saturation of the metal coordination sphere, which is attained by using bulky ligands. These criteria are met by 4n + 2 π-electron aromatic ligands such as various substituted and unsubstituted cyclopentadienyl anions (Cp'–), cyclooctatetraene dianions (COT'2–), and their analogues. These ligands are present in most organometallic complexes of lanthanides. It is evident that aromatic hydrocarbon dianions and their heteroatomic analogues perfectly meet both of the above criteria. Furthermore, owing to the higher negative charge, these ligands should provide the kinetic stability of REE compounds even at lower steric hindrance than the conventional Cp'– and COT'2– ligands. Nevertheless, this area of REE organometallic chemistry is still relatively little studied. The few studies of these compounds are mainly focused on complexes like [LnX2\(]_{2}^{ + }\)[(μ-L)]– [2–8], where X– is an auxiliary monoanion ligand and L2– is the dianion ligand. Such a metal–ligand system can be represented as a three-ion structure composed of two singly charged cations and anthracene (or another aromatic hydrocarbon) dianion. However, of much greater interest are systems such as [XLnL]0 [9–11] or [\({\text{X}}_{2}^{ - }\)Ln3+L2–]– [12–14]. The molecules or complex anions in these compounds contain simultaneously a strong Lewis acid (Ln3+) and a strong Lewis base (L2–). Therefore, they can be regarded as being analogous to sterically hindered frustrated Lewis pairs [15–17] and, hence, similar reactivity should be expected. Complexes based on the dianions of aromatic hydrocarbons such as anthracene, naphthalene, their analogues, etc. refer to this type of compounds. Although anthracene dianion has long been used in the REE organometallic chemistry [9, 13, 18, 19], complexes with this ligand have been little studied. Moreover, REE complexes with substituted anthracene dianion are unknown.
The purpose of this study is to elucidate characteristic features of coordination of the anthracene dianion containing bulky alkyl (tert-butyl) substituents and to identify the potential electronic effects influencing characteristics of the Ln–ligand interactions that may be induced by the introduction of these substituents, in comparison with analogous REE complexes containing unsubstituted anthracenide ligand.
EXPERIMENTAL
The synthetic operations were carried out in the purified argon atmosphere in anhydrous solvents using the SPEKS-GB2 glove box. Tetrahydrofuran was distilled from potassium/benzophenone. Hexane was distilled from potassium–sodium eutectics/benzophenone. Toluene was distilled from sodium/benzophenone. LuCl3(THF)3 was obtained by the procedure reported in [20]. Cyclopentadienyl sodium was prepared by the procedure described in [21], and KC8 was obtained by the procedure of [22]. 2,6-Di(tert-butyl)anthracene was synthesized as described in [23] and purified prior to use by sublimation in the dynamic vacuum of 5 × 10–2 mm Hg. Elemental analysis was carried out on a Thermo Scientific FLASH 2000 CHNS/O Analyzer. The content of lutetium was determined by complexometric titration with the xylenol orange indicator.
1H, 13C{1H}, 1H–1H COSY, 13C–1H HSQC, and 13C–1H HMBC spectra were recorded on a Bruker DRX500 instrument.
Synthesis of [(η5-C5H5)(η2-2,6-tBu2C14H8)Lu-(THF)2] (I). LuCl3(THF)3 (2 mmol, 995 mg) was suspended in THF (20 mL), and 2,6‑di(tert-butyl)anthracene (2 mmol, 580 mg) was added to the suspension. A solution of CpNa (2 mmol, 176 mg) in THF (10 mL) was added dropwise to the resulting mixture over a period of 5 min. During this period, the suspension dissolved to give a transparent pale yellow solution, which was stirred for 30 min. Then potassium graphite (KC8) (0.649 g, 4.8 mmol, 20% excess) was added in portions with stirring, the mixture was stirred for 12 h, and the precipitate was separated by centrifugation (6000 rpm, 15 min). The solution was evaporated in vacuum, the solid precipitate was dissolved in THF (10 mL), a layer of hexane (30 mL) was carefully added, and the mixture was left for crystallization for 5 days. This gave dark orange crystals, which were separated from the solution by decanting, and dried in vacuum. The yield of I was 1.215 g (90%).
For C35H47O2Lu | |||
Anal. calcd., % | Lu, 25.93 | C, 62.30 | H, 7.02 |
Found, % | Lu, 25.49 | C, 61.14 | H, 6.62 |
1H NMR (THF-d8; δ, ppm): 1.02 (s, t-Bu, 18H), 1.58 (THF-d7), 1.63 (m, THF, 8H), 3.41 (s, H9, H10, 2H), 3.43 (THF-d7), 3.47 (m, THF, 8H), 5.50 (s, Cp, 5H), 5.96 (d, J = 7.8 Hz, H4, H8, 2H), 6.11 (d, J = 1.9 Hz, H1, H5, 2H), 6.24 (dd, J = 7.8, 1.9 Hz, H3, H7, 2H). 13C{1H} NMR (THF-d8; δ, ppm): 22.0 (THF), 23.1 (THF-d8), 28.9 (t-Bu-CH3), 31.1 (Ct-Bu), 63.7 (C9, C10), 64.1 (THF), 65.0 (THF-d8), 107.1 (Cp), 114.2 (C1, C5), 115.1 (C3, C7), 116.7 (C4, C8), 133.2 (C2, C6), 135.6 (C9A, C10A), 140.5 (C4A, C8A).
The crystals suitable for X-ray diffraction were obtained by slow diffusion of hexane into a THF solution of I.
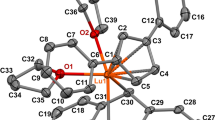
X-ray diffraction study of complex I was carried out on a Bruker SMART APEX II diffractometer (MoKα radiation, λ = 0.71073Å, graphite monochromator, ω‑scan mode). The reflection intensities were found using the SAINT software [24]. The absorption corrections were applied semiempirically on the basis of equivalent reflections using the TWINABS program [24]. The structures were solved by direct methods with the SHELXT program [25] and refined by the least squares method in the anisotropic full-matrix approximation on \(F_{{hkl}}^{2}\) using the SHELXL-2018 program [26]. One coordinated THF molecule was disordered over two positions (C(24)…C(27) atoms) with occupancy ratio of 0.71(4) : 0.29(4) (see Fig. 1b). One tert-butyl group (C(15)…C(18) atoms) was also disordered over two positions (0.79(1) : 0.21(1)). The disordered groups were refined with restraints on atomic displacements and position parameters (SADI and EADP instructions of SHELXL). The hydrogen atoms were calculated by the rigid body model (C–H distances: 0.950 Å for aromatic, 0.980 Å for methyl, 0.990 Å for methylene, and 1.000 Å for cyclopentadienyl hydrogen atoms) and refined in the relative isotropic approximation with Uiso(H) = 1.5Ueq(C) for methyl groups and Uiso(H) = 1.2Ueq(C) for other hydrogen atoms. The rotating methyl group model was used. The key crystallographic data and refinement details for compound I are summarized in Table 1. The Mercury program was used to minimize and calculate the mean-square deviations of atomic positions for structure comparison [27].

(a) Molecular structure of complex I with p = 50% without allowance for disorder; (b) more detailed structure of complex I with short Lu–C contacts and disordered coordinated THF molecule and tert-butyl group.
The atomic coordinates and other parameters of the structures were deposited with the Cambridge Crystallographic Data Centre (CCDC no. 2215512 (I), deposit@ccdc.cam.ac.uk or http://www.ccdc.cam.ac.uk/data_request/cif).
RESULTS AND DISCUSSION
The few known crystal structures of lanthanide anthracenide complexes (CCDC, version 2022.2.0 [21, 22]) can be conventionally divided into three groups depending on the type of coordination of the dianionic ligand: bis-allyl bridging μ2-η3:η3 type (A), bridging μ2‑η4:η4 or μ2‑η6:η6 type (B), and η2 type with the predominant two-center HOMO localization in the dianion (C); the complexes also contain the monoanionic X ligand (Scheme 1). Type A, \(\left\{ {\left[ {{\text{L}}{{{\text{n}}}^{{3 + }}}\left( {{{{\text{C}}}_{{\text{5}}}}{\text{M}}{{{\text{e}}}_{{\text{5}}}}} \right)_{2}^{ - }} \right]_{2}^{ + }} \right.\)(μ2‑η3:η3‑C14H10)2–\(\left. {_{{_{{}}^{{}}}}^{{_{{_{{}}^{{}}}}^{{}}}}} \right\}\), contains a planar bridging anthracene dianion and two bulky [Ln3+Cp\(_{2}^{{* - }}\)]+ complex cations (Ln = La, Sm; CCDC codes: NAGSOD [18] and WEVNAM [19]). The coordination between the Ln3+ cation and the dianion involves atoms located in positions 4, 4a, 10 and 8, 8a, 9; the mode of coordination of the dianion is best described as the bis-allyl coordination. In \(\left\{ {\left[ {{\text{L}}{{{\text{n}}}^{{3 + }}}{\text{X}}_{2}^{ - }} \right]_{2}^{ + }} \right.\)(C14H10)2–\(\left. {_{{_{{}}^{{}}}}^{{_{{_{{}}^{{}}}}^{{}}}}} \right\}\) complexes of type B, where \({\text{X}}_{2}^{ - }\) is either two monoanion ligands or one dianion ligand with spatially separated charges, Ln3+ is coordinated to positions 1, 2, 3, 4 and 8a, 9, 10, 10a (μ2-η4:η4) or 1, 2, 3, 4, 4a, 9a and 4a, 8a, 9, 9a, 10, 10a (μ2-η6:η6) on the opposite sides of the C14H102– dianion, the coordinated rings of which are bent or slightly bent along the straight lines that pass through the carbon atoms in positions 9, 10 and 1, 4 (UXUMIK [3], WIYLIZ [6], SIRRAO [5]). A similar coordination of planar anthracene is found in the ytterbium(II) complex {[Yb2+X–]\(_{2}^{ + }\)(μ2-η4:η4-C14H10)2–} (ABONIS [2], where X is a monoanion chelating ligand). In this type of complexes, encountered only for Sc3+, Y3+, and Yb2+ cations, the coordination of the anthracene dianion to the REE cation resembles, to some extent, the coordination of aromatic ligands in classical d-metal π-complexes.

Scheme 1 .
In the mononuclear [X–Ln3+(η2-C14H10)2–] complexes of type C (QOFFIB [11], UCOXEP [28], VUJDAF [9]) and in the [\({\text{X}}_{2}^{ - }\)Ln3+(η2‑C14H10)2–]– complex anions (NURLOE, NURLUK, NURMAR [12], YA-XSUL [13], YEMYEV [4]), the nonplanar dianion has mainly a two-center localization of HOMO (positions 9 and 10). In some cases, this η2-coordination is described in the literature as η4 or even unsymmetrical η6 coordination, because there are four short contacts with atoms of the central ring, two shorter (in positions 4a and 9a) and two longer ones (in positions 8a and 10a). Coordination types A and C are both observed in the scandium(III) anthracenide ate complex, [{(η5‑1,3‑Ph2C5H3)Sc(η2-C14H10)}2(μ2‑η3:η3‑C14H10)] (QIBKIY) [29].
The least studied type C, which is typical for Ln3+ cations with small ion radii (late lanthanides, Sc, and Y), is most interesting.
It is noteworthy that metal complexes based on substituted anthracene dianion have not been previously described in the literature except for magnesium complexes with 1,4-dimethylanthracene [30] and 9,10-bis(trimethylsilyl)anthracene [31, 32] dianions.
The [(η5-C5H5)Lu(η2-2,6-tBu2C14H8)(THF)2] complex (I) was prepared (Scheme 2) by the reaction of dipotassium 2,6-di(tert-butyl)anthracene derivative, generated in situ by treatment of 2,6-di(tert-butyl)anthracene with potassium graphite (2 equiv.), with [(C5H5)LuCl2(THF)3], generated in situ from lutetium chloride tetrahydrofuranate and a stoichiometric amount of sodium cyclopentadienide, similarly to the procedure used in our previous study to prepare type C lutetium anthracenide complexes [9, 11, 13]. The structure of I was determined by X-ray diffraction (Fig. 1).

Scheme 2 .
Complex I is constructed similarly to two other known complexes of this type: [(η5-C5H5)Lu(η2-C14H10)(THF)2] (II) (Z' = 2) [9] and [(η5-C5H4CH2CH2PPh2)Lu(η2-C14H10)(DME)]·(DME) (III) [11]. Due to the essential localization of HOMO and LUMO of the 2,6-di(tert-butyl)anthracene dianion on the C(9) and C(10) atoms and to the dianion interaction with the highly polarizing Lu3+ cation, the dianion is η2-coordinated to the metal via these atoms, with the corresponding bond lengths (Lu–C(9)/C(10), Table 2) being similar to those in II (2.43(1)–2.463(9) Å) and III (2.399(6), 2.436(7) Å). The dianion is folded along the C(9)–C(10) line, and the folding angle is 31.08(11)° in I (29.9°–38.7° in II and III). Like complexes II and III, the molecule of I has four short contacts with central ring atoms, including two shorter contacts (with C(8a) and C(10a), Table 2) and two longer contacts (with C(4a) and C(9a)). Analogous distances in II and III are in the ranges of 2.681–2.720 Å and 2.839–3.011 Å.
The Ln–CCp bond lengths in I (Table 2) are equal within the ESDs, while the Ln–Cp(centroid) distance and the Ln–Cp(plane) perpendicular length are equal, which is indicative of symmetric η5-coordination of the Cp ligand. While comparing the Lu–CCp, Lu–Cp(centroid), Lu–Cp(plane), and CCp–CCp distances in I, II, and III, it can be noticed that differences are present only for the CCp–CCp bond lengths in III (1.410(9)–1.437(9) Å), which are, on average, slightly (by 0.03 Å) elongated compared to those in I and II, because of the presence of the alkyl substituent in the ring. The Lu3+ coordination number is 7, like in II and III, but this is lower than the C.N. in most organolutetium compounds.
The Lu–O distances in I (Table 2) are in the range typical of coordinated σ-donor neutral solvent molecules (THF, DME, etc.). In particular, in II and III, these distances are 2.290(6)–2.364(7) Å.
A comparison of the conformations of the Lu(O–solv)2(C5–Cp)(C14–anthracene) crystallographic node in the series of lutetium complexes I, II, and III showed that they virtually do not change. Note that despite the postulated predominantly ionic Ln–ligand bond [33], the relative positions of the ligands, interatomic interactions, and bond lengths do not depend on the crystal packing effects, which are a priori different in non-isostructural compounds. The root-mean-square deviations (RMSD) for the positions of Lu atoms, anthracenide (C14) and cyclopentadienyl (C5) carbon atoms, and two oxygen atoms are rather low, amounting to 0.133 Å (22 pairs of atoms; the superimposition of the complexes is shown in Fig. 2). When oxygen atoms are not included, this value decreases to 0.109 Å (20 pairs of atoms). RMSD for I and for one molecule of II for the same 22 pairs of skeletal atoms is 0.100 Å.

Superposition of similar structural moieties of complexes I (dark gray) and III (light gray) without taking into account the disorder in I.
In order to find out whether or not the structure of I changes upon transition from the crystalline phase to a solution, compound I was studied by NMR spectroscopy in THF-d8. According to the 1H, 13C{1H}, 1H–1H COSY, and 13C–1H HSQC NMR data, the structure of the complex is retained in the solution.
The upfield shifts of the H(9) and H(10) proton signals and the C(9) and C(10) carbon signals in the 1H and 13C{1H} NMR spectra with respect to other signals of the anthracenide ligand indicate that the non-planar structure of the anthracenide ligand folded along the C(9)–C(10) line is retained in solution. The 2D 1H–1H COSY and 13C–1H HSQC NMR spectra (Fig. 3) fully confirm the assumed structure of complex I. This enables full assignment of signals of the anthracenide ligand. As expected, the 1H–1H COSY NMR spectrum shows cross-peaks corresponding to 3JHH for the H(3) (H7) and H(4) (H8) protons of the anthracenide ligand and for THF protons. In the case of H(1) (H5) and H(3) (H7) protons, relatively weak cross-peaks (4JHH) are observed. Only diagonal peaks are manifested for H(9) (H10).

(a) 1H–1H COSY NMR and (b) 13C–1H HSQC NMR spectra of complex I in THF-d8.
Thus, in this study, a REE complex with anthracenide ligand containing bulky alkyl substituents, [(η5-C5H5)Lu(η2-2,6-tBu2C14H8)(THF)2], was prepared for the first time. The structure of I in the crystal was studied by X-ray diffraction and the structural rigidity of the complex was demonstrated. It was shown by NMR spectroscopy that the structure of the complex is retained in solution.
REFERENCES
Evans, W.J., Polyhedron, 1987, vol. 6, p. 803.
Richardson, G.M., Douair, I., Cameron, S.A., et al., Chem.-Eur. J., 2021, vol. 27, p. 13144.
Huang, W., Khan, S.I., and Diaconescu, P.L., J. Am. Chem. Soc., 2011, vol. 133, p. 10410.
Huang, W., Dulong, F., and Wu, T., Nature Commun., 2013, vol. 4, р. 1448.
Huang, W., Abukhalil, P.M., Khan, S.I., and Diaconescu, P.L., Chem. Commun., 2014, vol. 50, p. 5221.
Fryzuk, M.D., Jafarpour, L., and Kerton, F.M., Angew. Chem., Int. Ed. Engl., 2000, vol. 39, p. 767.
Huang, W. and Diaconescu, P.L., Chem. Commun., 2012, vol. 48, p. 2216.
Huang, W. and Diaconescu, P.L., Eur. J. Inorg. Chem., 2013, p. 4090.
Roitershtein, D.M., Ellern, A.M., Antipin, M.Y., et al., Mendeleev Commun., 1992, vol. 2, p. 118.
Protchenko, A.V., Zakharov, L.N., Bochkarev, M.N., and Struchkov, Y.T., J. Organomet. Chem., 1993, vol. 447, p. 209.
Roitershtein, D.M., Romanenkov, A.V., Lyssenko, K.A., et al., Russ. Chem. Bull., 2007, vol. 56, p. 1749.
Ghana, P., Hoffmann, A., Spaniol, T.P., and Okuda, J., Chem.-Eur. J., 2020, vol. 26, p. 10290.
Roitershtein, D.M., Rybakova, L.F., and Petrov, E.S., J. Organomet. Chem., 1993, vol. 460, p. 39.
Cassani, M.C., Gun’ko, Yu.K., Hitchcock, P.B., et al., Organometallics, 1999, vol. 18, p. 5539.
Stephan, D.W. and Erker, G., Angew. Chem., Int. Ed. Engl., 2015, vol. 54, p. 6400.
Stephan, D.W., Science, 2016, vol. 354, p. aaf7229.
Jupp, A.R. and Stephan, D.W., Trends Chem., 2019, vol. 1, p. 35.
Thiele, K.-H., Bambirra, S., Schumann, H., and Hemling, H., J. Organomet. Chem., 1996, vol. 517, p. 161.
Evans, W.J., Gonzales, S.L., and Ziller, J.W., J. Am. Chem. Soc., 1994, vol. 116, p. 2600.
Edelmann, F. and Poremba, P., in Synthetic Methods of Organometallic and Inorganic Chemistry, Edelmann, F.T. and Herrmann, W.A., Eds., Stuttgart: Thieme Medical, 1997, p. 34.
Panda, T.K., Gamer, M.T., and Roesky, P.W., Organometallics, 2003, vol. 22, p. 877.
Ottmers, D.M. and Rase, H.F., Carbon, 1966, vol. 4, p. 125.
Wang, J., Wan, W., Jiang, H., et al., Org. Lett., 2010, vol. 12, p. 3874.
APEX-III, Madison: Bruker AXS Inc., 2019.
Sheldrick, G.M., Acta Crystallogr., Sect. A: Found. Adv., 2015, vol. A71, p. 3.
Sheldrick, G.M., Acta Crystallogr., Sect. C: Struct. Chem., 2015, vol. C71, p. 3.
Macrae, C.F., Sovago, I., Cottrell, S.J., et al., J. Appl. Crystallogr., 2020, vol. 53, p. 226.
Fedushkin, I.L., Bochkarev, M.N., Dechert, S., and Schumann, H., Chem.-Eur. J., 2001, vol. 7, p. 3558.
Ellis, J.E., Minyaev, M.E., Nifant’ev, I.E., and Churakov, A.V., Acta Crystallogr., Sect. C: Struct. Chem. 2018, vol. C74, p. 769.
Bogdanovic, B., Janke, N., and Kruger, C., Angew. Chem., Int. Ed. Engl., 1985, vol. 24, p. 960.
Lehmkuhl, H., Shakoor, A., Mehler, K., Kruger, C., et al., Chem. Ber., 1985, vol. 118, p. 4239.
Alonso, T., Harvey, S., Junk, P.C., et al., Organometallics, 1987, vol. 6, p. 2110.
Raymond, K.N. and Eigenbrot, C., Acs. Chem. Res., 1980, vol. 13, p. 276.
Funding
This study was supported by the Russian Science Foundation (grant no. 22-23-00711).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflicts of interest.
Additional information
Translated by Z. Svitanko
ADDITIONAL INFORMATION
This article is prepared for the memorial issue in tribute to the Corresponding Member of the Russian Academy of Sciences K.Yu. Zhizhin on his 50th birthday.
Rights and permissions
Open Access. This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Roitershtein, D.M., Lyssenko, K.A., Nifant’ev, I.E. et al. Lutetium Cyclopentadienyl Complex with the 2,6-Di-tert-Butylanthracene Dianion. Russ J Coord Chem 49, 369–376 (2023). https://doi.org/10.1134/S1070328423700586
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1070328423700586