
Abstract—
Despite the efforts to develop the strategies of tuberculosis control, this disease still takes more than a million lives annually. The development of tuberculosis infection can be considered as an imbalance between the immune response of the host organism and the growth of Mycobacterium tuberculosis bacteria. In order to gain a foothold successfully in an infected organism, M. tuberculosis must overcome the mechanisms of innate immunity, including those that aim at the recognition of alien nucleic acids. RIG-I-like receptors (RLR) is a system of intracellular receptors (sensors of alien RNA), which is involved in the recognition of viruses and bacterial pathogens. The RIG-I, MDA5, and LGP2 receptors interact directly with RNA in the cell cytoplasm and trigger a cascade of interactions, which leads to the synthesis of type I interferons and proinflammatory cytokines. To date, it has been proven that the activation of RLR during tuberculosis infection is the most important component of innate immunity. Their obvious role in the activation of type I interferons (which, however, can be not only protective, but also negative for the immune system) was demonstrated. The review considers the latest data on the functioning of RLR in tuberculosis on the example of model organisms and humans.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
CONTENTS
INTRODUCTION
STRUCTURAL CHARACTERISTICS OF RLR AND HOW THEY FUNCTION
RLR—BACTERIAL RNA SENSORS
TYPE I INTERFERON FAMILY AND BACTERIAL INFECTION
ROLE OF MYCOBATERIAL RNA IN HOST IMMUNE RESPONSE
CONCLUSIONS
REFERENCES
INTRODUCTION
The activation of innate immunity in response to bacterial infection begins with the process of recognition of special microbial structures called pathogen-associated molecular patterns (PAMP) that include secreted virulence factors, cell wall components, as well as bacterial nucleic acids recently considered as the most important PAMP [1].
Several cellular systems are responsible for the recognition of pathogenic RNA. The diversity of pattern recognition receptors (PRRs) provides the reliability of pathogen identification and immune response specificity. Among them, there are receptors with a single transmembrane domain of the family of Toll-like receptors (TLR) and a system of cytoplasmic sensory factors, consisting of the proteins RIG-I (retinoic gene-inducible I), MDA5 (melanoma differentiation-associated protein 5), and LGP2 (laboratory of genetics and physiology 2), a family of RIG-like receptors (RLR). Both types of detecting systems are able to induce the production of type I interferons (IFN) and proinflammatory cytokines [2–5]. In recent years, data on other systems of cellular RNA sensors, including the NLRs family (NOD-like receptor, nucleotide-binding oligomerization domain-like receptors) [6, 7] and extracellular RNA receptors SRas (class A scavenger receptors) [8], also appeared.
TLRs are mainly expressed in antigen-presenting cells (in macrophages and plasmacytoid dendritic cells). These receptors face inside the endosomes; therefore, they can be activated during pathogen degradation as a result of endocytosis or phagocytosis.
RLRs are present in almost all cell types and, unlike TLR, recognize RNA not in the endosomes, but directly in the cytoplasm. Activated RLR regulate the activity of one of the key proteins of the antiviral response system (MAVS) through a number of adapter proteins (TMEM173, TVK1, TRAF3), which leads to the activation of IRF3, IRF7 factors and stimulation of the production of type I IFN. Activated RLR receptors also trigger a signaling pathway, which leads to a release of cytochrome c from mitochondria and Bax-dependent apoptosis of the infected cell [9]. Initially, the proteins of these families were discovered as participants of the innate immunity against viral RNA. Later, a lot of data began to appear that RLR signaling pathways are also triggered with bacterial infections.
STRUCTURAL CHARACTERISTICS OF RLR AND HOW THEY FUNCTION
RIG-I and MDA5 have a similar location of domains and demonstrate a high degree of homology (Fig. 1a). Both receptors contain the main RNA helicase domain with ATPase activity, two N-terminal tandem caspase activation and recruitment domains (CARD), and a regulatory C-terminal domain (CTD). The main domain contains two helicase domains (Hel1, Hel2) and Hel2 insertion domain (Hel2i). CTD plays a critical role in the recognition of pathogen nucleic acids, while the helicase main domain enhances the ligand specificity through conformational changes. CARD domains belong to a subclass of protein motifs known as DExD/H domains and interact with the adapter proteins to initiate downstream signaling pathways, including the activation of transcription of IFN-regulatory factors 3 and 7 (IRF3, IRF7) and nuclear factor κB (NF-κB). In the absence of RNA ligand, RIG-I is in an inactive, closed conformation, which is provided by the interaction of CARD domains with Hel2i, which blocks subsequent signal transmission in the absence of a stimulus. When a ligand is detected, the helicase domain “wraps” around RNA, thus “opens” the closed CARD domains for their binding to the MAVS adapter protein, which initiates subsequent signal transmission [2] (Fig. 1b).

(a) RLR domain organization. The RIG-I, MDA5, and LGP2 proteins contain a central helicase domain and C-terminal CTD domain. N-terminal tandem CARD domains that are critical for downstream signal transmission are present only in the RIG-I and MDA5; (b) RIG-I activation. Binding of alien RNA leads to a change in the protein conformation and release of CARD domains.
Unlike RIG-I and MDA5, the LGP2 contains the main RNA helicase domain and CTD, but has no CARD. The LGP2 helicase domain also recognizes alien RNA molecules and is capable of ATP hydrolysis, but cannot initiate subsequent signal transmission. The CTD provides the specificity of RNA recognition and binding mechanism. The LGP2 is a regulator of the signaling mechanism of RLR [10].
RIG-I and MDA5 are activated by different groups of pathogens and bind to RNA with different properties [11, 12]. In addition, the length and type of nucleic acids, as well as 5'-phosphorylation, are critical for RLR-mediated innate immune signaling [13–16].
RIG-I recognizes and binds to single-stranded RNA (ssRNA), double-stranded RNA (dsRNA), and short dsRNA. A triphosphate at the “blunt” 5'-end of RNA is a specific signal (epitope) for RIG-I. RIG-I also preferentially binds to short RNA with a length of ~20 nt [17, 18]. RIG-I binds to negatively charged 5'‑ppp fragment through a gap present in the CTD domain, while adjacent dsRNA nucleotide fragments with a blunt end help to stabilize this binding. In addition, RIG-I can detect RNA containing a diphosphate group [13, 19]. However, binding of 5'-p dsRNA is difficult due to the emerging loop in the Hel2 domain [20]. Surprisingly, monophosphate dsRNAs have a stronger inhibitory effect on RIG-I than the 5'-OH dsRNAs lacking any phosphates [20]. It was demonstrated that poly-U/UC-specific motifs are required for RIG-I recognition and its optimal activation in RNA. After RNA binding, RIG-I groups into dimers, then into tetramers [21, 22].
MDA5 is activated by a longer dsRNA (often mRNA), which has no 2'-O-methylation and a cap structure at the 5'-end [23, 24]. MDA5 recognizes dsRNA of different sizes (0.5–7.0 kb) and binds to them in nonterminal RNA regions [25–27]. MDA5 is activated by short dsRNA (~100 nt in length) only in those cases if it is present in large amounts. The process of MDA5 activation by a longer RNA molecule (1–2 kb) is mediated by the formation of filamentous oligomers as a result of a joint assembly of helicase domains on the dsRNA molecule [25, 28]. MDA5 recognizes RNA fragments with monophosphates at the ends formed as a result of hydrolysis by RNase L [16, 29], which was first demonstrated in the model of parainfluenza virus 5 (PIV5) infection [30].
In the process of RNA recognition, RLR undergo ATP-dependent conformational changes that initiate the triggering of MAVS-mediated signaling cascades [31]. The MAVS forms huge oligomers by aggregation, which is a stimulus for the IRF3 activation and subsequent signal transmission in the infected cells [32]. This entire process leads to the formation of the IPS-1 signalosome, which triggers the activation of TANK-binding kinase 1 (TBK1) and IκB kinase ε (IKKε) [33]. Phosphorylated forms of IRF3 and IRF7 form homo- and heterodimers that are translocated to the nucleus and subsequently activate the transcription of type I IFN genes [34] (Fig. 2).
RLR—BACTERIAL RNA SENSORS
The activation of RLR by bacterial RNA was for the first time demonstrated for mouse bone marrow-derived macrophages (BMDM) upon infection with Legionella pneumophila [35] and Listeria monocytogenes [36], as well as upon infection of mouse dendritic cells with Helicobacter pylori bacteria [37]. The activation of the production of type I IFN during Mycobacterium tuberculosis (MTb) infection was for the first time demonstrated by Manzanillo et al. [38], but a DNA-induced signal was described in this work. The induction of type I IFN upon tuberculosis infection caused by pathogenic RNA was demonstrated upon infection of mouse BMDM and J774 macrophage cell line with MTb in the work of Andreu et al. [39]. The activation of transcription of the genes of RIG-I (Ddx58), LGP2 (Dhx58), and MDA5 (Ifih1) receptors, as well as the transcription factors mediated by them NF-κB, IRF7, IRF9, STAT1, STAT2, effectors of IFN-β and IL-1β, was proven. In the work of Ranjbar et al. [40], it was demonstrated that upon infection of human macrophages differentiated from peripheral blood mononuclear cells with MTb, the bacterial RNA activates the RLR/MAVS signal in the cytoplasm, which leads to the expression of type I IFN.
TYPE I INTERFERON FAMILY AND BACTERIAL INFECTION
The family of type I interferons (IFN type I) is a multigene family of cytokines that are activated in response to viral and bacterial infection [41]. It includes 14 subtypes of IFN-α with 75–100% amino acid sequence identity, IFN-β with 30% identity with IFN-α consensus sequence, as well as less studied IFN-ε, IFN-ω, and some others [42]. If in the case of a viral infection this activation has a protective effect [43], then with a bacterial infection the effect can be not only protective, but also negative for the immune system.
For example, IFNAR1–/– line mice live longer and have a lower bacterial load as compared with wild-type mice when infected with pathogenic Chlamydia muridarum [44]; a similar effect was observed upon Listeria monocytogenes infection [45]. Type I IFN did not limit the replication in Legionella pneumophila macrophages [46]. When mice were infected with Salmonella typhimurium, the expression of type I IFN was induced, which triggered the process of necroptosis in macrophages and allowed the pathogens to evade the immune response [47].
The role of type I IFN in tuberculosis is also controversial. Type I IFN (especially IFN-β) are involved in the development of tuberculosis infection, which was confirmed in many studies [48–54].
At the same time, the experimental evidence of the adverse effect of type I IFN on the course of the disease is numerous. When mice are infected, type I IFN have a detrimental effect on microorganisms; the severity of this effect depends on the host genotype [55, 56]. An increased level of type I IFN is caused by more virulent MTb strains [57] and is associated with a larger sensitivity to infection [58]. The analysis of blood cell transcriptomes in patients with an active phase of tuberculosis demonstrated a correlation between type I IFN activation and the disease severity, as well as a decreased sensitivity to the therapy [59, 60]. In another study, it was demonstrated that the activation of type I interferon response precedes the transition of infection from a latent to an active stage and is already registered 18 months before the diagnosis [61]. Similar data are summarized in the review of Moreira-Teixeira et al. [49].

RLR signaling pathway. As a result of infection, alien RNAs are present in the cell cytoplasm that are recognized by RIG-I and MDA5 sensor proteins. RNA binding leads to a conformational change and release of CARD domains that interact with CARD domains of the mitochondrial MAVS protein. CARD oligomerization contributes to the signal transduction into the nucleus, which leads to transcription of type I IFN genes.
Nevertheless, the facts of the successful use of IFN-α in the treatment of patients with active tuberculosis have been described [62]. The role of type I IFN (especially IFN-β) in the elimination of tuberculosis infection was demonstrated in mouse models [48]. Zhang et al. demonstrated that blocking the activation of type I IFN synthesis was efficient in the treatment of tuberculosis (on mouse models) at any stage of the disease [63]. The role of RIG-I, MDA5, MAVS, and RNA-dependent kinase PKR in a decrease in the bacterial load in tuberculosis was demonstrated [64]. Some proteins activated by type I IFN (such as GBP1, STAT1, and TAP1) can play a protective role in tuberculosis [60].
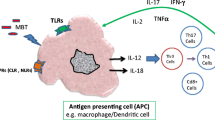
At present, the following idea on the role of type I IFN in tuberculosis was expressed [49] (Fig. 3): at the initial stage of infection or at low expression levels, type I IFN activates the synthesis of IL-12 and TNF-α, which is protective. However, persistently high level of type I IFN expression contributes to the activation of IL-10 synthesis and suppresses the production of protective cytokines IL-12, TNF-α, IL-1α, and IL-1β. IL-10 mediates an inhibitory feedback loop, contributing to a decrease in the production of IL-12 and TNF-α. Type I IFN also suppresses the sensitivity of myeloid cells to IFN-γ due to both IL-10-dependent and independent mechanisms, suppressing IFN-γ-dependent host-protective immune responses. In addition, type I IFN can contribute to the cell death in alveolar macrophages and the accumulation of permissive myeloid cells at the place of infection [55].

The effect of type I IFN expression on infected organism in tuberculosis. (a) Low level of type I IFN expression stimulates the production of protective cytokines IL-12 and TNF-α; (b) persistently high level of type I IFN expression contributes to the production of IL-10 and suppresses the production of protective cytokines IL-12, TNF-α, IL-1α, and IL-1β. IL-10 also mediates a feedback loop, contributing to a decrease in the production of IL-12 and TNF-α, which leads to the suppression of the protective immune processes; (c) in the absence of IFN-γ receptor, type I IFN inhibits the expression of Arg1, increasing the level of TNF-α and thus regulating M2-activation of macrophages. Type I IFN signal transmission can also contribute to the attraction, differentiation and/or survival of protective myeloid cells that control the pathology at the place of infection. Arg1, arginase 1; IFNγR, IFN-γ receptor; IL-10R, IL-10 receptor; TNFR, TNF-α receptor. Adapted according to [49].
Thus, depending on the stage of the disease, the activation of RLR by bacterial RNA leads to a modulation of the host immune response; moreover, it can have a negative effect on it. Based on this, data on the functioning of RLR in tuberculosis infection can be of practical use, since they open new possible targets for the effect of drugs.
ROLE OF MYCOBATERIAL RNA IN HOST IMMUNE RESPONSE
Thus, it became obvious in recent years that mycobacterial RNA in the cytosol of infected macrophages actively affects the immune response. In 2012, it was for the first time demonstrated that mycobacteria also secrete nucleic acids (in addition to proteins) into the cytoplasm of macrophages, namely, low-molecular-weight fragmented RNA. This RNA affects caspase-8-dependent, caspase-1-, and TNF-α-independent pathways of apoptosis, thus weakening the antibacterial protection. This is the first work in which a fundamental possibility of secretion of mycobacterial RNA and its effect on the infectious process was demonstrated [65]. Until recently, the question about how mycobacterial RNA appears in the cytosol of infected macrophages remained open. Sing et al. [66] suggested that Mtb RNA is released during the active macrophage infection, since some transcripts were found in the exosomes released from the infected mouse macrophages.
Cheng and Schorey demonstrated that RNA appears in the cytosol only in the presence of functional systems of SecA2 and Esx-1 secretion; at the same time, SecA2 is responsible for the release of RNA from the bacterium, while ESX-1 for the exit into the cytosol [67]. It is known that the SecA2 secretion system is involved in the process of phagosome maturation arrest through the protein secretion [68]. Currently, it is unknown how SecA2 performs a transport of RNA through the bacterial cell wall and what is the specificity of this process. The authors suggest the existence of RNA-binding proteins with chaperone-like functions that help to transport the bacterial RNA through the SecA2 secretion apparatus [69], and indicate that no DNA secretion through SecA2 was observed [67].
Recently, the role of extracellular vesicles in the transmission of an immunostimulatory signal from the infected cells to uninfected and immune cells was actively discussed. Data appear that such vesicles also contain nucleic factors (in addition to protein ones). Cheng et al. demonstrated that Mtb-infected macrophages release the vesicles that contain pathogenic RNA. The representation of this tuberculous RNA in the vesicles depends on the activity of the bacterial SecA2 secretion system. Such vesicles induce the expression of type I IFN in uninfected macrophages by triggering the RIG-I/MAVS/TBK1/IRF3 cascade [70].
Previously, it was known that Listeria monocytogenes infection induces a type I interferon response [71]; at the same time, RIG-I is activated by nucleic acids [36]. Later, it was found that during L. monocytogenes infection, the pathogen RNA is secreted in the vesicles, and noncoding RNA are mainly present there [72]. One of them (rli32) initiates the production of IFN-β and enhances the intracellular growth of bacteria [73]. It is unknown which RNAs act as RIG-1 ligands in tuberculosis. Cheng and Schorey demonstrated that the ppe11 and pol2A gene RNA bind to RIG-1, but not to the MDA5 receptor. There is no more information about the RNA interactome of the RIG-1 receptor.
CONCLUSIONS
Tuberculosis is still one of the leading causes of death due to bacterial infection [74]. The epidemic is exacerbated by the absence of efficient vaccination and the spread of new M. tuberculosis strains with an extensive drug resistance. The therapeutic approaches aimed at correcting the host organism immune response to infection (host-directed therapy) become an alternative to traditional antibacterial methods of treatment. In recent years, the role of RLR as components of the innate immunity system in tuberculosis has been actively studied [75]. Although many aspects of the effect on RNA-binding receptors are still not sufficiently clear, the first candidates have been already suggested: thus, it was demonstrated that the drug nitazoxanide, which enhances the signals activated by RLR receptors, can be used for the treatment of tuberculosis [40].
REFERENCES
Yamashiro, L.H., Oliveira, S.C., and Báfica, A., Microbes Infect., 2014, vol. 16, pp. 991–997. https://doi.org/10.1016/j.micinf.2014.09.006
Batool., M., Kim, M.S., and Choi, S., Med. Res. Rev., 2022, vol. 42, pp. 399–425. https://doi.org/10.1002/med.21845
Chattopadhyay, S. and Sen, G.C., J. Interferon Cytokine Res., 2014, vol. 34, pp. 427– 436. https://doi.org/10.1089/jir.2014.0034
Liu, H.M., IUBMB Life, 2021, vol. 74, pp. 180–189. https://doi.org/10.1002/iub.2551
Meylan, E., Tschopp, J., and Karin, M., Nature, 2006, vol. 442, pp. 39–44. https://doi.org/10.1038/nature04946
Martínez, I., Oliveros, J.C., Cuesta, I., de la Barrera, J., Ausina, V., Casals, C., de Lorenzo, A., García, E., García-Fojeda, B., and Garmendia, J., Front. Microbiol., 2017, vol. 8, p. 276. https://doi.org/10.3389/fmicb.2017.00276
Platnich, J.M. and Muruve, D.A., Arch. Biochem. Biophys., 2019, vol. 670, pp. 4–14. https://doi.org/10.1016/j.abb.2019.02.008
Semple, S.L., Vo, N.T., Poynter, S.J., Li, M., Heath, D.D., DeWitte-Orr, S.J., and Dixon, B., Dev. Comp. Immunol., 2018, vol. 89, pp. 93–101. https://doi.org/10.1016/j.dci.2018.08.010
Mehrbod, P., Ande, S.R., Alizadeh, J., Rahimizadeh, S., Shariati, A., Malek, H., Hashemi, M., Glover, K.K., Sher, A.A., and Coombs, K.M., Virulence, 2019, vol. 10, pp. 376–413. https://doi.org/10.1080/21505594.2019.1605803
Bruns, A.M., Pollpeter, D., Hadizadeh, N., Myong, S., Marko, J.F., and Horvath, C.M., J. Biol. Chem., 2013, vol. 288, pp. 938–946. https://doi.org/10.1074/jbc.M112.424416
Kato, H., Takeuchi, O., Sato, S., Yoneyama, M., Yamamoto, M., Matsui, K., Uematsu, S., Jung, A., Kawai, T., Ishii, K.J., Yamaguchi, O., Otsu, K., Tsujimura, T., Koh, C.S., Reis e Sousa, C., Matsuura, Y., Fujita, T., abd Akira, S., Nature, 2006, vol. 441, pp. 101–105. https://doi.org/nature04734
Loo, Y.M., Fornek, J., Crochet, N., Bajwa, G., Perwitasari, O., Martinez-Sobrido, L., Akira, S., Gill, M.A., Garcia-Sastre, A., Katze, M.G., and Gale, M., Jr., J. Virol., 2008, vol. 82, pp. 335–345. https://doi.org/JVI.01080-07
Goubau, D., Schlee, M., Deddouche, S., Pruijssers, A.J., Zillinger, T., Goldeck, M., Schuberth, C., Van der Veen, A.G., Fujimura, T., Rehwinkel, J., Iskarpatyoti, J.A., Barchet, W., Ludwig, J., Dermody, T.S., Hartmann, G., and Reis e Sousa, C., Nature, 2014, vol. 514, pp. 372–375. https://doi.org/10.1038/nature13590
Kowalinski, E., Lunardi, T., McCarthy, A.A., Louber, J., Brunel, J., Grigorov, B., Gerlier, D., and Cusack, S., Cell, 2011, vol. 147, pp. 423–435. https://doi.org/10.1016/j.cell.2011.09.039
Chiu, Y.H., Macmillan, J.B., and Chen, Z.J., Cell, 2009, vol. 138, pp. 576–591. https://doi.org/10.1016/j.cell.2009.06.015
Malathi, K., Dong, B., Gale, M., Jr., and Silverman, R.H., Nature, 2007, vol. 448, pp. 816–819. https://doi.org/nature06042
Schlee, M., Immunobiology, 2013, vol. 218, pp. 1322–1335. https://doi.org/10.1016/j.imbio.2013.06.007
Zust, R., Cervantes-Barragan, L., Habjan, M., Maier, R., Neuman, B.W., Ziebuhr, J., Szretter, K.J., Baker, S.C., Barchet, W., Diamond, M.S., Siddell, S.G., Ludewig, B., and Thiel, V., Nat. Immunol., 2011, vol. 12, pp. 137–143. https://doi.org/10.1038/ni.1979
Linehan, M.M., Dickey, T.H., Molinari, E.S., Fitzgerald, M.E., Potapova, O., Iwasaki, A., and Pyle, A.M., Sci. Adv., 2018, vol. 4, p. e1701854. https://doi.org/10.1126/sciadv.1701854
Ren, X., Linehan, M.M., Iwasaki, A., and Pyle, A.M., Cell Rep., vol. 26, pp. 2019–2027. https://doi.org/10.1016/j.celreP.2019.01.107
Peisley, A., Wu, B., Yao, H., Walz, T., and Hur, S., Mol. Cell, 2013, vol. 51, pp. 573–583. https://doi.org/10.1016/j.molcel.2013.07.024
Peisley, A., Wu, B., Xu, H., Chen, Z.J., and Hur, S., Nature, 2014, vol. 509, pp. 110–114. https://doi.org/10.1038/nature13140
Pichlmair, A., Schulz, O., Tan, C.P., Rehwinkel, J., Kato, H., Takeuchi, O., Akira, S., Way, M., and Schiavo, G., Reis e Sousa, C., J. Virol., 2009, vol. 83, pp. 10761–10769.
Kato, H., Takeuchi, O., Mikamo-Satoh, E., Hirai, R., Kawai, T., Matsushita, K., Hiiragi, A., Dermody, T.S., Fujita, T., and Akira, S., J. Exp. Med., 2008, vol. 205, pp. 1601–1610. https://doi.org/10.1084/jem.20080091
Berke, I.C. and Modis, Y., EMBO J., 2012, vol. 31, pp. 1714–1726. https://doi.org/10.1038/emboj.2012.19
Peisley, A., Lin, C., Wu, B., Orme-Johnson, M., Liu, M., Walz, T., and Hur, S., Proc. Natl. Acad. Sci. U. S. A., 2011, vol. 108, pp. 21010–21015. https://doi.org/10.1073/pnas.1113651108
Wu, B., Peisley, A., Richards, C., Yao, H., Zeng, X., Lin, C., Chu, F., Walz, T., and Hur, S., Cell, 2013, vol. 152, pp. 276–289. https://doi.org/10.1016/j.cell.2012.11.048
Huang, Y.H., Liu, X.Y., Du, X.X., Jiang, Z.F., and Su, X.D., Nat. Struct. Mol. Biol., 2012, vol. 19, pp. 728–730. https://doi.org/10.1038/nsmb.2333
Manivannan, P., Siddiqui, M.A., and Malathi, K., J. Virol., 2020, vol. 94, p. e00205-20. https://doi.org/10.1128/JVI.00205-20
Luthra, P., Sun, D., Silverman, R.H., and He, B., Proc. Natl. Acad. Sci. U. S. A., 2011, vol. 108, pp. 2118–2123. https://doi.org/10.1073/pnas.1012409108
Chiang, J.J., Davis, M.E., and Gack, M.U., Cytokine Growth Factor Rev., 2014, vol. 25, pp. 491–505. https://doi.org/10.1016/j.cytogfr.2014.06.005
Hou, F., Sun, L., Zheng, H., Skaug, B., Jiang, Q.X., and Chen, Z.J., Cell, 2011, vol. 146, pp. 448–461. https://doi.org/10.1016/j.cell.2011.06.041
Liu, Y., Olagnier, D., and Lin, R., Front. Immunol., 2016, vol. 7, p. 662. https://doi.org/10.3389/fimmu.2016.00662
Panne, D., Curr. Opin. Struct. Biol., 2008, vol. 18, pp. 236–242. https://doi.org/10.1016/j.sbi.2007.12.002
Monroe, K.M., McWhirter, S.M., and Vance, R.E., PLoS Pathog., 2009, vol. 5, p. e1000665. https://doi.org/10.1371/journal.ppat.1000665
Abdullah, Z., Schlee, M., Roth, S., Mraheil, M.A., Barchet, W., Bottcher, J., Hain, T., Geiger, S., Hayakawa, Y., Fritz, J.H., Civril, F., Hopfner, K.P., Kurts, C., Ruland, J., Hartmann, G., Chakraborty, T., and Knolle, P.A., EMBO J., 2012, vol. 31, pp. 4153–4164. https://doi.org/10.1038/emboj.2012.274
Rad, R., Ballhorn, W., Voland, P., Eisenacher, K., Mages, J., Rad, L., Ferstl, R., Lang, R., Wagner, H., Schmid, R.M., Bauer, S., Prinz, C., Kirschning, C.J., and Krug, A., Gastroenterology, 2009, vol. 136, pp. 2247–2257. https://doi.org/10.1053/j.gastro.2009.02.066
Manzanillo, P.S., Shiloh, M.U., Portnoy, D.A., and Cox, J.S., Cell Host Microbe, 2012, vol. 11, pp. 469–480. https://doi.org/10.1016/j.chom.2012.03.007
Andreu, N., Phelan, J., de Sessions, P.F., Cliff, J.M., Clark, T.G., and Hibberd, M.L., Sci. Rep., 2017, vol. 7, p. 42225. https://doi.org/10.1038/srep42225
Ranjbar, S., Haridas, V., Nambu, A., Jasenosky, L.D., Sadhukhan, S., Ebert, T.S., Hornung, V., Cassell, G.H., Falvo, J.V., and Goldfeld, A.E., iScience, 2019, vol. 22, pp. 299–313. https://doi.org/10.1016/j.isci.2019.11.001
Ivashkiv, L.B. and Donlin, L.T., Nat. Rev. Immunol., 2014, vol. 14, pp. 36–49. https://doi.org/10.1038/nri3581
Hertzog, P.J. and Williams, B.R., Cytokine Growth Factor Rev., 2013, vol. 24, pp. 217–225. https://doi.org/10.1016/j.cytogfr.2013.04.002
Crouse, J., Kalinke, U., and Oxenius, A., Nat. Rev. Immunol., 2015, vol. 15, pp. 231–242. https://doi.org/10.1038/nri3806
Qiu, H., Fan, Y., Joyee, A.G., Wang, S., Han, X., Bai, H., Jiao, L., Van Rooijen, N., and Yang, X., J. Immunol., 2008, vol. 181, pp. 2092–2102. https://doi.org/10.4049/jimmunol.181.3.2092
Auerbuch, V., Brockstedt, D.G., Meyer-Morse, N., O’Riordan, M., and Portnoy, D.A., J. Exp. Med., 2004, vol. 200, pp. 527–533. https://doi.org/10.1084/jem.20040976
Opitz, B., Vinzing, M., van Laak, V., Schmeck, B., Heine, G., Gunther, S., Preissner, R., Slevogt, H., N’Guessan, P.D., Eitel, J., Goldmann, T., Flieger, A., Suttorp, N., and Hippenstiel, S., J. Biol. Chem., 2006, vol. 281, pp. 36173–36179. https://doi.org/10.1074/jbc.M604638200
Robinson, N., McComb, S., Mulligan, R., Dudani, R., Krishnan, L., and Sad, S., Nat. Immunol., 2012, vol. 13, pp. 954–962. https://doi.org/10.1038/ni.2397
Kaufmann, S.H. and Dorhoi, A., Curr. Opin. Immunol., 2013, vol. 25, pp. 441–449. https://doi.org/10.1016/j.coi.2013.05.005
Moreira-Teixeira, L., Mayer-Barber, K., Sher, A., and O’Garra, A., J. Exp. Med., 2018, vol. 215, pp. 1273–1285. https://doi.org/10.1084/jem.20180325
Manca, C., Tsenova, L., Freeman, S., Barczak, A.K., Tovey, M., Murray, P.J., Barry, C., and Kaplan, G., J. Interferon Cytokine Res., 2005, vol. 25, pp. 694–701. https://doi.org/10.1089/jir.2005.25.694
Mayer-Barber, K.D., Andrade, B.B., Oland, S.D., Amaral, E.P., Barber, D.L., Gonzales, J., Derrick, S.C., Shi, R., Kumar, N.P., Wei, W., Yuan, X., Zhang, G., Cai, Y., Babu, S., Catalfamo, M., Salazar, A.M., Via, L.E., and Barry, C.E., 3rd, and Sher, A., Nature, 2014, vol. 511, pp. 99–103. https://doi.org/10.1038/nature13489
Robinson, C.M., Jung, J.Y., and Nau, G.J., Cytokine, 2012, vol. 60, pp. 233–241. https://doi.org/10.1016/j.cyto.2012.06.012
Stanley, S.A., Johndrow, J.E., Manzanillo, P., and Cox, J.S., J. Immunol., 2007, vol. 178, pp. 3143–3152. https://doi.org/10.4049/jimmunol.178.5.3143
Teles, R.M., Graeber, T.G., Krutzik, S.R., Montoya, D., Schenk, M., Lee, D.J., Komisopoulou, E., Kelly-Scumpia, K., Chun, R., Iyer, S.S., Sarno, E.N., Rea, T.H., Hewison, M., Adams, J.S., Popper, S.J., Relman, D.A., Stenger, S., Bloom, B.R., Cheng, G., and Modlin, R.L., Science, 2013, vol. 339, pp. 1448–1453. https://doi.org/10.1126/science.1233665
Dorhoi, A., Yeremeev, V., Nouailles, G., Weiner, J., 3rd, Jorg, S., Heinemann, E., Oberbeck-Muller, D., Knaul, J.K., Vogelzang, A., Reece, S.T., Hahnke, K., Mollenkopf, H.J., Brinkmann, V., and Kaufmann, S.H., Eur. J. Immunol., 2014, vol. 44, pp. 2380–2393. https://doi.org/10.1002/eji.201344219
Moreira-Teixeira, L., Stimpson, P.J., Stavropoulos, E., Hadebe, S., Chakravarty, P., Ioannou, M., Aramburu, I.V., Herbert, E., Priestnall, S.L., Suarez-Bonnet, A., Sousa, J., Fonseca, K.L., Wang, Q., Vashakidze, S., Rodriguez-Martinez, P., Vilaplana, C., Saraiva, M., Papayannopoulos, V., and O’Garra, A., Nat. Commun., 2020, vol. 11, p. 5566. https://doi.org/10.1038/s41467-020-19412-6
Wiens, K.E. and Ernst, J.D., PLoS Pathog., 2016, vol. 12, p. e1005809. https://doi.org/10.1371/journal.ppat.1005809
Manca, C., Tsenova, L., Bergtold, A., Freeman, S., Tovey, M., Musser, J.M., Barry, C.E., 3rd, Freedman, V.H., and Kaplan, G., Proc. Natl. Acad. Sci. U. S. A., 2001, vol. 98, pp. 5752–5757. https://doi.org/10.1073/pnas.091096998
Berry, M.P., Graham, C.M., McNab, F.W., Xu, Z., Bloch, S.A., Oni, T., Wilkinson, K.A., Banchereau, R., Skinner, J., Wilkinson, R.J., Quinn, C., Blankenship, D., Dhawan, R., Cush, J.J., Mejias, A., Ramilo, O., Kon, O.M., Pascual, V., Banchereau, J., Chaussabel, D., and O’Garra, A., Nature, 2010, vol. 466, pp. 973–977. https://doi.org/10.1038/nature09247
Zak, D.E., Penn-Nicholson, A., Scriba, T.J., Thompson, E., Suliman, S., Amon, L.M., Mahomed, H., Erasmus, M., Whatney, W., Hussey, G.D., Abrahams, D., Kafaar, F., Hawkridge, T., Verver, S., Hughes, E.J., Ota, M., Sutherland, J., Howe, R., Dockrell, H.M., Boom, W.H., Thiel, B., Ottenhoff, T.H.M., Mayanja-Kizza, H., Crampin, A.C., Downing, K., Hatherill, M., Valvo, J., Shankar, S., Parida, S.K., Kaufmann, S.H.E., Walzl, G., Aderem, A., and Hanekom, W.A., Lancet, 2016, vol. 387, pp. 2312–2322. https://doi.org/S0140-6736(15)01316-1
Scriba, T.J., Fiore-Gartland, A., Penn-Nicholson, A., Mulenga, H., Kimbung, MbandiS., Borate, B., Mendelsohn, S.C., Hadley, K., Hikuam, C., Kaskar, M., Musvosvi, M., Bilek, N., Self, S., Sumner, T., White, R.G., Erasmus, M., Jaxa, L., Raphela, R., Innes, C., Brumskine, W., Hiemstra, A., Malherbe, S.T., Hassan-Moosa, R., Tameris, M., Walzl, G., Naidoo, K., Churchyard, G., and Hatherill, M., Lancet Infect. D, vol. 21, no. is. 2021, pp. 354–365. https://doi.org/10.1016/S1473-3099(20)30914-2
Zarogoulidis, P., Kioumis, I., Papanas, N., Manika, K., Kontakiotis, T., Papagianis, A., and Zarogoulidis, K., J. Chemother., 2012, vol. 24, pp. 173–177. https://doi.org/10.1179/1973947812Y.0000000005
Zhang, L., Jiang, X., Pfau, D., Ling, Y., and Nathan, C.F., J. Exp. Med., 2021, vol. 218, p. e20200887. https://doi.org/10.1084/jem.20200887
Ranjbar, S., Haridas, V., Jasenosky, L.D., Falvo, J.V., and Goldfeld, A.E., Cell Rep., vol. 13, pp. 874–883. https://doi.org/10.1016/j.celreP.2015.09.048
Obregon-Henao, A., Duque-Correa, M.A., Rojas, M., Garcia, L.F., Brennan, P.J., Ortiz, B.L., and Belisle, J.T., PLoS One, 2012, vol. 7, p. e29970. https://doi.org/10.1371/journal.pone.0029970
Singh, P.P., Li, L., and Schorey, J.S., Traffic, 2015, vol. 16, pp. 555–571. https://doi.org/10.1111/tra.12278
Cheng, Y. and Schorey, J.S., J. Exp. Med., 2018, vol. 215, pp. 2919–2935. https://doi.org/10.1084/jem.20180508
Sullivan, J.T., Young, E.F., McCann, J.R., and Braunstein, M., Infect. Immun., 2012, vol. 80, pp. 996–1006. https://doi.org/10.1128/IAI.05987-11
Miller, B.K., Zulauf, K.E., and Braunstein, M., Microbiol. Spectr., 2017, vol. 5, pp. 2–5. https://doi.org/10.1128/microbiolspec.TBTB2-0013-2016
Cheng, Y. and Schorey, J.S., EMBO Rep., 2019, vol. 20, p. e46613. https://doi.org/10.15252/embr.201846613
O’Connell, R.M., Saha, S.K., Vaidya, S.A., Bruhn, K.W., Miranda, G.A., Zarnegar, B., Perry, A.K., Nguyen, B.O., Lane, T.F., Taniguchi, T., Miller, J.F., and Cheng, G., J. Exp. Med., 2004, vol. 200, pp. 437–445. https://doi.org/10.1084/jem.20040712
Vdovikova, S., Luhr, M., Szalai, P., Nygard, SkalmanL., Francis, M.K., Lundmark, R., Engedal, N., Johansson, J., and Wai, S.N., Front. Cell Infect. Microbiol., 2017, vol. 7, p. 154. https://doi.org/10.3389/fcimb.2017.00154
Frantz, R., Teubner, L., Schultze, T., La Pietra, L., Muller, C., Gwozdzinski, K., Pillich, H., Hain, T., Weber-Gerlach, M., Panagiotidis, G.D., Mostafa, A., Weber, F., Rohde, M., Pleschka, S., Chakraborty, T., and Abu, MraheilM., mBio, 2019, vol. 10, p. e01223-19. https://doi.org/10.1128/mBio.01223-19
Harding, E., Lancet Respir. Med., 2020, vol. 8, p. 19. https://doi.org/S2213-2600(19)30418-7
Burkert, S. and Schumann, R.R., Vaccines (Basel), 2020, vol. 8, p. 67. https://doi.org/10.3390/vaccines8010067
Funding
This work was supported by the Russian Science Foundation (grant No. 22-14-00235, https://rscf.ru/project/22-14-00235/).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflict of interest. This article does not contain any studies involving animals or human participants performed by any of the authors.
Additional information
Translated by A. Barkhash
The article is dedicated to the memory of Academician of the Russian Academy of Sciences Vadim Tikhonovich Ivanov.
Abbreviations: dsRNA, double-stranded RNA; BMDM, bone marrow-derived macrophages; CARD, caspase activation and recruitment domain; CTD, C-terminal domain; IFN, interferon; MDA5, melanoma differentiation-associated protein 5; MTb, Mycobacterium tuberculosis; RIG-I, retinoic acid-inducible gene I; RLR, RIG-I-like receptors; IFNAR, IFN-α receptor.
Rights and permissions
Open Access. This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Skvortsova, Y.V., Bychenko, O.S. & Azhikina, T.L. Role of RIG-I-Like Receptors in the Activation of Innate Immunity in Tuberculosis. Russ J Bioorg Chem 49, 742–750 (2023). https://doi.org/10.1134/S1068162023040192
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1068162023040192