
Abstract
Possessing unique physicochemical properties, phthalocyanines are widely used as active components of supramolecular ensembles and nanomaterials. The functional properties of phthalocyanine-based materials are governed by not only the structure of their discotic molecules, but also the character of their intermolecular interactions, which determine both the self-assembly mechanism and the structure of such systems. This review discusses the experimental approaches, which are based on the notions of colloid and coordination chemistry that enable one to control intermolecular interactions in low-dimensional supramolecular ensembles based on phthalocyanines and metallocomplexes thereof. Using double-decker crown-substituted lanthanide phthalocyaninates as an example, it is shown how one- and two-dimensional nanomaterials with different properties can be obtained from the same type of building blocks employing a set of colloid-chemical methods. Such materials are, in particular, capable for controlled absorption of visible light in ultrathin films and can be employed as conducting one-dimensional components of planar elements for organic electronics.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
Phthalocyanines are macroheterocyclic compounds that are of great interest for many fields of modern science and technology, such as organic electronics [1–4], solar power engineering [5, 6], catalysis [7–10], medicine [11–15], and sensorics [16]. The wide application of phthalocyanines is due to their ability to efficiently absorb electromagnetic radiation, semiconducting and magnetic properties, and high thermal and chemical stability [17]. Owing to the active development of synthetic methods during the entire history of studying phthalocyanines, it is, today, possible to finely tune their properties and functions by varying molecular structure via, e.g., the incorporation of metal ions into macrocycle cavities and attachment of peripheral and axial substituents. For example, cobalt- and iron phthalocyanines catalyze reduction of oxygen and carbon dioxide [7–10], while zinc-containing complexes are used in photodynamic therapy of cancer [5, 18]. The sensitivity of sensors based on cobalt phthalocyaninate with respect to different biologically significant molecules may be varied by changing its side substituents [19]. Thus, phthalocyanines are promising building blocks for creating functional materials.
At the same time, when passing from individual molecules to molecular ensembles, the properties of resulting materials begin to depend on not only the structure of the building blocks (molecules), but also on the character of their interactions [20, 21]. In the case of phthalocyanines, their discotic molecules with developed conjugated π-electron systems are highly prone to aromatic stacking. This leads, as a rule, to the formation of lengthy aggregates, whose molecules are oriented at an angle to the stacking direction [22, 23]. On the one hand, the strong intermolecular stacking ensures efficient transport of charges along aggregates and stability of the formed structures; on the other hand, it hinders other types of intermolecular interactions and reduces the solubility of phthalocyanines. This fact makes difficult the use of “wet” chemistry methods for producing functional materials, because these methods are based on dissolution of components. Moreover, the low solubility of phthalocyanines makes it impossible to obtain large defectless aggregates (molecular single crystals) under mild conditions, thus deteriorating the characteristics of semiconductor materials. In particular, the problem of polycrystallinity is critical when developing phthalocyanine-based transistors. In [23], it has been shown that electron mobility in single crystals of halogen-substituted phthalocyanines obtained by physical precipitation from a gas phase may be as high as 1.1 cm2/(V s), while this value is decreased by two orders of magnitude in polycrystalline samples be-cause of charge recombination at grain boundaries [2].
The predominance of aromatic stacking over other types of intermolecular interactions (van der Waals, hydrogen, and electrostatic forces) limits the diversity of the structures formed by phthalocyanines, and, hence, the spectrum of the functional characteristics of materials based thereon. At the same time, overcoming the tendency of these molecules to stacking is of fundamental significance for the development of phthalocyanine-based drugs for the photodynamic therapy of cancer, because the efficiency of formation of singlet oxygen directly depends on the accessibility of the metal center. One of the methods for suppressing aromatic stacking is the incorporation of bulky substituents that create steric hindrances for the contacts between macrocycles [5, 11, 12, 18]; however, this approach requires great efforts on the development of procedures for the synthesis of large molecules. At the same time, the discoidal shape of molecules and the orientation of the dipole moment in the macrocycle plane, make phthalocyanines promising for creating anisotropic materials [24–26] and liquid crystals [27]. Therewith, if we possess methods for the control over orientation and aggregation of molecules, we can produce materials with different properties on the basis of the same compound. Thus, the search for approaches to the control over arrangement of phthalocyanines in condensed phases is an important problem to be solved for complete unveiling potential of phthalocyanines for developing modern functional materials.
This review reports the methods of colloid and coordination chemistry that enable one to control orientation of molecules in phthalocyanine-based materials with different dimensionalities. Especial attention is focused on the works performed by the scientific group headed by M.A. Kalinina (Dr. Habil. in Chemistry, Frumkin Institute of Physical Chemistry and Electrochemistry, Russian Academy of Sciences) dealing with supramolecular hybrid materials. These works obviously show how the employment of colloid-chemical approaches enables one to produce low-dimensional ensembles with different structures and properties using the same type of phthalocyanine complexes as initial building blocks.
CONTROL OVER ARRANGEMENT OF PHTHALOCYANINES IN ULTRATHIN FILMS
The use of disperse systems containing phases with different physicochemical properties and interfaces between them makes it possible to control both the solubility of phthalocyanines and their organization due to the structuring action of an interface, the interaction with which may appear to be more energetically advantageous than intermolecular stacking. A striking example is the preparation of phthalocyanine-based ultrathin ordered films at an air/water interface by the Langmuir–Blodgett (LB) method. Such coatings are used as active layers in field-effect transistors [3, 4, 28]. In individual monolayers, phthalocyanines are mainly assembled into lengthy aggregates, in which molecular planes are oriented perpendicularly (H-aggregates) or at an angle (J-aggregates) to an interface [29, 30]. The main advantage of the assembly on an aqueous subphase is the possibility of preparing ordered aggregates with nanometer thicknesses on planes with large areas. At the same time, the LB method can be employed to assemble monolayer phthalocyanine coatings with a planar arrangement [31]. In such films, a phthalocyanine macrocycle is oriented parallel to an interface and is not in contact with aromatic rings of adjacent molecules. There are several approaches to stacking suppression in such monolayers: (i) increasing the amphiphility of molecules by introducing peripheral substituents containing large polar groups [32–39]; (ii) the incorporation of metal centers capable of coordinating small molecules in the axial position [34]; (iii) the use of mixed monolayers containing simple aliphatic compounds adsorbable on macrocycle (fatty acids, fatty alcohols, octadecyl-substituted urea, octadecyl-substituted imidazole, etc.) [35, 36, 40].
In bulk aqueous solutions, phthalocyanine aggregation is prevented by the method, which is classical for colloid chemistry, i.e., solubilization in solutions of micelles and polymer globules [14, 15]. Since this approach not only preserves the photoactivity of aromatic molecules, but also increases their solubility in aqueous media, it is widely used to produce drugs for photodynamic therapy of III-generation cancer [13]. Another highly interesting method is the functionalization of metal nanoparticles with phthalocyanines. The combination of the electron systems of a large aromatic phthalocyanine ring and a nanoparticle enhances the photophysical and photochemical properties of such hybrids (increases the lifetime of the triplet state of singlet oxygen and intensifies its generation) [41, 42]. Colloidal solutions of functionalized nanoparticles are used for photodynamic antibacterial protection [41, 43, 44].
ASSEMBLY OF COORDINATION POLYMERS FROM PHTHALOCYANINES BY COORDINATION CHEMISTRY METHODS
The self-assembly of phthalocyanines via coordination interactions with metal ions, metal clusters, or small organic molecules makes it possible to obtain one-, two-, and three-dimensional coordination-type polymers with a preset molecular structure. The combination of the highly ordered structure of coordination polymers with the unique physicochemical properties of phthalocyanines gives wide possibilities of producing functional materials for photocatalysis, photonics, and organic electronics [45–49]. Of special interest is the development of electroconducting materials based on such structures. Charge transfer along an aggregate is known to occur only provided that molecules in an ensemble are oriented perpendicularly to the stacking direction, because, at such arrangement, the overlap of the π-orbitals of adjacent molecules gives rise to the appearance of a conduction band [50]. At the same time, in the case of simple aromatic stacking, molecules of an aggregate are, as a rule, oriented at an angle to the stacking direction, and such ensembles are dielectrics. The combination of stacking and coordination interactions in the same structure enables one to create a necessary molecular arrangement. For the first time, such organization of phthalocyanines was realized in coordination polymers of the so-called “shish kebab” structure. Such ensembles consist of macrocycles connected by bidentate ligands via ligand–central phthalocyanine ion coordination bonds [51–54]. The electrical conductivity of such structures is based on the overlap of electron orbitals as a result of coordination of oxygen or fluorine atoms with phthalocyanine metal centers [51, 52] or the narrowing of the band gap in ensembles with macrocycle bridges (pyrazine and its derivatives) [53–55]. In spite of the qualitative manifestation of the semiconductor properties, the quantitative conductivity characteristics of the shish kebab structures are low. The conductivity of such one-dimensional wires can reach values typical for covalently bonded conducting polymers (up to 650 S/cm) only after being doped due to partial oxidation of phthalocyanines [51, 56] or a change in macrocycle orientation in an aggregate [57]. At present, the principle of combining coordination and stacking interactions in the same structure has found its development in the assembly of bulky porous metal-organic frameworks (MOFs). In this case, phthalocyanines are assembled into a two-dimensional network via coordination bonding between side substituents of phthalocyanines and metal ions, while the layers are held together by means of π–π stacking interactions. In spite of high ordering at the molecular level and the structure, which is “true” for the appearance of electrical conduction, the maximum conductivity of such MOFs is no higher than 10–2 S/cm [48, 49]. As in the above-described cases, the low conductivity is caused by small sizes of single-crystalline domains and high losses resulting from the charge transfer through intergrain interfaces in a macroscopically defective material.
Thus, the use of the approaches based on the concepts of colloid and coordination chemistry makes it possible to efficiently control the arrangement of phthalocyanines in ensembles, thereby varying the properties of resulting materials. At the same time, the production of defectless materials by “mild” methods still remains an urgent problem because of the low solubility of phthalocyanines.
DOUBLE-DECKER CROWN-SUBSTITUTED PHTHALOCYANINATES OF RARE-EARTH METALS AS UNIVERSAL BUILDING BLOCKS FOR ASSEMBLING FUNCTIONAL MATERIALS
The above-described arsenal of physicochemical methods employed to control intermolecular interactions in ensembles of phthalocyanines yields the potential opportunities for producing materials with different properties using a single type of building blocks. This will reduce the volumes of the efforts and resources expended for the synthesis of initial phthalocyanine derivatives, with this fact being of special importance from the viewpoint of scientific and technical society aimed at the universal implementation of “green” technologies.
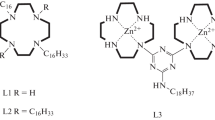
From this point of view, a promising class of derivatives is represented by double-decker tetra-(15-crown-5)-substituted phthalocyanines of rare-earth metals (LnL2) (Fig. 1a) [58]. Each structural element of these metallocomplexes is multifunctional and may realize different functions depending on the structure of a resulting material. The large π-conjugated system of a macrocyclic ring can efficiently absorb visible light and, in the case of aromatic stacking between rings, provide the charge transfer along an aggregate. On the one hand, crown-ether groups increase the hydrophilicity of phthalocyanines, thus facilitating the formation of Langmuir monolayers based thereon [59–61], while, on the other hand, they can coordinate metal ions. The structure of an obtained complex depends on the ratio between the radii of a metal ion and the cavity of a crown-ether ring. When the cation radius is larger than the crown-ether radius, one cation binds two ethers to form a sandwich-type complex (Fig. 1). For (15-crown-5)-ether (1.7–2.2 Å), this case is realized in the presence of potassium (2.66 Å) and rubidium (2.96 Å) ions. The use of double-decker complexes may ensure the growth of one-dimensional coordination-type polymers, provided that one potassium ion coordinates two crown ethers belonging to different molecules, while single-decker crown-substituted phthalocyanines form only dimers. The presence of unpaired electrons in double-decker complexes [Pc2−]Ln3+[Pc−•] increases the conductivity relative to that of 1 : 1 MPc metallocomplexes [62, 63]. Moreover, the interdeck distance in a phthalocyanine molecule can be regulated by changing the central metal ion. This makes it possible, first, to tune the absorption spectrum of a metallocomplex and, second, to vary the type of coordination from the intermolecular one at a small radius of a lanthanide ion (Lu3+ and Ce4+) (Fig. 1b) [1, 64] to the intramolecular type, which is realized upon increasing the size of a metal center (Tb3+, Tm3+, La3+, and Ce3+) (Fig. 1c) [1, 65].

(a) Structural formula of double-decker tetra-(15-crown-5)-substituted phthalocyanines of rare-earth metals (LnL2) and (b) intermolecular and (c) intramolecular types of coordination of potassium ions with crown-ether groups.
SELF-ASSEMBLY OF ONE-DIMENSIONAL COORDINATION POLYMERS BASED ON LnL2
Such properties as a high electrical conductivity and the feasibility to obtain one-dimensional coordination polymers via the intermolecular coordination of potassium ions with crown-ether groups make LnL2 promising for producing conducting nanowires from them.
The complexation between LnL2 and potassium ions began to be studied in 1990s by the research group headed by Simon, who used double-decker lutetium crown-phthalocyaninate (LuL2) as a building block [64, 66–70]. Using static light scattering, they revealed that the addition of a potassium salt to a LuL2 solution in chloroform gives rise to the formation of short rods consisting of, on average, 260 molecules [64]. Then, Martynov employed spectrophotometric titration of solutions of several LnL2 metallocomplexes (Ln = Tm, Tb, La) with different potassium salts to show that, as lanthanide radius increases, the intermolecular incorporation of potassium is replaced by intramolecular one [65]. The tendency to this or that type of incorporation depends not only on the size of a metal center, but also on the nature of potassium salt anions [64, 65]. In particular, the delocalized negative charge of the anion in potassium tetraphenylborate destabilizes K+/BPh4 ion pairs in a bipolar aprotic medium, thereby facilitating the LnL2-K+−LnL2 intermolecular bonding.
The group headed by Kalinina was the first to propose a procedure for obtaining one-dimensional micron-sized coordination polymers that could be used as nanowires [1]. The nanowires were assembled using an approach based on the addition of seeds to a reaction solution to control the amount of crystallization sites. The K+-LnL2 aggregates were assembled by injecting a potassium tetraphenylborate solution in acetonitrile into a LnL2 solution in chloroform at a KBPh4-to-LnL2 molar ratio of 4 : 1. The main difference of the proposed procedure from those reported previously is the use of a saturated salt solution. An increase in the salt concentration leads to more than 250-fold elongation of the aggregates (from 170 nm to 50 µm) (Fig. 2). The efficiency of this experimentally simple procedure is associated with a local KBPh4 supersaturation arising upon mixing the solvents because of the insolubility of the salt in chloroform. Under these conditions, a small amount of KBPh4 microcrystals are formed and act as seeds for the rapid growth of supramolecular aggregates. Thus, variations in the number of seed centers make it possible to tune the length of nanowires in a wide range from nano- to microscale.

AFM and SEM images of coordination polymers (K+-LnL2) obtained by adding potassium tetraphenylborate solutions with concentrations of (а) 1 × 10–3 and (b, c) 1 × 10–2 M (saturated solution) to a LnL2 solution [1]. Printed on the authority of the American Chemical Society ©2021.
Since the radius of a metal center predetermines the type of complexation (intra- or intermolecular), which will be realized upon mixing the solutions, the properties of the nanowires can be controlled by selecting a metallocomplex with a necessary intermolecular distance. The comparative study of the structural and electrophysical characteristics of the nanowires obtained from LnL2 complexes with different metal centers (Ln = Lu, Ce, and Tb) has shown that, in the case of LuL2 with an actually minimum interdeck distance, only the intermolecular coordination of potassium ions takes place. This type of assembly yields completely conjugated nanowires with a conductivity of 11.4 S/cm, which is today maximum for undoped ensembles of phthalocyanines. The large sizes of cerium and terbium ions leads, in turn, to the formation of mixed intra- and intermolecular K+-CeL2 and K+-TbL2 phases with low conductivities (4 × 10−5 and 5 × 10−8 S/cm, respectively).
The possibility to vary the morphology and physicochemical properties of nanowires with the use of the same basic procedure of assembly makes it easier to include such structures into the industrial production of electronic nanodevices. In addition, coordination polymers have one more important advantage as compared with classical covalent ones, i.e., the reversibility of their self-assembly. While the nanowires are stable in water and the majority of organic solvents, they easily dissociate into monomers in a 1 : 1 chloroform/acetonitrile mixture. Owing to different solubilities in these organic solvents, the components can be separated and repeatedly used to produce functional materials. This makes it possible not only to solve the problem concerning utilization of the components of electronic devises, but also to return valuable lanthanide-containing complexes to the production cycle.
ULTRATHIN ORDERED FILMS OBTAINED FROM ONE-DIMENSIONAL SUPRAMOLECULAR AGGREGATES BASED ON LnL2
In order to integrate nanowires into multicomponent electronic devices, it is necessary to be able to preset the arrangement of such structures in films on solid substrates. Commonly, ultrathin coatings are formed from one-dimensional molecular structures by casting from solutions (cast films) [71, 72] and spreading from solutions on rotating substrates (spin-coating) [73, 74]. However, these procedures enable one to control only layer thickness rather than its structure. An exact orientation of nanowires on a substrate may be preset with the help of template synthesis [75, 76] and lithographic methods [77, 78], whose wide application is, however, limited by high cost and laboriousness.
An original approach to obtaining layers with a parallel arrangement of nanowires based on LuL2 reported in [1] employs the ability of such coordination polymers to undergo electrophoresis, i.e., the movement and orientation in the direction from an anode to a cathode under the action of an electric field (Fig. 3a). This occurs due to the dipoles arising as a result of a displacement of the anion “coat” consisting of [BPh4]– relative to the positively charged aggregate with quiescent Lu3+ and K+ metal ions (Fig. 3b). The process of the arrangement and deposition lasts nearly 20 min at a preset voltage of 80 V. At the same time, the packing density of the layer can be controlled by varying the concentration of a colloidal solution of nanowires (Figs. 3c, 3d). In the proposed procedure, the electrodes are applied directly onto glass substrates immersed in a suspension of nanowires in chloroform; however, the electrodes may be, potentially, located outside of the vessel with the colloidal solution, thereby eliminating limitations imposed on the characteristics of the substrates and widening the applicability of the method.

(a) Schematic representation of experimental setup for electrophoretic assembly of ordered coatings from K+-LuL2 nanowires, (b) schematic representation of the formation of dipole under an applied electric field, (c) optical micrograph of a film, which is composed of nanowires and located between two electrodes, and (d) SEM micrograph of K+-LuL2 nanowires deposited from a dilute colloidal solution between two electrodes [1]. Printed on the authority of the American Chemical Society ©2021.
Another approach proposed by Kalinina et al. for assembling ordered coatings from K+-LnL2 nanowires is based on the classical method for the formation of ultrathin films, i.e., the Langmuir–Blodgett technique [79]. Since the application of a pure suspension of nanowires onto the surface of an aqueous subphase does not lead to the formation of uniform densely packed coatings (Fig. 4a), it was proposed, for obtaining dense films, to introduce, at the stage of the synthesis, into the reaction mixture an additional component, which facilitates spreading of the aggregates over the water surface. The addition of tert-butylamine to a LuL2 solution not only provides the uniform spreading of nanowires on the interface, but also makes it possible to regulate their sizes by varying the phthalocyaninate-to-tert-butylamine ratio in the system. Using atomic force microscopy (AFM), it was shown that, as the fraction of tert-butylamine is increased, the size of nanowires grows, while the morphology of the films varies from a threadlike continuous structure to separately lying nanowires (Figs. 4b–4d).

AFM images and corresponding profiles for surfaces of LB films based on K+-LuL2 coordination polymers obtained in systems with different contents of tert-butylamine (t-BA). LuL2 : t-BA molar ratios are (a) 1 : 0, (b) 1 : 2, (c) 1 : 5, and (d) 1 : 10 [79]. Printed on the authority of ISUCT Publishing ©2021.
CONTROL OVER ORIENTATION OF LnL2 MOLECULES IN MONOLAYERS ON LIQUID AND SOLID SURFACES
The presence of eight crown-ether groups imparts LnL2 complexes with surface-active properties; therefore, they can be used to obtain monolayer phthalocyanine coatings by the Langmuir–Blodgett method. It is known that, in a loosely packed monolayer on the surface of an aqueous subphase (the state of a two-dimensional gas and a liquid-expanded phase), LnL2 molecules occur in a planar orientation, which changes into a tilted one upon a subsequent increase in the surface pressure (Fig. 5a) [59, 80, 81]. In principle, the transfer of the monolayers at different surface pressures makes it possible to obtain films with both planar and tilted orientations. At the same time, in work [80] devoted to studying CeL2 monolayers, it has been shown that the transfer of a monolayer from a pure water surface onto a mica substrate at a surface pressure corresponding to the planar orientation of molecules gives rise to the formation of a monolayer with a tilted orientation of molecules on the solid surface (Fig. 5b). Molecular orientation was determined by AFM from the film thickness, which was equal to 2 nm, thereby corresponding to the diameter of a CeL2 molecule. This value implies that, in the course of the transfer, a spontaneous phase transition occurs in the monolayer because of its contact with the solid substrate. In this case, two competitive processes, i.e., the aggregation due to the π-stacking and the interaction of the crown ethers of the lower deck of the complex with potassium ions present on the mica surface take place, with the aggregation being more energetically advantageous.

(a) Surface pressure versus molecular area isotherm for CeL2 monolayer on the surface of an aqueous subphase. Points 1 and 2 denote the surface pressures at which the monolayers were transferred onto mica substrates. (b, c) AFM images and corresponding profiles for surfaces of monolayers transferred onto mica at surface pressures of 9 and 1 mN/m, respectively. (d) Scheme for the formation of a CeL2 monolayer with the planar orientation of molecules via the interaction of CeL2 molecules with K+ cations present on mica surface [80]. Printed on the authority of Elsevier ©2016.
Monolayers with the planar orientation can be obtained via substrate-induced condensation [82] of loosely packed monolayers on solid substrates doped with potassium cations. The transfer of a monolayer at a surface pressure of ≤1 mN/m onto a slowly moving mica substrate results in the spontaneous formation of a densely packed monolayer, in which molecules are oriented parallel to the solid surface (Fig. 5c). The thickness of such a film is ~0.75 nm, which corresponds to the height of a molecule occurring in the planar orientation. Under such conditions, the main process is the complexation between crown ethers and potassium ions present on the substrate (Fig. 5d), thus providing the formation of a uniform coating with a small amount of microdefects.
Different arrangements of molecules in a monolayer cause differences in the optical properties of the resulting coatings. First, the passage from the planar to tilted orientation of molecules in a film changes the positions and intensity ratios of absorption bands. This is due to an increase in the hydrophobicity of the local environment of aromatic rings because of a decrease in the area of the contact between molecules and an aqueous phase, with this decrease leading to partial oxidation of cerium ions. Second, in spite of the fact that the specific concentration of molecules in the films with a tilted orientation is nearly 1.5-fold higher than that at the planar orientation, the absorption intensity is almost the same for the films of both types. This is due to the fact that the light absorption by molecules occurring in the planar orientation is more intense because of the coincidence between the electric-field vector of the incident wave and the dipole moments of the electron transitions in CeL2, which lies in the ring plane [83].
At the same time, the integration of the monolayers of both types into ultrathin composite films with gold nanoparticles, in which the resonance enhancement of visible light absorption occurs in a specified spectral range [84], has shown that, for such systems, the key parameter is the specific concentration of molecules in a film rather than their orientation. The absorption intensities of composite films based on CeL2 monolayers with tilted and planar orientations differ by two times. Thus, the feasibility of regulating orientation of CeL2 molecules in monolayers gives the possibility of producing materials with different optical properties.
CONCLUSIONS
The generalization of the information reported in the review leads to the conclusion that, in spite of the active use of phthalocyanines as components of functional materials intended for organic electronics and photonics, the production of ordered ensembles with a controlled structure based thereon remains to be rather difficult for researchers. The main problem in this field is associated with the pronounced tendency of phthalocyanines to aromatic stacking, which decreases the solubility of such aromatic molecules and hinders the formation of structures defectless in the micron-level scale. Taking into account the tendency to a decrease in the sizes of electronic devices, producing of actually defectless ensembles of phthalocyanines with a specified structure is a first-priority problem, whose solution will make it possible to widen the possibilities of using phthalocyanines in modern materials science. There are two ways to solving this problem: (i) the search for universal approaches to creating ensembles that would be adopted for different types of phthalocyanines and other classes of organic compounds and (ii) the reasonable selection of building blocks that could be used to produce materials with different properties. Colloid chemistry gives wide possibilities for suppressing phthalocyanine aggregation by actively employing interfaces. An interface may serve as a mechanical barrier hindering aggregation. This approach is realized in the case of phthalocyanine solubilization in micellar solutions. At the same time, an interface may play the role of an active component of a system, when the interaction of phthalocyanines with this component becomes more energetically advantageous than the intermolecular stacking. This concept underlies the formation of multilayer coatings with the planar orientation by the Langmuir–Blodgett technique including the use of the effect of substrate-induced condensation.
The methods based on the notions of coordination chemistry enable one, while remaining stacking of molecules preserved, vary their mutual orientation, thereby affecting the properties of ensembles. A change in the orientation from a tilted one, which results from stacking, to the perpendicular orientation, which results from coordination of metal ions with side substituents or bidentate ligands with the metal center of phthalocyanine, leads to an increase in the electrical conductivity of phthalocyanine ensembles.
The combination of the methods of colloid chemistry and coordination chemistry when assembling functional materials enables one to control their structure at all levels of material organization from the molecular level to the macroscopic one. Therewith, since the assembly is a spontaneous process, these approaches do not require high expenditures of energy, thus predetermining the promise of their use in real production processes.
The active widening of the spectrum of the methods employed to compose ensembles of phthalocyanines gives the opportunity for realizing the new combinatorial approach to producing nanomaterials based thereon with the use of the same type of building blocks to obtain structures of different functional destinations. The success of this approach is a consequence of two components: the great diversity of the methods for the assembly and the ability of a selected phthalocyanine derivative to form intermolecular bonds. The above-described example of implementation of this approach with the use of double-decker crown-substituted lanthanide phthalocyaninates shows that, being involved in different intermolecular interactions, such multifunctional building blocks may form ordered structures possessing different properties.
Thus, the information accumulated to date in the field of composing ensembles of phthalocyanines makes it possible to solve a number of problems relevant to the control over the arrangement of such macrocyclic molecules. At the same time, the further search for universal molecular building blocks and methods for the assembly of ordered nanostructures based thereon is necessary for the valuable employment of the potential of phthalocyanines when constructing functional materials.
REFERENCES
Zvyagina, A.I., Aleksandrov, A.E., Martynov, A.G., et al., Inorg. Chem., 2021, vol. 60, no. 20, pp. 15509–15518.
Ling, M.-M., Bao, Z., and Erk, P., Appl. Phys. Lett., 2006, vol. 89, no. 16, p. 163516.
Xiao, K., Liu, Y., Huang, X., et al., J. Phys. Chem., vol. 107, no. 35, pp. 9226–9230.
Chaure, N.B., Cammidge, A.N., Chambrier, I., et al., Sci. Technol. Adv. Mater., 2011, vol. 12, no. 2, p. 025001.
Javier Ramos, F., Ince, M., Urbani, M., et al., Dalton Trans., 2015, vol. 44, no. 23, pp. 1084–10851.
Urbani, M., Ragoussi, M.-E., Nazeeruddin, M.K., et al., Coord. Chem. Rev., 2019, vol. 381, pp. 1–64.
Wang, M., Torbensen, K., Salvatore, D., et al., Nat. Commun., 2019, vol. 10, no. 1, p. 3602.
Yang, J., Tao, J., Isomura, T., et al., Carbon, 2019, vol. 145, pp. 565–571.
Chen, K., Liu, K., An, P., et al., Nat. Commun., 2020, vol. 11, no. 1, p. 4173.
Boutin, E., Wang, M., Lin, J.C., et al., Angew. Chem., Int. Ed., 2019, vol. 58, no. 45, pp. 16172–16176.
Spesia, M.B. and Durantini, E.N., The Chemical Record, 2022, vol. 22, no. 4, p. e202100292.
Halaskova, M., Rahali, A., Almeida-Marrero, V., et al., ACS Med. Chem. Lett., 2021, vol. 12, no. 3, pp. 502–507.
Borzęcka, W., Domiński, A., and Kowalczuk, M., Nanomaterials, 2021, vol. 11, no. 9, p. 2426.
Setaro, F., Wennink, J.W.H., Mäkinen, P.I., et al., J. Mater. Chem. B., vol. 8, no. 2, pp. 282–289.
Pinto, B.C.S., Ambrósio, J.A.R., Marmo, V.L.M., et al., Photodiagn. Photodyn. Ther., 2022, vol. 38, p. 102850.
Klyamer, D., Bonegardt, D., and Basova, T., Chemosensors, 2021, vol. 9, no. 6, p. 133.
Koifman, O.I., Ageeva, T.A., Beletskaya, I.P., et al., Macroheterocycles, 2020, vol. 13, no. 4, pp. 311–467.
Makhseed, S., Machacek, M., Alfadly, W., et al., Chem. Commun., 2013, vol. 49, no. 95, p. 11149.
Demir, E., Silah, H., and Uslu, B., Crit. Rev. Anal. Chem., 2022, vol. 52, no. 2, pp. 425–461.
Goldshleger, N.F., Lapshina, M.A., Baulin, V.E., et al., Russ. Chem. Bull., 2020, vol. 69, no. 7, pp. 1223–1244.
Antipin, I.S., Alfimov, M.V., Arslanov, V.V., et al., Russ. Chem. Rev., 2021, vol. 90, no. 8, pp. 895–1107.
Zeis, R., Siegrist, T., and Kloc, C., Appl. Phys. Lett., 2005, vol. 86, no. 2, p. 022103.
Jiang, H., Ye, J., Hu, P., et al., Sci. Rep., 2015, vol. 4, no. 1, p. 7573.
Ruan, L., Tong, J., Luo, F., et al., Appl. Surf. Sci., 2022, vol. 585, pp. 152445.
Anzai, Y., Higashi, T., Kajii, H., et al., Org. Electron., 2018, vol. 60, pp. 16–21.
Ohmori, M., Nakatani, M., Kajii, H., et al., Jpn. J. A-ppl. Phys., 2018, vol. 57, no. 3, p. 03EH10.
Basova, T., Hassan, A., Durmuş, M., et al., Coord. Chem. Rev., 2016, vol. 310, pp. 131–153.
Cao, Y., Wei, Z., Liu, S., et al., Angew. Chem., Int. Ed., 2010, vol. 49, no. 36, pp. 6319–6323.
Valli, L., Adv. Colloid Interface Sci., 2005, vol. 116, nos. 1−3, pp. 13–44.
Roy, D., Das, N.M., Shakti, N., et al., RSC Adv., 2014, vol. 4, no. 80, pp. 42514–42522.
Palacin, S., Adv. Colloid Interface Sci., 2000, vol. 87, nos. 2–3, pp. 165–181.
Fujiki, M., Tabei, H., and Kurihara, T., Langmuir, 1988, vol. 4, no. 5, pp. 1123–1128.
Nakahara, H., Sun, K.Z., Fukuda, K., et al., J. Mater. Chem., 1995, vol. 5, no. 3, p. 395.
Fouriaux, S., Armand, F., Araspin, O., et al., J. Phys. Chem., 1996, vol. 100, no. 42, pp. 16984–16988.
Palacin, S., Ruaudel-Teixier, A., and Barraud, A., J. Phys. Chem., 1989, vol. 93, no. 20, pp. 7195–7199.
Palacin, S., Lesieur, P., Stefanelli, I., et al., Thin Solid Films, 1988, vol. 159, nos. 1–2, pp. 83–90.
Yan, W., Zhou, Y., Wang, X., et al., J. Chem. Soc., Chem. Commun., 1992, no. 12, pp. 873–875.
Porteu, F., Palacin, S., Ruaudel-Teixier, A., et al., J. Phys. Chem., 1991, vol. 95, no. 19, pp. 7438–7447.
Palacin, S., Ruaudel-Teixier, A., and Barraud, A., J. Phys. Chem., 1986, vol. 90, no. 23, pp. 6237–6242.
Gregory, B.W., Vaknin, D., Gray, J.D., et al., J. Phys. Chem., vol. 101, no. 11, pp. 2006–2019.
Dube, E., Oluwole, D.O., Nwaji, N., et al., Spectrochim. Acta, Part A, 2018, vol. 203, pp. 85–95
Rapulenyane, N., Antunes, E., and Nyokong, T., New J. Chem., 2013, vol. 37, no. 4, p. 1216.
Magadla, A., Oluwole, D.O., Managa, M., et al., Polyhedron, 2019, vol. 162, pp. 30–38.
Mapukata, S., Nwahara, N., and Nyokong, T., J. Photochem. Photobiol., A, 2020, vol. 402, no. July, pp. 112813.
Zhang, M., Si, D., Yi, J., et al., Small, 2020, vol. 16, no. 52, p. 2005254.
Wang, M., Shi, H., Zhang, P., et al., Adv. Funct. Mater., 2020, vol. 30, no. 30, p. 2002664.
Singh, A., Roy, S., Das, C., et al., Chem. Commun., 2018, vol. 54, no. 35, pp. 4465–4468.
Meng, Z., Aykanat, A., and Mirica, K.A., J. Am. Chem. Soc., 2019, vol. 141, no. 5, pp. 2046–2053.
Nagatomi, H., Yanai, N., Yamada, T., et al., Chem.— Eur. J., 2018, vol. 24, no. 8, pp. 1806–1810.
Ukei, K., Acta Crystallographica Section B Structural Crystallography and Crystal Chemistry, 1973, vol. 29, no. 10, pp. 2290–2292.
Dirk, C.W., Mintz, E.A., Schoch, K.F., et al., Journal of Macromolecular Science: Part A—Chemistry, 1981, vol. 16, no. 1, pp. 275–298.
Linsky, J.P., Paul, T.R., Nohr, R.S., et al., Inorg. Chem., 1980, vol. 19, no. 10, pp. 3131–3135.
Schneider, O. and Hanack, M., Angew. Chem., Int. Ed. Engl., 1980, vol. 19, no. 5, pp. 392–393.
Keppeler, U., Deger, S., Lange, A., et al., Angew. Chem., Int. Ed. Engl., 1987, vol. 26, no. 4, pp. 344–345.
Litvinov, A.L., Kuzmin, A.V., Yudanova, E.I., et al., Eur. J. Inorg. Chem., 2016, vol. 2016, no. 35, pp. 5445–5448.
Petersen, J.L., Schramm, C.S., Stojakovic, D.R., et al., J. Am. Chem. Soc, 1977, vol. 99, no. 1, pp. 286–288.
Schramm, C.J., Stojacovic, D.R., Hoffman, B.M., et al., Science, 1978, vol. 200, no. 4337, pp. 47–48.
Gorbunova, Y.G., Martynov, A.G., and Tsivadze, A.Y., Crown-substituted phthalocyanines: From synthesis towards materials, in Handbook of Porphyrin Science: With Applications to Chemistry, Physics, Materials Science, Engineering, Biology and Medicine, World Scientific Book, 2012, vol. 24, pp. 271–388.
Selektor, S.L., Shokurov, A.V., Arslanov, V.V., et al., J. Phys. Chem., vol. 118, no. 8, pp. 4250–4258.
Arslanov, V.V., Gorbunova, Y.G., Selektor, S.L., et al., Russ. Chem. Bull., 2004, vol. 53, no. 11, pp. 2532–2541.
Selektor, S.L., Shokurov, A. V., Raitman, O.A., et al., Colloid J., 2012, vol. 74, no. 3, p. 334–345.
Pushkarev, V.E., Tomilova, L.G., and Nemykin, V.N., Coord. Chem. Rev., 2016, vol. 319, pp. 110–179.
Jiang, J. and Ng, D.K.P., Acc. Chem. Res., 2009, vol. 42, no. 1, p. 79–88.
Toupance, T., Benoit, H., Sarazin, D., et al., J. Am. Chem. Soc, 1997, vol. 119, no. 39, pp. 9191–9197.
Martynov, A.G., Gorbunova, Y.G., and Tsi-vadze, A.Y., Prot. Met. Phys. Chem. Surf., 2011, vol. 47, no. 4, pp. 465–470.
Toupance, T., Ahsen, V., and Simon, J., J. Chem. Soc., Chem. Commun., 1994, vol. 1, no. 1, pp. 75–76.
Toupance, T., Ahsen, V., and Simon, J., J. Am. Chem. Soc, 1994, vol. 116, no. 12, pp. 5352–5361.
Steybe, F. and Simon, J., New J. Chem., 1998, vol. 22, no. 12, pp. 1305–1306.
Toupance, T., Plichon, V., and Simon, J., New J. Chem., 1999, vol. 23, no. 10, pp. 1001–1006.
Thami, T., Chassenieux, C., Fretigny, C., et al., J. Porphyrins Phthalocyanines, 2002, vol. 06, no. 09, pp. 563–570.
Zhang, R., Li, B., Iovu, M.C., et al., J. Am. Chem. Soc, 2006, vol. 128, no. 11, pp. 3480–3481.
Sauvé, G. and McCullough, R.D., Adv. Mater., 2007, vol. 19, no. 14, pp. 1822–1825.
Klauk, H., Halik, M., Zschieschang, U., et al., J. Appl. Phys., 2002, vol. 92, no. 9, pp. 5259–5263.
Niles, E.T., Roehling, J.D., Yamagata, H., et al., J. Phys. Chem. Lett., 2012, vol. 3, no. 2, pp. 259–263.
Son, M., Park, K.H., Shao, C., et al., J. Phys. Chem. Lett., 2014, vol. 5, no. 20, pp. 3601–3607.
Pan, L., Qiu, H., Dou, C., et al., Int. J. Mol. Sci., 2010, vol. 11, no. 7, pp. 2636–2657.
Wang, P., Li, Z., Zhang, L., et al., Opt. Lett., 2013, vol. 38, no. 7, pp. 1040–1042.
Laza, S.C., Polo, M., Neves, A.A.R., et al., Adv. Mater., 2012, vol. 24, no. 10, pp. 1304–1308.
Zvyagina, A.I., Naumova, A.D., Kuzmina, N.V., et al., Macroheterocycles, 2021, vol. 14, no. 1, pp. 59–64.
Zvyagina, A.I., Meshkov, I.N., Ezhov, A.A., et al., Colloids Surf., A, 2016, vol. 509, no. September, pp. 376–383.
Shokurov, A.V., Selektor, S.L., Arslanov, V.V., et al., Macroheterocycles, 2012, vol. 5, nos. 4–5, pp. 358–365.
Kalinina, M. A., Colloid J., 2015, vol. 77., no. 5, pp. 537–555.
McKeown, N.B., Phthalocyanine Materials: Synthesis, Structure, and Function, Cambridge: University Press, 1998.
Zvyagina, A.I., Ezhov, A.A., Meshkov, I.N., et al., J. Mater. Chem., 2018, vol. 6, no. 6, pp. 1413–1420.
Funding
This work was supported by the Russian Science Foundation (project no. 20-13-00279).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The author declares that she has no conflicts of interest.
Additional information
Translated by A. Kirilin
Rights and permissions
Open Access. This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zvyagina, A.I. Controlled Self-Assembly of Low-Dimensional Supramolecular Systems Based on Double-Decker Lanthanide Phthalocyaninates. Colloid J 84, 633–641 (2022). https://doi.org/10.1134/S1061933X22700090
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1061933X22700090