
Abstract
Rhizogenic, callus, and suspension cultures in vitro were obtained for Digitalis lanata and their growth, cytophysiological and biochemical characteristics were investigated. The obtained cultures were characterized by good growth characteristics (growth indexes I in the range of 5–13). Suspension cell culture had a specific growth rate μ within 0.2–0.3 days–1 and it was characterized by a two-phase growth curve (growth retardation during the exponential phase). In the obtained cultures, a study of the qualitative and quantitative composition of secondary metabolites by UPLC-ESI-MS and HPLC-ESI-MS showed the absence of cardiac glycosides. At the same time, phenylethanoids and steroidal glycosides of the furostanol type were found in all studied cultures. The total content of phenylethanoids in callus and suspension cultures was approximately 0.5% of the dry biomass. Based on the results of mass spectrometry, ten phenylethanoid structures, including digiciliside A, digiciliside B, maxoside, purpureaside E, and their methyl derivatives and isomers, and also seven furostanol glycosides with aglycones tigogenin and gitogenin were identified. It has been shown that the composition of secondary metabolites depends on the degree of cell differentiation: furostanol glycosides were prevalent in a rhizogenic culture consisting mainly of differentiated cells, while the diversity of phenylethanoids significantly increases in callus and suspension cell cultures consisting of dedifferentiated cells. The results of the study confirm the hypothesis put forward in our previous works about the specificity of secondary metabolism and its high intensity in plant cell cultures.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
INTRODUCTION
Currently, despite advances in synthetic chemistry, plants are still used to create many pharmaceuticals and dietary supplements. At least 25% of all drugs in industrialized countries contain phytochemical compounds, while in developing countries about 75% of the population relies exclusively on plant origin medicinal products [1]. Due to the fact that the populations of medicinal wild-growing plants are extremely often reduced due to unregulated procurement of raw materials and many species are rare and endangered, there is a search for alternative sources of plant biologically active compounds. Plant cell, organ and tissue cultures can be considered a promising method for obtaining plant fresh materials with target substances [2].
Digitalis lanata Ehrh., commonly known as woolly foxglove, belongs to the family Plantaginaceae and is found in Italy, Hungary, and the Balkans. Genus Digitalis is well known in medicine from the 18th century due to the presence in these plants of cardiac glycosides (cardenolides), which were used to treat cardiovascular diseases [1, 3]. Digoxin (the most commonly used cardiac glycoside in medicine) is obtained from dried leaves D. lanata because its chemical synthesis is economically unprofitable. At the present, cardenolides of foxgloves do not play a major role in the treatment of cardiovascular pathologies, but they are actively considered as promising agents in the treatment of a number of oncological diseases and viral infections [1, 4].
Cell culture study of Digitalis spp. as a source of cardiac glycosides began over 50 years ago. It was noted in many experimental works that if cardenolides are even present in small concentrations in callus and suspension cultures at the initial stages of cultivation, then their synthesis completely disappears during long-term cultivation [3, 5, 6]. In turn for leaf and root cultures in vitro D. lanata, the production of cardiac glycosides was noted, which indicates a relationship between the biosynthesis of these compounds with the processes of morphological differentiation [5, 7, 8]. In addition, there is evidence that embryogenic cultures can also accumulate a significant amount of these compounds [9]. From the analysis of available sources, it follows that dedifferentiated cells in vitro foxgloves are incapable of synthesizing cardenolides, despite the fact that the necessary enzymatic systems function in them, which confirms their ability to biotransformation target compounds when introduced into the media of various substrates [5, 10].
In almost all works on the study of secondary metabolism in D. lanata cell cultures, the main attention is only paid to the presence of cardiac glycosides in them [3, 5]. At the same time, it is known that intact plants of this species contain a wide range of secondary metabolites of different groups—steroidal glycosides, digitanols, phenylethanoids, anthraquinones, flavonoids, many of which have biological activity [4].
The aim of this work was to obtain in vitro Digitalis lanata cultures with various degrees of differentiation (rhizogenic, callus and suspension) and study specificity of their growth and synthesis of secondary metabolites of varied structural groups.
MATERIALS AND METHODS
To obtain in vitro D. lanata cultures, we used aseptic seedlings grown from D. lanata seeds taken from plants from the Botanical Garden, Faculty of Biology, Moscow State University.
Seeds were sterilized with a mixture of Tween-20 detergent (Sigma, Germany) and 50% sodium hypochlorite solution (Belizna, Russia), washed with sterile distilled water, and plated on Petri dishes with Murashige and Skoog (MS) agar medium [11] without adding growth regulators. Petri dishes were placed on racks under fluorescent lamps and germinated at 25 ± 1°C and 16 h of light.
Rhizogenesis was induced on MS agar medium with α-naphthylacetic acid (α-NAA) and kinetin (Merck, Germany) (concentration range from 1.0 to 2.0 mg/L) growth regulators using cotyledon leaves or hypocotyls of aseptic seedlings as explants.
Callus culture was obtained on rhizogenic culture using growth regulators: α-NAA, 2,4-dichlorophenoxyacetic acid (2,4-D), kinetin, and 6-benzylaminopurine (BAP) (Merck) (concentration range from 0.1 to 2.5 mg/L).
Cultivation of callus and rhizogenic cultures was carried out on Petri dishes with a diameter of 60 or 90 mm in the dark at 25 ± 1°C.
To obtain a suspension cell culture, callus culture was used as explants. Calli were placed in 250 mL flasks with 33–40 mL of liquid medium, which were placed on a rotary shaker (100 rpm) and cultured in the dark at 25 ± 1°C.
For growing the obtaining cultures in vitro, we used MS medium supplemented with casein hydrolyzate (0.5 g/L), inositol (0.1 g/L), 3% sucrose, and growth regulators (α-NAA, 2,4-D, kinetin, BAP). When growing callus and suspension cell cultures, casein hydrolyzate was not used.
Micrographs of the suspension cell culture were made using a ToupCam SCMOS 0.3 Mpix digital camera (China).
To characterize the growth and physiological state of D. lanata cultures, we used the level of accumulation of fresh and dry biomass, concentration (number of cells in 1 mL of suspension) and viability of cells.
The viability of the suspension culture was determined using the vital dye phenosafranine (0.1% solution) (Merck) by counting live (unstained) and dead (stained) cultured units under a microscope [12].
To determine the cell concentration (the number of cells in 1 mL of suspension) in the suspension culture, we used cell counting in a Fuchs-Rosenthal hemocytometer after incubation of the suspension in a 20% chromic acid solution at 60°C for 20–30 min [12].
To determine the fresh and dry biomass of callus and rhizogenic cultures, callus or roots separated from the nutrient medium were used. For the growth curve, the weight of one callus or a part of the root culture was used with a transplant weight of 150 mg ± 30% of fresh biomass. For each point, at least three calli or root culture parts were used.
For a suspension cell culture, a fixed volume of suspension (at least 10 mL in two biological replicates) was filtered under vacuum through a paper filter using a Buchner funnel [12].
Biomass of all obtained in vitro cultures was dried in an oven at 50°C for 24 h.
Based on the obtained results, the growth parameters of the suspension culture, such as growth index (I), specific growth rate (μ), biomass doubling time (τ), economic coefficient (Y), and biomass productivity (P), were calculated. The following formulas were used for calculations [12]:
(1) I = Xmax/X0, where Xmax and X0 are the maximum and initial values of the growth criterion, respectively (dry weight, fresh weight, cell concentration).
(2) µ = (lnX2 – lnХ1)/(t2 – t1), where X2 and X1 are the values of the growth criterion (dry weight, fresh weight, cell concentration) at a point in time t2 and t1, respectively (calculated for the exponential growth phase).
(3) τ = ln2/μ.
(4) Y = (Xmax – Xo)/So, where Xmax and Xo are maximum and initial concentration of dry biomass (g/L), respectively, and So is the initial concentration of the substrate (sucrose) in the medium (g/L of the medium).
(5) P = (Xi – Xo)/(ti – to), where Xo and Xi are the amount of dry biomass at the beginning of cultivation and at the time ti, respectively.
For a qualitative and quantitative analysis of the composition of secondary metabolites in the studied cultures, we used ultrahigh-performance liquid chromatography coupled with electrospray ionization mass spectrometry (UPLC-ESI-MS) and high-performance liquid chromatography coupled with electrospray ionization mass spectrometry (HPLC-ESI-MS) respectively.
For sample preparation, 100–300 mg dried plant material (cell or root biomass) was extracted three times with 70% (by volume) ethyl alcohol for 30 min under the action of ultrasound (Sapfir ultrasound bath, Russia) at room temperature and then centrifuged (MiniSpin plus centrifuge, Eppendorf, Germany) at 9660 g for 5 min and the supernatant was taken into a pear-shaped flask. The combined ethanolic extracts were evaporated under vacuum at 40°C, suspended in 3 mL of 5% (by volume) aqueous acetic acid solution, and loaded onto a Supelclean ENVI-18 solid-phase extraction cartridge (Supelco, United States). The cartridge was washed with 3 mL of 5% aqueous acetic acid solution and analytes were washed into a pear-shaped flask with 3 mL of 70% ethanol. The resulting extract was evaporated to dryness under vacuum at 40°C and used for chromatographic analysis.
For UPLC-ESI-MS the samples were dissolved in a mixture of acetonitrile : water (1 : 1, by volume) and filtered using a nylon filter with 0.2 μm pores (Acrodisc, Pall Corporation, United States). UPLC-ESI-MS-analysis was performed on a Waters Acquity UPLC chromatograph (Waters, United States) equipped with a Xevo QTof hybrid quadrupole time-of-flight mass spectrometer (Waters, United States). A sample in a volume of 1 μL was loaded onto an ACQUITY UPLC BEH Phenyl column (50 × 2.1 mm, 1.7 μm; Waters, United States). The column temperature was 40°С, the volumetric flow rate of the mobile phase was 0.4 mL/min. A 0.1% (by volume) solution of formic acid in water (solvent A) and a 0.1% (by volume) solution of formic acid in acetonitrile (solvent B) were used as the mobile phase. Chromatographic separation was performed in the gradient elution mode. During the analysis, the composition of the mobile phase changed as follows (B, % by volume): 15% at 0–1 min, 15 → 30% at 1–5 min, 30 → 38% at 5–15 min, 38 → 45% at 15–15.5 min, 45% at 15.5–23 min, 45 → 95% at 23–23.5 min. The analysis was carried out in the mode of detecting positive and negative ions (range m/z 100–1200). Ionization source parameters: ionization source temperature of 120°С, desolvation temperature of 250°С, capillary voltage of 3.0 kV, sample injection cone voltage of 30 V, nitrogen (desolvation gas) flow rate of 600 L/h. The results were processed using the MassLynx software (Waters, United States). The identification of the compounds was carried out on the basis of interpretation of the mass spectra and comparing the chromatographic and mass spectrometric behavior of the detected compounds with the literature data [1, 13–18].
Before the HPLC-ESI-MS analysis, all samples were dissolved in a mixture of acetonitrile : water (1 : 1, by volume) and filtered into glass vials using Teflon filters with 0.45 μm pores CAMEO 17F (GVSS.pA, Italy). The analysis was carried out on an Agilent 1260 Infinity instrument (Agilent Technologies, United States) equipped with a mass selective detector (6100, Agilent Technologies). We used a Poroshell 120 EC-C18 column (100 mm × 3 mm, 2.7 μm, Agilent, United States) with a temperature of 43°C and mobile phase flow rate of 0.4 mL/min. The injection volume is 0.5 μL. A 0.05% (by volume) solution of formic acid (Fluka, United States) in water (solvent A) and acetonitrile (solvent B) were used as the mobile phase. During the analysis, the composition of the mobile phase changed as follows (B, % by volume): 5 → 13% at 0–7.5 min, 13 → 21% at 7.5–9 min, 21 → 31% at 9–19 min, 31 → 95% at 19–19.5 min, 95% at 19.5–21.5 min. The analysis was carried out with the detection of negative ions (range m/z 100–1300, fragmentor of 90) in the mode of recording signals for certain ions (SIM mode—“selected-ion monitoring”). Ionization source parameters were the following: 100°С quadrupole temperature, 150°С carrier gas temperature (nitrogen), 12.5 L/min nitrogen supply rate (desolvation gas), 2484 Torr nitrogen pressure, 4 kV capillary voltage.
The quantitative determination of the content of individual phenylethanoid glycosides was carried out on the basis of HPLC-ESI-MS analysis by external calibration against a standard sample of echinacoside (Sigma).
To determine the sensitivity of the HPLC-ESI-MS method, we used standard samples of lanatoside C (Sigma), convallatoxin, digitoxin and digoxin (ChromaDex, United States) when screening cardiac glycosides in the samples. The minimum detectable concentration for solutions of standard samples of cardiac glycosides was in the range of 0.4–0.8 μg/mL.
Determination of fresh and dry biomass and cell viability was carried out in two to three biological replicates. The number of cells was counted in two to three biological and two analytical replicates. The qualitative and quantitative determination of secondary metabolites was carried out in an individual extract without taking into account biological and analytical replicates. When quantifying the content of individual phenylethanoid glycosides by the HPLC-ESI-MS method, the relative standard deviation for the retention times of the compounds did not exceed 5%, while that for the peak areas of echinacoside was 10%. Data analysis was performed using the Excel program included in the Microsoft Office 2010. The graphs show the means and their standard errors.
RESULTS
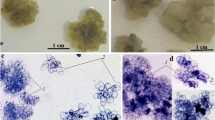
The first stage of the work was to obtain a rhizogenic culture of D. lanata. As explants, we used cotyledon leaves and hypocotyls of aseptic seedlings obtained from sterile seeds. Cotyledon leaves or hypocotyls were removed from seedlings and placed on Petri dishes with MS agar medium supplemented with 2 mg/L α-NAA and 2 mg/L kinetin. The formation of a rhizogenic culture was usually observed after 10–14 days. In the presence 2 mg/L α-NAA and 2 mg/L kinetin, a stable and well-growing rhizogenic culture was formed in D. lanata (Fig. 1). It should be noted that intensive rhizogenesis was characteristic mainly of leaf explants, while necrosis of the obtaining culture was observed for hypocotyls.

(a) Rhizogenic, (b) callus, and (c) suspension cultures of D. lanata.
Callus cell cultures were initiated from the rhizogenic culture of D. lanata. To induce callusogenesis, MS media with various combinations of growth regulators were used: 1 mg/L α-NAA and 2.5 mg/L BAP, 1 mg/L 2,4-D and 0.1 mg/L kinetin, 2 mg/L 2,4-D and 1 mg/L kinetin, 1 mg/L 2,4-D and 0.5 mg/L BAP. The MS medium with 1 mg/L 2,4-D and 0.5 mg/L BAP was found to be optimal for the formation of a callus cell culture. The obtaining callus culture had a brown color and a fragile structure (Fig. 1). It was not possible to induce callusogenesis on other combinations of growth regulators; necrosis of the root culture was observed in most cases.
Suspension cell culture of D. lanata was obtained from a callus culture more than 1 year old (11 cultivation cycles). Callus cells (2–4 g of fresh biomass) were placed in flasks with a liquid nutrient medium MS supplemented with 1 mg/L 2,4-D and 0.5 mg/L BAP. The flasks were placed on a rotary shaker, and a primary suspension cell culture was obtained after 18–22 days of cultivation. As a result of work on optimizing the growing mode of the obtaining suspension, it was found that the following cultivation cycle is optimal: 21 days and with a ratio of inoculum : fresh medium during transplantation of 1 : 8. The suspension had a yellow-brown color, and contained large aggregates of meristem-like and parenchyma-like cells as well as many single meristem-like, parenchyma-like and elongated abnormal cells. Most of the single cells in suspension were not viable (Fig. 1).
After 1.5–2.5 years of cultivation (15–25 cultivation cycles) of the obtained callus and rhizogenic cultures, a study of their growth characteristics was carried out. It was found that the growth curves of callus and rhizogenic cultures of D. lanata did not yet reach the stationary phase by 42–43 days of cultivation. At the same time, a lag phase for 7 days was characteristic for the root culture, and a slowdown in growth was noted for a callus cell culture after 29 days of cultivation. For rhizogenic culture of D. lanata on the 42nd day of cultivation, a ninefold increase in fresh biomass and an eightfold increase in dry biomass was noted. For the callus cell culture on the 43rd day of cultivation, there was an increase of five times for both dry and fresh biomass (Fig. 2).

Growth curves in the standard and semilogarithmic coordinate systems of (a, b) rhizogenic and (c, d) callus cultures of D. lanata: (1) fresh biomass; (2) dry biomass. Numerical data are indicated per callus or part of rhizogenic culture.
Growth characteristics of D. lanata suspension culture were determined after seven cultivation cycles (5 months of cultivation after obtaining) (Fig. 3). When analyzing the growth curves, the absence of a lag phase for the indicator of cell concentration and its presence for 6–8 days for the accumulation of fresh and dry biomass was noted. It is also worth noting that a period of growth retardation was observed in the cultivation cycle during an exponential phase of 4 days, which divides the cultivation cycle into two phases. After 21 days of cultivation, growth slows down and the culture transitions to the stationary growth phase, which lasts approximately 4 days. In addition, for the D. lanata suspension, a relatively low indicator of cell viability was recorded it was at the level of 60–70% during the entire cultivation cycle and approached 50% on the 28th day. Such dynamics can be associated with a large number of single dead cells and the presence of the majority of living cells mainly in large aggregates.

Growth curves in (a) standard and (b) semilogarithmic coordinate systems and cell viability in D. lanata suspension culture: (1) cell concentration; (2) fresh biomass; (3) dry biomass; (4) cell viability.
Table 1 shows the growth derived parameters of suspension culture: growth index (I), specific growth rate (μ), biomass doubling time (τ), maximum accumulation of dry biomass (Mmax), economic coefficient (Y), and maximum productivity (P).
From the presented growth parameters, it follows (Table 1) that the D. lanata suspension culture with a relatively low cell viability was a well-growing culture. Growth indexes for cell concentration and fresh and dry biomass were 12–13, the specific growth rate was within 0.2–0.3 days–1, and the maximum accumulation of dry biomass was 9.4 g/L. In addition, a fairly good economic coefficient was noted for growing a culture, which is 0.29, from which it follows that almost 30% of the sucrose of the nutrient medium is spent on construction the cell biomass.
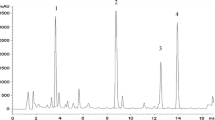
To study the qualitative composition of secondary metabolites in the rhizogenic culture of D. lanata, we used the UPLC-ESI-MS method. The chromatogram of the ethanol extract from the biomass of the root culture recorded in the total ion current mode (positively charged ions detection) and the mass spectra (positive ions) of some peaks of the identified compounds are shown in Fig. 4.

UPLC-ESI-MS-chromatogram (a, recorded in the total ion current mode, positive ions detection) of an ethanolic extract from the biomass of a rhizogenic culture of D. lanata and mass spectra (positive ions) of peaks with a retention time of 2.03 (b, tentative identification—phenylethanoid demethyl purpureaside E), 3.12 (c, tentative identification—furostanol glycoside with aglycone gitogenin), 4.01 (d, tentative identification—furostanol glycoside with aglycone tigogenin). The peak numbers correspond to Table 2.
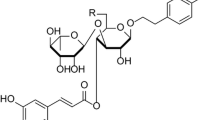
In rhizogenic culture, at least ten peaks of compounds were found with retention times on the column within 1–5 min. Phytochemical analysis showed the absence of cardiac glycosides, the main secondary metabolites of foxgloves, in the extract from the biomass of rhizogenic culture, but phenylethanoids and steroidal glycosides of the furostanol type were found and their structures are shown in Fig. 5 and Table 2.

The results of mass spectrometry indicate that the detected phenylethanoid glycosides correspond to digiciliside A, maxoside, and demethyl purpureaside E and furostanol glycosides are derivatives of two aglycones: gitogenin and tigogenin. The rhizogenic culture contains at least seven furostanol glycosides and three phenylethanoids.
The data obtained by the UPLC-ESI-MS method were used for targeted (performed using HPLC-ESI-MS) qualitative and quantitative analysis of phenylethanoids as well as qualitative analysis of steroidal glycosides in the biomass of callus and suspension cultures of D. lanata. To increase the sensitivity, HPLC-ESI-MS analysis was performed by detecting negatively charged ions [20] and detection certain selected ions (SIM mode—“selected-ion monitoring”) [21]. Overview HPLC-ESI-MS (SIM mode) chromatograms of ethanol extracts from biomass of cell cultures are represented in Fig. 6. The list of phenylethanoid ions (under the proposed conditions, HPLC-ESI-MS were detected as deprotonated ions [M–H]–) and steroidal glycosides (decorated as adduct ions with a component of the mobile phase—formic acid, [M–H + HCOOH]–) is presented in Table 3.

HPLC-ESI-MS-chromatograms (the detection of negative ions, SIM mode; the list of ions for which the analysis was carried out and their identification is presented in Table 3) ethanol extracts from biomass of cell cultures of D. lanata: (a) callus cell culture, (b) suspension cell culture. The peak numbers correspond to Table 3.
In suspension cell culture of D. lanata, at least ten phenylethanoids and four furostanol glycosides were found, while in callus culture there were nine phenylethanoids and four furostanol glycosides. HPLC-ESI-MS analysis showed an increase in the spectrum of phenylethanoid glycosides and a decrease in the diversity of steroidal glycosides in dedifferentiated cell cultures of D. lanata. It should be noted that the rhizogenic culture contained furostanol glycosides having tigogenin and gitogenin as aglycones, while only tigogenin derivatives were found in callus and suspension cultures.
Quantitative HPLC-ESI-MS (SIM mode) analysis of phenylethanoid glycosides in callus and suspension cell cultures of D. lanata showed that the content of these compounds reached 0.5% of the dry weight in the presented samples (Table 4).
It is noted that the main compounds in cultures of D. lanata consisting mainly of dedifferentiated cells (callus and suspension) are maxoside and demethyl purpureaside E (these compounds were also found in root culture but in insignificant amounts). At the same time, purpureaside E, digiciliside A, isomer of digiciliside A, and dimethyl-digiciliside B in callus and suspension cell cultures are present mainly in trace amounts, while the intensity of the chromatographic peak of digiciliside A (judging by UPLC-ESI-MS analysis) was quite high in rhizogenic culture.
DISCUSSION
Digitalis lanata is considered one of the most studied foxglove species in terms of secondary metabolism, both in an intact plant and in a cell, organ and tissue cultures in vitro [5]. Most of the data indicate that cardiac glycosides are usually either absent or present in extremely low concentrations at the initial stages of production and cultivation in callus and suspension cell cultures of foxgloves; however, they disappear completely with prolonged cultivation as a rule. The relationship of cardenolide biosynthesis Digitalis spp. with morphological differentiation is emphasized by the results of a number of studies with different types of cultures in vitro [3, 5]. It is known that organ cultures (leaves and roots) as well as callus culture with embryogenic structures of D. lanata retained the ability to form cardiac glycosides [5, 7–9, 22].
The results of this work confirm the absence of cardenolide synthesis in dedifferentiated D. lanata cell cultures. Despite the available data that these compounds are found in shoot and root cultures in vitro foxgloves [5, 7, 8], they could not be found within the framework of the presented study in rhizogenic culture of D. lanata. It should be said that, judging by the available literary sources, rhizogenic cultures of foxglove are not capable of producing cardiac glycosides in all cases [5]. Digitalis spp. cardenolides are formed in intact plants and accumulate mainly in leaves; their majority is concentrated in the mesophyll, therefore, morphogenetic processes associated with the formation of shoot and leaf cultures are most preferable for their synthesis [23].
The formation of phenylethanoids and steroidal glycosides of the furostanol type in in vitro D. lanata cultures indicates the specificity of their secondary metabolism and changes in the groups and spectrum of compounds depending on the degree of differentiation of cells in cultures in vitro. Furostanol glycosides prevalent in a rhizogenic culture, consisting mainly of differentiated cells, while the variety of phenylethanoids significantly increases in callus and suspension cell cultures consisting of dedifferentiated cells. It should be noted that there is information in the literature about the formation of anthraquinones in dedifferentiated D. lanata cells [24, 25]; in turn, the presence of phenylethanoids and steroidal glycosides in cells in vitro foxglove has been previously noted for callus cultures of D. purpurea [5, 26].
The results of the study confirm the hypothesis put forward in our previous works about the specificity of secondary metabolism and its high intensity in plant cell cultures. However, the mechanisms that underlie the patterns of synthesis of specific groups of secondary metabolites in cell, organ and tissue cultures in vitro for plants of the genus Digitalis require further study.
REFERENCES
Kreis, W., The foxgloves (Digitalis) revisited, Planta Med., 2017, vol. 83, p. 962. https://doi.org/10.1055/s-0043-111240
Nosov, A.M., Application of cell technologies for production of plant-derived bioactive substances of plant origin, Appl. Biochem. Microbiol., 2012, vol. 48, p. 609. https://doi.org/10.1134/S000368381107009X
Verma, S.K., Das, A.K., Cingoz, G.S., and Gurel, E., In vitro culture of Digitalis L. (foxglove) and the production of cardenolides: an up-to-date review, Ind. Crops Prod., 2016, vol. 94, p. 20. https://doi.org/10.1016/j.indcrop.2016.08.031
Clemente, E.S., Muller-Uri, F., Nebauer, S.G., Segura, J., Kreis, W., and Arrillaga, I., Digitalis, in Wild Crop Relatives: Genomic and Breeding Resources: Legume Crops and Forages, Kole, C., Ed., Berlin: Springer-Verlag, 2011, p. 73. https://doi.org/10.1007/978-3-642-21201-7_5
Rucker, W., Digitalis spp.: in vitro culture, regeneration, and the production of cardenolides and other secondary products, in Medicinal and Aromatic Plants IV, B-ajaj, Y.P.S., Ed., Berlin: Springer-Verlag, 1988, p. 388. https://doi.org/10.1007/978-3-642-73026-9_21
Hagimori, M., Matsumoto, T., and Kisaki, T., Studies on the production of Digitalis cardenolides by plant tissue culture I. Determination of digitoxin and digoxin contents in first and second passage calli and organ redifferentiating calli of several Digitalis species by radioimmunoassay, Plant Cell Physiol., 1980, vol. 21, p. 1391. https://doi.org/10.1093/pcp/21.8.1391
Lui, J. and Staba, E., Effects of precursors on serially propagated Digitalis lanata leaf and root cultures, Phytochemistry, 1979, vol. 18, p. 1913. https://doi.org/10.1016/S0031-9422(00)82701-6
Lui, J. and Staba, E., Effects of age and growth regulators on serially propagated Digitalis lanata leaf and root cultures, Planta Med., 1981, vol. 41, p. 90. https://doi.org/10.1055/s-2007-971682
Kuberski, Ch., Scheibner, H., Steup, C., Diettrich, B., and Luckner, M., Embryogenesis and cardenolide formation in tissue cultures of Digitalis lanata, Phytochemistry, 1984, vol. 23, p. 1407. https://doi.org/10.1016/S0031-9422(00)80475-6
Spieler, H., Alfermann, A.W., and Reinhard, E., Biotransformation of β-methyldigitoxin by cell cultures of Digitalis lanata in airlift and stirred tank reactors, Appl. Microbiol. Biotechnol., 1985, vol. 23, p. 1. https://doi.org/10.1007/BF02660109
Murashige, T. and Skoog, F., A revised medium for rapid growth and bio-assays with tobacco tissue cultures, Physiol. Plant., 1962, vol. 15, p. 473. https://doi.org/10.1111/j.1399-3054.1962.tb08052.x
Nosov, A.M., Methods for assessment and characteristics of growth of higher plants cell cultures, in Molekulyarno-geneticheskie i biokhimicheskie metody v sovremennoi biologii rastenii (Molecular Genetics and Biochemical Methods in Current Plant Biology), Kuznetsov, Vl.V, Kusnetsov, V.V, and Romanov, G.A, Eds., Moscow: Binom. Laboratoriya Znanii, 2012, p. 386.
Skhirtladze, A., Kemertelidze, E., Nebieridze, V., and Ganzera, M., Phenylethanoid glycosides from the roots of Digitalis ciliata Trautv., Helv. Chim. Acta, 2016, vol. 99, p. 241. https://doi.org/10.1002/hlca.201500288
Perrone, A., Plaza, A., Bloise, E., Nigro, P., Hamed, A.I., Belisario, M.A., Pizza, C., and Piacente, S., Cytotoxic furostanol saponins and a megastigmane glucoside from Tribulus parvispinus, J. Nat. Prod., 2005, vol. 68, p. 1549. https://doi.org/10.1021/np0502138
Skhirtladze, A.V., Kopaliani, T.A., Nebieridze, V.G., Kemertelidze, E.P., and Ganzera, M., New steroidal glycosides from pericarp of Digitalis ferruginea, Chem. Nat. Compd., 2017, vol. 53, p. 1083. https://doi.org/10.1007/s10600-017-2206-x
Calis, I., Akbay, P., Kuruuzum, A., Yalcin, F.N., Sahin, P., and Pauli, G.F., Phenylethanoid and cardioactive glycosides from Digitalis ferruginea, Pharmazie, 1999, vol. 54, p. 926. https://doi.org/10.1002/chin.200010190
Zhou, B.N., Bahler, B.D., Hofmann, G.A., Mattern, M.R., Johnson, R.K., and Kingston, D.G., Phenylethanoid glycosides from Digitalis purpurea and Penstemon linarioides with PKCα-inhibitory activity, J. Nat. Prod., 1998, vol. 61, p. 1410. https://doi.org/10.1021/np980147s
Jin, Q., Jin, H.G., Shin, J.E., Hong, J., and Woo, E.R., Phenylethanoid glycosides from Digitalis purpurea L., Bull. Korean Chem. Soc., 2011, vol. 32, p. 1721. https://doi.org/10.5012/BKCS.2011.32.5.1721
Kirmizibekmez, H., Kusz, N., Karaca, N., Demirci, F., and Hohmann, J., Secondary metabolites from the leaves of Digitalis viridiflora, Nat. Prod. Commun., 2017, vol. 12, p. 59.
Liigand, P., Kaupmees, K., Haav, K., Liigand, J., Leito, I., Girod, M., Antoine, R., and Kruve, A., Think negative: finding the best electrospray ionization/MS mode for your analyte, Anal. Chem., 2017, vol. 89, p. 5665. https://doi.org/10.1021/acs.analchem.7b00096
Muller, M. and Volkel, W., The use of liquid chromatography/mass spectrometry (LC/MS) in biological monitoring, in The MAK-Collection Part IV: Biomonitoring Methods, Chichester: Wiley, 2007, vol. 11, p. 3. https://doi.org/10.1002/3527600418.bilcmsmonite0011
Luckner, M. and Diettrich, B., Formation of cardenolides in cell and organ cultures of Digitalis lanata, in Primary and Secondary Metabolism of Plant Cell Cultures, Neumann, K.H., Barz, W., and Reinhard, E., Eds., Berlin: Springer-Verlag, 1985, p. 154. https://doi.org/10.1007/978-3-642-70717-9_15
Hagimori, M., Matsumoto, T., and Obi, Y., Studies on the production of Digitalis cardenolides by plant tissue culture II. Effect of light and plant growth substances on digitoxin formation by undifferentiated cells and shoot-forming cultures of Digitalis purpurea L. grown in liquid media, Plant Physiol., 1982, vol. 69, p. 653. https://doi.org/10.1104/pp.69.3.653
Furuya, T. and Kojima, H., 4-Hydroxydigitolutein, a new anthraquinone from callus tissue of Digitalis lanata, Phytochemistry, 1971, vol. 10, p. 1607. https://doi.org/10.1016/0031-9422(71)85033-1
Furuya, T., Kojima, H., and Katsuta, T., 3-Methylpurpurin and other anthraquinones from callus tissue of Digitalis lanata, Phytochemistry, 1972, vol. 11, p. 1073. https://doi.org/10.1016/S0031-9422(00)88455-1
Matsumoto, M., Koga, S., Shoyama, Y., and Nishioka, I., Phenolic glycoside composition of leaves and callus cultures of Digitalis purpurea, Phytochemistry, 1987, vol. 26, p. 3225. https://doi.org/10.1016/S0031-9422(00)82474-7
Funding
The work was carried out on the basis of the “Scientific and Production Biotechnological Complex for the Study, Preservation, and Practical Application of Cultured Cells and Organs of Higher Plants and Microalgae” with the financial support of the Megagrant of the Government of the Russian Federation (Agreement no. 075-15-2019-1882).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interests. The authors declare that they have no conflicts of interest.
Statement on the welfare of humans or animals. This article does not contain any studies involving humans or animals performed by any of the authors.
Additional information
Abbreviations: HPLC-ESI-MS—high-performance liquid chromatography coupled with electrospray ionization mass spectrometry; SIM—selected-ion monitoring; UPLC-ESI-MS—ultrahigh-performance liquid chromatography coupled with electrospray ionization mass spectrometry.
Rights and permissions
Open Access. This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tomilova, S.V., Kochkin, D.V., Tyurina, T.M. et al. Specificity of Growth and Synthesis of Secondary Metabolites in Cultures in vitro Digitalis lanata Ehrh.. Russ J Plant Physiol 69, 25 (2022). https://doi.org/10.1134/S1021443722020200
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1134/S1021443722020200