
Abstract
Thermoresponsive diblock copolymers (DCs) were prepared by two-stage reversible addition-fragmentation chain transfer/macromolecular design by interchange of xanthate (RAFT/MADIX) polymerization of N-vinylcaprolactam and N-vinylimidazole (VI). The poly(N-vinylcaprolactam) (PVCL) blocks were first synthesized and used as macro-chain transfer agent in VI polymerization. The temperature behavior of PVCL and DCs in aqueous media has been studied by static and dynamic light scattering. It has been shown that the phase separation temperature of both PVCLs and DCs depends on the length of the PVCL chain and the composition of aqueous solvent. The temperature range above the PVCL θ temperature and below the cloud point is characterized by the conformational rearrangements leading to the formation of mesoglobules. The study of catalytic activity of DCs in the hydrolysis reaction of p-nitrophenyl propionate has shown that their activity substantially increases in this transitional temperature region owing to the formation of highly developed hydrophilic–hydrophobic interfaces inside the mesoglobules.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
INTRODUCTION
Stimuli-responsive or smart amphiphilic copolymers are capable of changing their morphology in response to various external stimuli, such as temperature, quality of the solvent, pH, and ionic strength [1–5]. Among them, thermosensitive copolymers demonstrating a lower critical solution temperature (LCST) near physiological temperatures, which stratify from aqueous solutions upon heating and recover their solubility after cooling, have received considerable attention owing to potential practical application in targeted drug delivery systems or as nanocontainers for chemical and biochemical reactions [6–9].
An introduction of imidazole units into polymer chains turns them into efficient water-soluble catalysts of esterolysis via the formation of a catalyst–substrate complex mimicking the biological catalysts enzymes. Imidazole-containing thermosensitive polymers poly(N-isopropylacrylamide-co-N-vinylimidazole) (P(NIPA-VI)) and poly(N-vinylcaprolactam-co-N-vinylimidazole) (P(VCL-VI)) are known to demonstrate a switchable catalytic activity [2, 10]. At temperatures above the LCST, these copolymers form globular aggregates and exhibit enhanced catalytic activity in esterolysis of amphiphilic substrate in water–2-propanol mixture. The ability of thermosensitive polymers to form globular aggregates named mesoglobules was substantiated theoretically and shown experimentally in previous works [11–15]. An increase of the reaction rate due to concentration of the substrate at the hydrophobic–hydrophilic interface of aggregates is in agreement with the results of theoretical analysis by Vasilevskaya et al. [16] of the role of this interface in catalytic properties of miniemulsions.
In [17–19], the protein-like fractions were isolated from P(VCL-VI) and P(NIPA-VI) copolymers prepared by radical polymerization in aqueous mixtures above the phase transition temperatures. These fractions did not precipitate from aqueous solutions upon heating, but underwent cooperative coil-to-globule transition with formation of globular aggregates. This nonprecipitating P(VCL-VI) fraction in protein-like conformation demonstrated an enhanced catalytic activity in esterolysis of p-nitrophenyl propionate (NPP) [20].
The use of thermoresponsive molecularly homogeneous diblock copolymers as tunable catalysts may be more advantageous in terms of adjustable hydrophilic–hydrophobic balance and, as a consequence, the formation of well-defined supramolecular structures. A successful example of RAFT/MADIX synthesis of PNIPA-b-PVI copolymer demonstrating a transition to micellar aggregates above the LCST of PNIPA was published by Z. Ge et al. [21]. It was shown that, above the critical micellization temperature, DCs become more effective catalysts in the hydrolysis of amphiphilic p-nitrophenyl acetate. The formation of micellar aggregates revealing an enhanced catalytic activity was shown additionally in water–methanol mixtures of specific composition.
However, the use of PNIPA derivatives, in particular, PNIPA-b-PVI copolymers, in medicine, pharmacology, and other biotechnology industries is very limited because of their toxicity. In comparison with PNIPA, poly(N-vinylcaprolactam) (PVCL) has an undeniable advantage because it does not form toxic low molecular weight amines during hydrolysis, and therefore, the application of PVCL is preferable as compared to other thermosensitive polymers, including PNIPA [22]. In light of these considerations, the aim of the present study is to synthesize PVCL-b-PVI DCs which will be able to demonstrate enhanced catalytic activity in esterolysis reaction within a specific temperature range as a result of their coil-to-globule transformation.
DCs of VCL were studied not so intensively because reversible deactivation radical polymerizations of unconjugated monomers, including VCL, is still a challenge owing to the high activity of growth radicals. Recently, the successful controlled synthesis of PVCL was carried out by reversible addition–fragmentation chain transfer and macromolecular design via the interchange of xanthates (RAFT/MADIX) polymerization [23–27]. Wan et al. reported that the polymerization of VCL in bulk in the presence of 2‑diphenylthiocarbamoylsulfanyl-2-methyl-propionic acid, ((O-ethylxanthyl)methyl)benzene, and 1-(O-ethylxanthyl)ethyl)benzene (EXEB) as chain transfer agents (CTAs) proceeds in a controlled manner [23]. The most active CTA was EXEB. The successful redox-initiated RAFT/MADIX polymerization of VCL at room temperature with O-ethyl-S-(1-methoxycarbonyl)ethyldithiocarbonate as CTA in water–ethanol mixtures was reported in [24]. Liu et al. prepared the high molecular weight PVCL (Mn = 1.5 × 105) with narrow MWD (Mw/Mn = 1.1) via RAFT/MADIX polymerization in the presence of O-ethyl-S-(1-methoxycarbonyl)ethyl dithiocarbonate [25]. The same CTA proved to be effective for the synthesis of thermosensitive DCs of VCL with N-vinylpyrrolidone (NVP) [26] and of amphiphilic random copolymers of VCL and vinyl acetate in dioxane [27]. For the first time, the preparation of PVCL-b-PVI DCs by RAFT/MADIX polymerization using EXEB as a low molecular CTA was described in our short communication [28].
In the present study, we describe the results of further detailed investigation of PVCL-b-PVI synthesis in a wider range of PVCL molar mass and the main features of RAFT/MADIX polymerization of each block, as well as the results of detailed study of their thermoresponsive behavior. The temperature-responsive behavior of prepared DCs is studied by static and dynamic light scattering (SLS-DLS), and unusual structural transformations resulting in mesoglobular structures below the phase separation temperature are demonstrated. The catalytic activity of DCs in the hydrolysis of amphiphilic NPP is studied over a wide temperature range, and its peculiarities are discussed in light of structural transformations of DCs. The studied thermoresponsive, nontoxic, biocompatible water-soluble DCs may be of interest for biomedical applications such as targeted drug delivery and catalysis.
EXPERIMENTAL
Materials
N-Vinylcaprolactam (VCL, Aldrich, 98%) and N-vinylimidazole (VI, Aldrich, ≥99%) were distilled under reduced pressure before use. 2,2'-Azobis(isobutyronitrile) (AIBN, Aldrich, >99%) was purified by recrystallization twice from methanol. EXEB was synthesized as described in [29]. 1Н NMR (δH, ppm, CDCl3) for EXEB: 1.42 (t, 3H, CH3), 1.77 (d, 3H, CH3), 4.66 (q, 2H, CH2), 4.96 (q, 1H, CHPh), 7.37 (m, 5H, Ph). 13С NMR (δC, ppm, CDCl3) for EXEB: 213.36, 141.80, 128.63, 127.56, 127.53, 69.77, 49.26, 21.77, 13.78 (Figs. S1 and S2).
Water was used from a Millipore Milli-Q water purification system. The solvents were cleaned in accordance with generally accepted methods. Other chemicals were purchased from Aldrich and used as received.
Synthesis of PVCL-CTAs
PVCL-CTAs of different molar masses were synthesized by RAFT/MADIX polymerization of VCL in bulk in the presence of EXEB ([EXEB] = 5.6 × 10‒2 mol/L) with addition of a radical initiator ([AIBN] = 1.0 × 10–2 mol/L) for 17.5–24.5 h. All polymerizations were conducted in Schlenk tubes. Before polymerization, the reaction mixture was deaerated by multiple freezing and thawing under vacuum, and then the Schlenk tubes were immersed in a thermostatic oil bath at 60°C. After the desired time, the tubes were cooled with liquid nitrogen, and the reaction mixtures were diluted with tetrahydrofuran (THF), followed by dropping into a large amount of diethyl ether and collected by centrifugation (11 000 rpm, 10 min). The purification of PVCLs was made by repeating dissolution in THF and precipitation into ether. Finally, the polymers were dried under vacuum at 40°C up to constant weight. The obtained PVCL-CTAs were stored in a dark place at 2–4°C in order to save their activity [26]. The conditions of VCL polymerization and molecular weight characteristics of the resulting polymers are shown in Table 1.
Synthesis of PVI-Based Macromolecular Xanthate Agents—PVI-CTAs
PVI-CTA was prepared by RAFT/MADIX polymerization of VI in mass in the presence of AIBN ([AIBN] = 1.1 × 10–2 mol/L) and EXEB ([EXEB] = 3.0–3.1 × 10–2 mol/L) at 60°C for 20 h. Polymerization was carried out according the method similar to that for PVCL-CTAs. After polymerization, the reaction mixture was cooled with liquid nitrogen and added dropwise to the mixture of hexane and diethyl ether. The polymer was isolated by centrifugation (11 000 rpm, 10 min). The conditions of synthesis of PVI-CTA and its molecular weight characteristics are shown in Table 1.
Synthesis of PVCL-b-PVI Copolymers
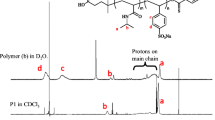
PVCL-b-PVI copolymers were prepared by RAFT/MADIX polymerization of VI in the presence of PVCL-CTAs as described in detail in [30]. The polymerization was conducted in DMF, THF, water and THF/water (50/50 mol %) with an initial monomer concentration of 1.2–3.2 mol/L at 60°C with AIBN as the initiator. The products were purified by dialysis through a Spectrapor membrane of 6–8 kDa (Spectrum Medical Industries, Inc., USA) against deionized water for 7 days and isolated by lyophilization. PVCL-b-PVI copolymers with different lengths of PVCL blocks and contents of VI units were synthesized (Tables 2 and 3). The composition of PVCL-b-PVI copolymers was estimated by 1H NMR (Fig. 1a). It should be noted that polymerization of VI in DMF at 60°C in the presence of EXEB and AIBN did not occur at [VI] = 0.44 and 3.2 mol/L, [AIBN] = 2.2 and 2.5 × 10–3 mol/L, and [EXEB] = 2.0 and 3.0 × 10‒2 mol/L; i.e., the formation of a homopolymer by the RAFT/MADIX polymerization under the conditions of synthesis of DC was unlikely.

1Н NMR spectra of (a) VCL62VI10, (b) PVCL62, and (c) PVI35 prepared by RAFT/MADIX polymerization D2О. Color figures can be viewed in the electronic version.
Characterization
The molecular weight characteristics (Mn, Mw, and Mw/Mn) of (co)polymers were determined by two methods: GPC and 1H NMR.
GPC analysis was carried out using the Agilent 1200 Series Chromatography system equipped with an isocratic pump, refractive index detector, and PLmixC column. As the eluent, 0.03 M LiCl in N-methylpyrrolidone filtered through 0.45 μm Fluoropore membrane and degassed was used at flow rate 0.5 mL/min and 50°C. A series of monodispersed polystyrene (PS) standards was used for calibration. The chosen eluent dissolves both homopolymers of VCL and VI and their copolymers and provides the size exclusion chromatography mode for all the polymers under study. The overestimation of calculated polydispersities of PS standards due to the band broadening in the chromatography system was 15%.
1H NMR measurements were recorded using a Bruker Avance 600 spectrometer operating at 600.22 MHz (1H). D2O and CDCl3 were used as solvent. The accuracy of the determination of chemical shifts was not worse than 0.001 ppm.
The value of Mn (NMR) for PVCL was determined by the method of 1H NMR spectroscopy, under the assumption that all polymer chains contain the terminal phenyl group of EXEB, according to the equation
where HPVCL and HEXEB are the integral intensities of one proton of –CH groups of the main polymer chain at 4.0–4.5 ppm and five protons of C6H5 group of EXEB at 6.9–7.6 ppm, and МVCL is the molecular weight of VCL. The 1H NMR spectrum of PVCL62 is shown in Fig. 1b.
The theoretical \(M_{{\text{n}}}^{{{\text{th}}}}\) values were calculated under the assumption that one molecule of EXEB leads to controlled growth of one polymer chain according to the equation
where MEXEB and MVCL are the molar masses of EXEB and VCL, [EXEB]0 and [VCL]0 are the initial concentrations of EXEB and VCL, and \({v}\) is the monomer conversion.
The synthesized DCs were labeled as VCLnVIm, where “VCL” and “VI” denote VCL and VI units, respectively; the degree of polymerization of PVCL block n = Mn(NMR)/139, where 139 is the molecular weight of VCL and Mn(NMR) is the number-average molecular weight of PVCL determined by 1H NMR; the degree of polymerization of PVI block m was calculated taking into account the content of VI units in copolymer determined by 1H NMR.
The polymerization rate of VCL is calculated by the following equation:
where t is the polymerization time, \({v}\) is the conversion of VCL, and \(M_{{\text{n}}}^{{{\text{th}}}}\) is the theoretical Mn at \({v}\) = 100%.
Static and Dynamic Light Scattering
Static and dynamic light scattering measurements (SLS-DLS) were performed using a PhotoCor Complex spectrometer (PhotoCor Instruments, Russia) equipped with a He-Ne laser (λ = 633 nm, 10 mW) as the light source and a pseudo cross-correlation system of photon counting, which made it possible to avoid distortions of the correlogram as a result of afterpulsing of the photomultiplier at small lag times and to measure Rh values down to approximately 1 nm. The real-time correlator was employed in the logarithmic configuration. Measurements were performed in dilute solutions within the range of scattering angles of 30°–140°. Distributions over decay time τ and hydrodynamic radius were obtained by means of a nonlinear regularized inverse Laplace transformation method (CONTIN). If the distribution function was described by a sum of two separated components (fast and slow modes), apparent self-diffusion coefficients D were determined for each mode using the relation D = 1/τq2, where τ is the relaxation time, q = (4πn/λ)sin(θ/2) is the scattering wave vector, n is the solvent refractive index, and θ is the scattering angle. The corresponding hydrodynamic radii Rh were calculated from Stokes–Einstein equation Rh = kT/6Dπη, where k is the Boltzmann constant and η is the solvent viscosity. The radius of gyration of aggregates Rg was calculated according to the Zimm or Guinier [31] relations from angular dependences of scattered light intensity normalized to the relative contribution of the slow mode. Apparent molar masses Mapp were determined according to the same relations using the total intensities, total concentrations and dn/dc values for PVCL at different temperatures (0.181 at T = 25°С and 0.232 at 37°С) [15].
Cloud points Tcp of thermoresponsive polymers were determined from the temperature dependences of light scattering as the onset of a sharp increase in scattered light intensity I. These curves were measured under the scattering angle of 90° with a temperature interval of 1°С at a slow heating rate (0.3 K/min); the measurements were carried out after 10 min for equilibration of temperature in the measuring cell to reach a constant LS intensity, and therefore, the values of Tcp may be considered to be close to the phase separation temperatures or points of binodal.
Hydrolytic Decomposition of NPP
The hydrolytic decomposition of NPP in the presence of PVCL-b-PVI copolymers was studied by measuring the optical density of the colored product p‑nitrophenol (408 nm) in the interval 25‒45°С depending on time. The kinetic measurements were carried out at NPP concentrations of 0.2 mM in 0.05 М tris(hydroxymethyl)aminomethane (Tris)/HCl buffer solution at рН 7.4. The copolymer concentration was varied from 0.29 to 1.38 mM relative to the content of imidazole groups.
The calculation of reaction rate was performed using Eq. (4) [22, 25]:
where dD/dt is the initial slope of optical density variation with time, ε is the extinction coefficient of p‑nitrophenol in deprotonated form (ε = 18 500 L/(mol cm)), and F is the fraction of p-nitrophenol in deprotonated form at 7.4 (F = 0.6, as was found by spectrophotometry).
RESULTS AND DISCUSSION
RAFT/MADIX Polymerization of VCL and VI Mediated by EXEB
The PVCL-b-PVI copolymers were prepared by two-stage RAFT/MADIX copolymerization of VCL and VI. At the first stage, the PVCL-CTAs were synthesized by RAFT/MADIX polymerization of VCL in the presence of AIBN as initiator and EXEB as a low-molecular CTA. EXEB was chosen for two reasons. Firstly, it was previously shown that, in the presence of EXEB, polymerization of VCL initiated by AIBN at the molar ratio [EXEB]/[AIBN]= 5/1 proceeds in a controlled manner with the formation of PVCL having a rather narrow molecular weight distribution (MWD), Mw/Mn = 1.24 [23]. Secondly, PNIPA-CTA with terminal 1-(O-ethylxanthyl)ethyl group is known as an effective macro-CTA of chain transfer in RAFT/MADIX polymerization of VI [21].
RAFT/MADIX polymerization of VCL (Scheme 1) was performed in bulk at 60°C. The AIBN concentration was maintained constant and equal to 1 × 10–2 mol/L. The EXEB concentration was varied from 1.8 to 3.1 × 10–2 mol/L in order to synthesize the PVCL-CTAs with various molar masses. The variation of the molar mass (chain length) of the PVCL block in DCs can allow, on one hand, controlling the phase transition temperature of the DCs and, on the other hand, finding the most efficient PVCL-CTA in RAFT/MADIX polymerization of VI.

Scheme 1.
Figure 1b shows the 1H NMR spectrum of PVCL62. The signals with chemical shifts at 4.46–3.96 ppm (1), 2.77–3.46 ppm (4), 2.63–2.02 ppm (3), and 1.90–1.17 ppm (2, 5) are attributed to one proton of –CH group, two protons of –COCH2 group, two protons of –NCH2 group, six protons of –NCCH2CH2CH2 group of caprolactam ring, and two protons of –CH2 group of the main polymer chain. The signals of five protons of EXEB phenyl group appear at 7.31–6.82 (11). Thus, 1H NMR spectroscopy demonstrated an incorporation of EXEB fragments in the PVCL chains. 1H NMR was employed to determine the Mn of PVCLs using Eq. (1) (Table 1). As can be seen, the Mn(NMR) of PVCLs are close to the \(M_{{\text{n}}}^{{{\text{th}}}}\). These values are used in what follows for description of block length in DCs.
Additionally, the molecular weights (Mw and Mn) and polydispersity coefficients (Mw/Mn) of prepared PVCLs were determined by GPC and are shown in Table 1. The MWD curves of PVCL-CTAs synthesized at various molar ratios [EXEB]/[AIBN] = 1.8, 2.2, and 3.1 are presented in Fig. 2. As shown in Table 1 and Fig. 2, all PVCLs are characterized by rather low values of polydispersity coefficients (Mw/Mn = 1.52–1.66, GPC) as compared to those for PVCL prepared by free-radical polymerization (Table S1 in supplementary materials) in the absence of EXEB, but Mn(GPC) are lower than the theoretical values. The underestimation of molar masses determined by GPC was additionally confirmed by the comparison of Mw(GPC) with Mw values determined by SLS for several PVCL samples prepared by radical polymerization (Table S1). This tendency to underestimation of Mn(GPC) determined using PS standards was repeatedly specified earlier for PVCL and its block copolymers [23, 32, 33].

MWD curves of (1) PVCL62, (2) PVCL209, and (3) PVCL255 synthesized at various EXEB concentrations.
As can be seen from Fig. S3, the dependence of experimental Mn(NMR) of PVCLs on the ratio [VCL]/[EXEB] at approximately the same conversion v ≈ 50% is linear (correlation coefficient 0.95), as is predicted theoretically in accordance with Eq. (2). Thus, all these observations allow suggesting that the polymerization of VCL with EXEB as CTA proceeds in a living manner.
To choose the appropriate type of macro-CTA for preparation of DCs, some polymerizations of VCL were carried out using a PVI sample prepared by RAFT/MADIX polymerization, as shown in Table 1. We found that the polymerization of VCL initiated by AIBN was inhibited by PVI-CTA. The inability of the PVI-CTA to reinitiate the polymerization of VCL is probably due to the high stability of PVI intermediates to fragmentation [34]. This supposition is in agreement with the fact that, to the best of our knowledge, PVI was never used in previous investigations as a macro-CTA for the preparation of DCs with various comonomers such as NIPA [21] and styrene [35]. Therefore, in this work, we used PVCL samples as macro-CTAs for synthesis of PVCL-b-PVI copolymers.
Synthesis of PVCL-b-PVI Copolymers by RAFT/MADIX Polymerization of VI
The PVCL-b-PVI copolymers were synthesized by RAFT/MADIX polymerization of VI and VI/VCL mixture in the presence of AIBN and PVCL-CTAs of various molar masses in DMF, THF, and water and in THF/water mixtures according to Scheme 2. Tables 2 and 3 show the reaction conditions and the characteristics of the resulting products.

Scheme 2.
The molecular characteristics of DCs prepared in DMF at 60°С presented in Table 2 and the MWD curves (Fig. 3) allow us to conclude that, under the chosen conditions, polymerization of VI with PVCL62 and PVCL209 as CTAs proceeds as a pseudo-living process. Apparent MWD curves of VI polymerization products are unimodal and symmetric, but their maxima are only slightly shifted to higher molar masses relative to those of macro-CTAs. Since GPC curves are based on the hydrodynamic volume of macromolecules, they are affected by chain conformations and solvent quality. If the DC has a more compact conformation as compared with homopolymers, we can obtain underestimated apparent MM values. As is seen from Tables 2 and 3, Mn(GPC) of DCs are substantially lower than Mn(NMR) determined as described below. The possible reason of compactization of DCs under study will be discussed in the next section devoted to temperature-induced self-assembly.

The comparison of MWD curves for PVCLs used as macro-CTA agents and PVCL-b-PVI copolymers prepared with them: (a) (1) PVCL62 and (2) (VCL62VI20)D; (b) (1) PVCL209 and (2) (VCL209VI40)D.
The 1H NMR spectrum of VCL62VI10 is presented in Fig. 1a as an example. The comparison with the 1H NMR spectra of PVCL (Fig. 1b) and PVI (Fig. 1c) shows that the PVCL and PVI blocks can be separated by the signals appearing at 4.46–3.96 ppm (1) for the initial PVCL block and at 7.3–6.5 ppm (8, 9, 10) for the added PVI block. Thus, the integrals of imidazole ring protons of VI units and one proton of –CH group of PVCL were used to estimate the DC composition in mol % of each monomer:
where HVI and HVCL are the integral intensities of the signals characterizing three protons of imidazole group and one –CH– proton of PVCL backbone. These data were used for calculation of Mn(NMR) of PVI blocks and DCs (Tables 2, 3), as well as the number average degrees of polymerization of blocks VCLnVIm used for identification of DCs in what follows.
A comparison of the samples VCL62VI10 and VCL62VI20 shows that doubling the monomer concentration in the initial reaction mixture while maintaining other reaction conditions approximately the same results in doubling the PVI chain length. Under similar conditions, the RAFT/MADIX соpolymerization of VI and VCL at [VCL] : [VI] = 68 : 32 mol % in the reaction mixture also proceeds with the formation of VCL62-P(VCL-co-VI) copolymer characterized by a unimodal MWD curve. However, if the relative amount of AIBN in VI polymerization was increased to ≥ 3 × 10–3 mol/L, bimodal MWD curves were observed (Fig. S4) containing a relatively high molecular weight peak, which, probably, was related to PVI produced by radical polymerization.
The VI polymerization in the presence of PVCL-CTA proceeds with a significant induction period with a duration not less than 16 h, as was often observed in RAFT/MADIX processes [23]. Indeed, we found that VI was hardly consumed in the reaction for 16 h. The specific feature of DC synthesis under study is the limited achievable monomer conversion. The maximum VI conversion achieved in 24 h with the use of PVCL62 as macro-CTA was 30–31% ((VCL62VI10)D and (VCL62VI20)D). Moreover, the achievable VI conversion decreased with the increase in macro-CTA chain length, so that, when PVCL209 was used, 23% conversion was achieved in 24 h (copolymer (VCL209VI43)D). Increasing the duration of VI polymerization from 24 to 46 h does not affect the conversion of the monomer and, consequently, the composition of the resulting DC, as is seen from the comparison of copolymers (VCL209VI43)D and (VCL209VI40)D. The further increase in Mn(NMR) to 35.5 × 103 (PVCL255) reduced VI conversion to 1% at a maximum. The polymerization of VI in the presence of PVCL255 was carried out in the [AIBN] range from 1.5 to 2.0 × 10‒2 mol/L for 3–24 h. In all cases, the content of VI units in reaction products did not exceed 1 mol %. Table 2 shows the conditions for preparation and characteristics of copolymer (VCL255VI3)D as an example.
For comparison, we carried out a series of VI poly-merizations in slightly polar THF (ε = 7.4), in aqueous media (ε = 81.0), and in THF : water mixtures (approximately 50 : 50 mol %) using C209 as macro-CTA. The use of aqueous media for the preparation of polymers instead of volatile organic solvents is considered as preferable from the point of view of environmental protection. It was found, however, that the nature of the reaction media hardly affected the composition of PVCL-b-PVI copolymers (Table 3). The content of VI units remained equal to 15 ± 2% in any product.
One of the possible reasons for the observed retardation in the rate of VI polymerization at the increased molar mass of PVCL-CTAs, as well as the found limitation in the conversion of PVI monomer in any solvent used, may be an associative behavior of PVCL-CTAs and PVCL-b-PVI copolymers in polymerization media. To get an idea of the possible structural transformations in the reaction media, a series of model experiments were performed using the DLS method. The dilute solutions (2 mg/mL) of PVCL and copolymers (VCL209VI43)T and (VCL62VI20)D were prepared in the solvents used for VI polymerization and were analyzed by DLS. In polar organic solvent DMF, we observed a molecularly dispersed solution of initial PVCL-CTAs and partially aggregated solutions of PVCL-b-PVI copolymers at both 25 and 55°C (Fig. S5). DCs were insoluble in both THF and THF : H2O (50 : 50 mol %), which are not solvents for PVI at room and at elevated temperatures. PVCL-CTA, in contrast, is molecularly soluble in these media. In pure water, both PVCL and PVCL-b-PVI copolymers were partially aggregated in dilute solutions at 25°C and subjected to phase separation above 40°C, as will be shown in the next section. Thus, all the solvents used lead to specific inhomogeneity at the transition from the initial PVCL to DC, which favors the assumption about the microheterogeneity of the reaction mass.
It is clear that the composition of model solutions in DLS experiments was not identical to the polymerization media: the concentration of polymers during RAFT/MADIX polymerization of VI was approximately two orders of magnitude higher; there was additionally some amount of monomer VI. Therefore, the analysis of the structure of the reaction mass and its correlation with the limitations of the polymerization process should be a subject for a special study.
Temperature-Induced Self-Assembly in Dilute Aqueous Solutions of PVCL and PVCL-b-PVI Copolymers. Light Scattering Study
In this section, we will consider a temperature-induced self-assembly of PVCL and DCs synthesized above and the nature of their structural rearrangement phenomena. The temperature-responsive behavior of PVCL and DCs was studied in aqueous and water-salt buffer solutions using the SLS-DLS method.
First of all, we consider the influence of the length of PVCL and PVI blocks and the solvent composition on the phase transition temperature, which is estimated as cloud points. These values for PVCL and DCs prepared on them are shown in Table 4; the examples of intensity curves are shown in Figs. 4a and 5a. The Tcp values of both PVCLs and DCs decrease with increasing solution concentration, which is characteristic of the left branch of the binodal. As is seen from Table 4, the molecular weight dependence of Tcp for PVCL demonstrates a tendency to a decrease upon an increase in Mn in accordance with type I miscibility behavior. A substantial decrease in Tcp is observed when going from PVCL29 to PVCL62; the further increase in chain length from PVCL62 to PVCL209 only slightly changes the value of Tcp. Earlier, it was repeatedly established that end groups originating from CTA may exert an influence on the thermo-responsive behavior of short-chain polymers with LCST [36–39]. So, highly hydrophobic triphenylmethyl, octadecyl, or benzyl end groups were shown to lead to an inverse molecular weight dependence of Tcp (its increase upon increase in Mn) [37, 40, 41]. We can see that the hydrophobicity of the end groups originating from EXEB used in our work is not enough to change the normal direction of Tcp dependence, but it may make this dependence weaker by lowering Tcp of the shortest chains.

Temperature dependences of (a) scattered light intensity and (b) the hydrodynamic radius of the scattering particles (aggregates) in solutions of (1) PVCL62, (2) (VCL62VI10)D, and (3) VCL62P(VCL-co-VI) in 0.05 M Tris/HCl, pH 7.4, concentration of 1 mg/mL. The inset shows the change in scattered light intensity on a large scale.

Temperature dependences of (a) scattered light intensity and (b) the hydrodynamic radius of the scattering particles in solutions of (1) (VCL62VI10)D and (2) (VCL62VI20)D in 0.05 М KH2PO4/Na2HPO4, рН 7.0, concentration of 0.25 mg/mL. The inset shows the change in scattered light intensity on a large scale.
As is seen from Table 4 and Fig. 4a, the values of Tcp of DCs based on PVCL62 are 1–2 degrees higher as compared with PVCL, from which they were prepared. For the longer PVCL209 block, no difference in Tcp between PVCL and its DC was found. No changes in Tcp with an increase in the length of the PVI block were observed in the pair (VCL62-VI20)D and (VCL62-VI10)D. The comparison of Tcp values determined for some DCs in various solvents shows that this temperature depends on the solvent composition, decreasing in the sequence water ≥ Tris/HCl ≥ KH2PO4/Na2HPO4. This difference is consistent with the published data on the destabilizing effect of salts on PVCL-hydrate complexes [42], where it was shown that the \({{{\text{H}}}_{{\text{2}}}}{\text{PO}}_{4}^{ - }\) anion is the strongest destabilizing agent.
Let us consider the temperature-induced self-assembly in solutions of PVCL and DCs in more detail using the SLS-DLS approach. In the insets in Figs. 4a and 5a, one can see the two-step changes in scattering intensity that are a characteristic feature of all the studied PVCL and DCs (additional examples are given in Figs S6). The first stage of limited growth of LS intensity below Tcp and above the θ temperature of PVCL is accompanied by the increase in Rh of scattering particles (Figs. 4b, 5b). At concentration c ≥ 1 mg/mL (Fig. 4b), Rh values of aggregates were used at all temperatures; at lower concentration, 0.25 mg/mL (Fig. 5b), no aggregation was detected before the first step of intensity growth, and Rh of macromolecules are used on the graph in this temperature region. At temperatures Tcp, a sharp growth of Rh values was observed, which was replaced by a decrease in this value (Figs. 4b, 5b) simultaneously with the further growth in scattering intensity. This decrease may be attributed to several reasons: to compaction of aggregating particles and, additionally, to a phenomenon of multiple light scattering in opaque systems and/or gradual deposition of heavier particles.
The distributions of scattered intensity over particle size shown in Figs. 6 and 7 illustrate the structural transformations in solutions at different temperatures: (1) at 25°C, (2) at the temperature corresponding to the first step of structural transformation below Tcp, and (3) above Tcp. The size distributions at 25°C are bimodal and consist of two modes corresponding to individual macromolecules and their aggregates, whose Rh values differed by approximately a factor of 10–30. The values of Rh of different species and the ratios of scattered intensity from macromolecules and aggregates Amol/Aagg are shown in Table 5. (Size distributions for PVCL29 at 25°C could not be measured because of overly low scattering intensity in solutions of this short-chain polymer). One should note that, given the large difference in sizes of macromolecules and aggregates, the mass fraction of aggregates wagg, whose contribution to scattered intensity is high, may be very small because the intensity of scattered light is related to the particle size by the relation I ~ w × Ra, where 1 < a ≤ 3 depending on the particle structure.

Size distributions of the scattering particles of PVCL62 in 0.05 M Tris/HCl, pH 7.4, depending on the temperature: (1) at 25°C; (2) in the region of globular aggregates at 36°С; near the phase separation temperature of (3) 40 and (4) 44°C. c = 1 mg/mL.

Size distributions of the scattering particles of DC (VCL62VI10)D in 0.05 M Tris/HCl, pH 7.4, depending on the temperature: (1) at 25°С; (2) in the region of globular aggregates at 36°С; near the phase separation temperature of (3) 40 and (4) 44°C. c = 1 mg/mL.
Note that the tendency toward partial aggregation in dilute solutions of PVCL, PNIPA, and other polymers with LCST and their block-copolymers at room temperature is not a unique feature of our polymers, and it was often observed earlier [39, 43–45]. For rather short-chain polymers, this was attributed to the impact of hydrophobic end groups [39, 43]. If we compare the Amol/Aagg ratios for PVCLs of different chain length (PVCL62 and PVCL209, Table 5), we can see that, for the longer PVCL chain, the impact of aggregates on light scattering is smaller. This allows us to suppose an influence of ethylbenzene groups originating from EXEB on the hydrophobic–hydrophilic balance of macromolecules. Comparing the aggregative behavior of PVCL and DCs at room temperature, one can see that the values of \(R_{{\text{h}}}^{{{\text{mol}}}}\) of DCs are somewhat smaller than those values of corresponding PVCLs (Table 5, PVCL62 and (VCL62VI10)D, PVCL209 and (VCL209VI43)T in Tris buffer). This peculiarity may presumably be attributed to the thermodynamic incompatibility between the two water-soluble blocks, resulting in more compact conformation of block copolymer. The similar interpretation was proposed in [44, 46, 47], where the behavior of solutions of double hydrophilic DCs PEO-b-PVCL was studied.
At the first transition step below the cloud point, the contribution of macromolecules to scattering sharply decreases; \(R_{{\text{h}}}^{{{\text{mol}}}}\) values decrease also as compared with those values at room temperature (Table 5, Figs. 6 and 7). At the same time, we observe an increase in \(R_{{\text{h}}}^{{{\text{agg}}}}\). All this indicates a contraction of partially dehydrated PVCL chains in the vicinity of LCST and an additional aggregation of collapsed chains and aggregates until the balance of the attractive interactions and steric stabilization provided by hydrophilic segments present on the outer surface of particles is reached. Similar polymolecular objects are usually named mesoglobules, and they may coexist in dilute solutions with collapsed molecular globules [11].
To obtain more detailed information about the structure of scattering particles and their conformational transformations, the dimensions of the aggregates Rg and Rh extrapolated to zero angle and asymmetry factor Rg/Rh for several DCs were determined at selected temperatures below the first transition temperature (25°C) and at the first step of scattering intensity growth below Tcp (Table 5). Since the size of rather low molecular weight macromolecules is too small relative to the light wavelength, it is impossible to estimate the conformations of macromolecules and their changes using Rg/Rh values. Therefore, the analysis of the changes taking place in the system is mostly based on the data on various dimensional characteristics of supramolecular particles. Unlike the primary aggregates at 25°C, supramolecular particles in the transition temperature region are characterized by an increased size and a more compact structure with an asymmetry factor Rg/Rh ≤ 1. Additionally, no angular dependence of apparent Rh values was found, which is characteristic only of spherical particles in the case of large particle dimensions (Rg ≈ q–1). The data obtained indicate a conformational rearrangement leading to the formation of more compact mesoglobules that are close in characteristics to a hard sphere. This process is accompanied by additional aggregation, which manifests itself in particle size growth, in an increase in apparent molar mass values (\(M_{{\text{w}}}^{{{\text{app}}}}\), Table 5), and in a substantial decrease in the contribution of macromolecules to light scattering. This aggregation and structural rearrangement process is reversible; i.e., when the solution is cooled, its characteristics approach the initial ones, but do not coincide completely. This tendency is due to the well-known influence of the kinetic factor on the formation and disintegration of mesoglobules [11].
The structural characteristics of DC mesoglobules in the transitional temperature region below Tcp determined in our study are in agreement with those reported earlier by Aseyev et al. [15]. The detailed structural characterization of mesoglobules of PNIPA, PVCL, and poly(vinyl methyl ether) was performed by the SLS-DLS method. It was shown that these three polymers form similar stable approximately spherical aggregates with asymmetry factor Rg/Rh about 0.77, which are characterized by a complex foamlike morphology and fractal dimension Df = 2.7.
The phenomenon of coil to globule transition in highly diluted solutions of high molecular weight PNIPA and PVCL near the θ point, which preceded the phase separation [48–52], and the phenomenon of temperature-induced formation of mesoglobules in highly diluted solutions of thermally sensitive polymers and block copolymers [15, 21, 33, 53–55] were repeatedly observed earlier and justified theoretically [11–13]. Namely, two-step assembly behavior of thermosensitive block copolymers based on PVCL and a second block composed of statistical copolymers with hydrophilic comonomer N-vinylpyrrolidone or N-methyl-N-vinylacetamide was observed by LS and turbidity methods in [53, 54]. The first transition temperature to micellar solution Tcp1 was close to Tcp of the PVCL block and appeared as a stepwise increase in scattering intensity and a transition from a partially aggregated solution with loose aggregates to micellar systems with more compact particles, and the second cloud point Tcp2 identified as a sharp loss of transmittance was close to Tcp of the copolymer from which DC was prepared. 1H NMR spectra confirmed a partial dehydration of the PVCL block above Tcp1.
The main distinction of our results from those in the cited works are as follows: we observed the two-step structural transition not only for DCs but also for PVCL solutions, and the first transition temperature to mesoglobular solution was in all cases below Tcp of the PVCL block, but close to the PVCL θ temperature.
The behavior of solutions, similar in its main features to that detected by us, was described in [44] for PEO44-b-PNIPA95 copolymer prepared by RAFT polymerization. At room temperature, diluted solutions of DC were partially aggregated. A gradual increase in the apparent Mw value began close to the PNIPA θ temperature, which was replaced by a sharp increase at Tcp (42°C). At this translational stage, the structural reorganization was accompanied with a sharp decrease in the size of aggregates and a gradual decrease in asymmetry factor Rg/Rh, which finally reached the values characteristic of spherical particles above Tcp. As a main factor that determined these features, the authors discussed the incompatibility between PNIPA and PEO blocks. This interpretation is in agreement with the results reported in [47], where the combination of fluorescence spectroscopy with pyrene as a probe, small-angle neutron scattering, and DLS was used for studying PNIPA-polyethylene glycol (PEG) DCs. The authors concluded that, even in the temperature region below the LCST of the PNIPA block, water starts to behave as a selective solvent for the PEG block and redistribution of hydrating water from the PNIPA to PEG blocks leads to a contraction of the PNIPA blocks and microphase separation. FTIR spectroscopy in combination with correlation analysis performed in [46] for PEO-b-PVCL copolymer led to a similar conclusion: the hydrophilic PEO shell plays the role of a water-absorbing sponge layer and captures expelled water from the PVCL core.
In some publications, a stepwise temperature-induced self-assembly of DCs was attributed to the influence of end groups. For example, in [43], C12-PEO-b-PNIPA copolymer with rather short blocks demonstrated two steps on LS intensity curves in contrast to DCs with a long PNIPA block because of the influence of C12 alkyl end group. This conclusion was confirmed by the cleavage of this group. From this point of view, the polymers studied in our work have not very high degrees of polymerization, and an impact of end groups on their specific structural features seems quite realistic. A unique example of complicated influence of end groups of thermosensitive α,ω-hydrophobically modified PNIPA was recently reported in [40] by high-sensitivity differential scanning calorimetry (HS-DSC). It was found that, in contrast to α,ω-di-n-octadecyl-PNIPA, solutions of α,ω-hydrophobically modified PNIPA with polycyclic terminal groups exhibited bimodal thermograms.
When considering the behavior of PVCL and its derivatives, it is useful to take into account that the thermoresponsive behavior of PVCL is more complicated as compared with PNIPA owing to its inability to self-associate with hydrogen bonding and also its reduced conformational mobility. The specific feature of this polymer is a wide temperature region of phase transition as compared with PNIPA, which is well known from differential scanning calorimetry (DSC) studies. In [56], HS-DSC revealed continuous changes in hydration structure of PVCL in the pre-transition region, which were interpreted as the noncooperative formation of a hydrophobic core of macromolecules. IR and two-dimensional correlation spectroscopy analysis performed in [57] led to the conclusion that PVCL mesoglobules would form a “spongelike” structure, which can further continuously expel water molecules upon increasing temperature. The peculiar two-step character of phase transition was clearly manifested by pressure perturbation calorimetry [58]. It was shown that the coefficient of thermal expansion underwent a sharp decrease at the onset of phase transition, then passed through a maximum, and decreased continuously with increasing temperature. These peculiarities of PVCL should influence the properties of its derivatives, including DCs.
In summary, the phenomena of temperature induced coil-to-globule transition and phase separation in PVCL and its DCs observed in our work are most likely influenced by all the factors of the molecular structure, including end groups and the nature and the length of different blocks, and also by the external environment. So, these phenomena are of interest for further research using other techniques like HS-DSC and pressure perturbation calorimetry.
Catalytic Properties of PVCL-b-PVI Copolymers
As was shown in many previous investigations, imidazole-containing polymers are able to catalyze ester hydrolysis reactions. The idea of controlling the catalytic activity of imidazole-bearing copolymers is based on the ability of thermosensitive polymers and copolymers to change their conformations in response to temperature change. Earlier [28], we reported that the rate of NPP hydrolysis (Scheme 3) in aqueous medium in the presence of VCL29VI13 and VCL29VI29 increases with temperature and exhibits the deviation from Arrhenius plot near the phase separation temperature. Similar deviations were observed for PNIPA-b-PVI copolymers in [21].

Scheme 3.
The thermoinduced structural and conformational rearrangements of synthesized DCs detected by SLS-DLS allowed us to suggest an increase in their catalytic activity in reactions involving amphiphilic organic substances in the temperature region preceding the phase separation (mesoglobule region).
The reaction rate of NPP hydrolysis was studied using various concentrations of copolymers (VCL62VI20)D and (VCL62VI10)D in Tris buffer at pH 7.4 and compared with the reaction rate in the absence of catalyst. The rate plots in Arrhenius coordinates are shown in Fig. 8 and Fig. S7. The plots indicate a sharp increase in reaction rate at temperatures about 30–35°C coinciding with the region of conformational rearrangements of DCs found by SLS-DLS. The results of linear fittings of these plots with correlation coefficients from 0.54 to 0.89 confirm a nonlinear type of this dependence, whereas the blank experiment without any catalyst shows a high correlation of the linear plot (correlation coefficient of 0.993).

NPP hydrolysis rate vs. reciprocal temperature (1) in the absence and in the presence of (VCL62VI20)D copolymer at concentrations of VI units of (2) 0.29, (3) 057, and (4) 1.38 mM in 0.05 Tris/HCl, рН 7.4.
The sharp increase in catalytic activity of DCs observed in our work in the temperature range predicting the phase separation temperature is rather similar to the features described earlier in [10], where catalytic properties of P(NIPA-VI) and P(VCL-VI) random copolymers in the p-nitrophenyl acetate (NPA) hydrolysis reaction were studied in a water/2-propanol (9/1 by volume) medium at temperatures close to LCST of copolymers. A sharp increase in the reaction rate and a sharp deviation from the Arrhenius plot were observed in the temperature region which was characterized by the formation of aggregates with Rh of about 100–200 nm. The statistical analysis of the rate curves in accordance with the linear model showed rather low correlation (correlation coefficients of 0.94–0.95), whereas for both 1-methylimidazole and PVI, the correlation coefficients were close to 0.99. Moreover, in the cited work, the analysis of reaction rate versus substrate concentration was performed, and the results were found to be successfully described by Michaelis–Menten type kinetics. This type of kinetics is characteristic of enzymatic catalysis, which proceeds through the formation of a catalyst–substrate complex.
The substrate concentration dependences are not tested in our study, but a considerable increase in catalytic activity together with a sharp deviation from the Arrhenius plot in the temperature region of mesoglobules may indicate a decreased activation energy due to some specific coordination of surface-active substrate molecules at core–shell interfaces of unimolecular or supramolecular nanoparticles of DCs. An additional factor of enhanced catalytic activity is concentration of the surface-active substrate near the highly developed core–shell boundary inside the DC particles.
CONCLUSIONS
A series of well-defined thermoresponsive PVCL-b-PVI diblock copolymers for the first time were synthesized by two-stage RAFT/MADIX polymerization. The PVCL blocks were first prepared by RAFT/MADIX polymerization of VCL in bulk using EXEB as CTA and AIBN as initiator, and further were used as macro-CTAs in preparation of PVCL-b-PVI copolymers in DMF, THF, water, and THF–water mixtures. The maximum achievable relative content of VI units in PVCL-b-PVI copolymers depends on the molecular weight of PVCL-CTA; namely, the highest fraction of VI units of about 25 mol % was achieved with PVCL62, but VI was hardly polymerized using as a macro-CTA the longest PVCL255. It is assumed that these features are caused by associative behavior of PVCL-b-PVI copolymers in the reaction medium.
In aqueous media, PVCL and diblock copolymers exhibit conformational rearrangements with the formation of micelle-like mesoglobules in the temperature range preceding the phase separation temperature. This phenomenon is explained by the change in the balance of hydrophobic interactions and hydration near the PVCL θ conditions, which contributes to the collapse and aggregation of PVCL blocks. This process is likely influenced by the factors of the molecular structure including end groups and the length of different blocks. The pronounced increase in the catalytic activity of copolymers toward the hydrolysis of amphiphilic NPP is observed in this temperature range, which is induced by the structural rearrangement of the block copolymer and the appearance of highly developed hydrophilic–hydrophobic interfaces inside the mesoglobules. Thus, the prepared thermoresponsive biocompatible PVCL-b-PVI DCs with temperature-controlled catalytic properties may be considered as prospective for biomedical applications such as targeted drug delivery and biocatalysis.
Change history
28 September 2021
An Erratum to this paper has been published: https://doi.org/10.1134/S0965545X21340010
REFERENCES
P. G. Khalatur and A. R. Khokhlov, Adv. Polym. Sci. 195, 1 (2006).
I. M. Okhapkin, E. E. Makhaeva, and A. R. Khokhlov, Adv. Polym. Sci. 195, 177 (2006).
C. M. Papadakis, P. Müller-Buschbaum, and A. Laschewsky, Langmuir 35, 9660 (2019).
Y. Mai and A. Eisenberg, Chem. Soc. Rev. 41, 5969 (2012).
I. Dimitrov, B. Trzebicka, A. H. E. Müller, A. Dworak, and C. B. Tsvetanov, Prog. Polym. Sci. 32, 1275 (2007).
H. Cabral, K. Miyata, K. Osada, and K. Kataoka, Chem. Rev. 118, 6844 (2018).
A. Kyritsis, A. Laschewsky. and C. M. Papadakis, in Thermodynamics and Biophysics of Biomedical Nanosystems, Ed. by C. Demetzos and N. Pippa (Springer, Singapore, 2019), pp. 397–444.
V. Kozlovskaya and E. Kharlampieva, ACS Appl. Polym. Mater. 2, 26 (2020).
N. A. Cortez-Lemus and A. Licea-Claverie, Prog. Polym. Sci. 53, 1 (2016).
I. M. Okhapkin, L. M. Bronstein, E. E. Makhaeva, V. G. Matveeva, E. M. Sulman, M. G. Sulman, and A. R. Khokhlov, Macromolecules 37, 7879 (2004).
K. A. Dawson, A. V. Gorelov, E. G. Timoshenko, Y. A. Kuznetsov, and A. Du Chesne, Phys. A (Amsterdam, Neth.) 244, 68 (1997).
E. G. Timoshenko and Y. A. Kuznetsov, J. Chem. Phys. 112, 8163 (2000).
E. G. Timoshenko, R. Basovsky, and Y. A. Kuznetsov, Colloids Surf., A 190, 129 (2001).
M. H. Siu, H. Y. Liu, X. X. Zhu, and C. Wu, Macromolecules 36, 2103 (2003).
V. Aseyev, S. Hietala, A. Laukkanen, M. Nuopponen, O. Confortini, F. E. Du Prez, and H. Tenhu, Polymer 46, 7118 (2005).
V. V. Vasilevskaya, A. A. Aerov, and A. R. Khokhlov, Dokl. Phys. Chem. 398, 258 (2004).
V. I. Lozinskii, I. A. Simenel, E. A. Kurskaya, V. K. Kulakova, V. Y. Grinberg, A. S. Dubovik, I. Y. Galaev, B. Mattiasson, and A. R. Khokhlov, Dokl. Chem. 375, 273 (2000).
V. I. Lozinsky, I. A. Simenel, V. K. Kulakova, E. A. Kurskaya, T. A. Babushkina, T. P. Klimova, T. V. Burova, A. S. Dubovik, V. Y. Grinberg, I. Y. Galaev, B. Mattiasson, and A. R. Khokhlov, Macromolecules 36, 7308 (2003).
V. I. Lozinsky, A. Simenel, M. G. Semenova, L. E. Belyakova, M. M. Il’in, V. Y. Grinberg, A. S. Dubovik, and A. R. Khokhlov, Polym. Sci., Ser. A 48, 435 (2006).
V. I. Lozinskii, I. A. Simenel, and A. R. Khokhlov, Dokl. Chem. 410, 170 (2006).
Z. Ge, D. Xie, D. Chen, X. Jiang, Y. Zhang, H. Liu, and S. Liu, Macromolecules 40, 3538 (2007).
H. Vihola, A. Laukkanen, L. Valtola, H. Tenhu, and J. Hirvonen, Biomaterials 26, 3055 (2005).
D. Wan, Q. Zhou, H. Pu, and G. Yang, J. Polym. Sci., Part A: Polym. Chem. 46, 3756 (2008).
I. Van Nieuwenhove, S. Maji, M. Dash, S. Van Vlierberghe, R. Hoogenboom, and P. Dubruel, Polym. Chem. 8, 2433 (2017).
J. Liu, C. Detrembleur, M. C. De Pauw-Gillet, S. Mornet, E. Duguet, and C. Jérôme, Polym. Chem. 5, 799 (2014).
X. Liang, V. Kozlovskaya, C. P. Cox, Y. Wang, M. Saeed, and E. Kharlampieva, J. Polym. Sci., Part A: Polym. Chem. 52, 2725 (2014).
L. Etchenausia, A. Khoukh, E. Deniau Lejeune, and M. Save, Polym. Chem. 8, 2244 (2017).
A. I. Barabanova, I. V. Blagodatskikh, O. V. Vyshivannaya, T. P. Klimova, N. V. Grinberg, T. V. Burova, A. V. Muranov, V. I. Lozinskii, V. Y. Grinberg, A. S. Peregudov, and A. R. Khokhlov, Dokl. Chem. 465, 253 (2015).
P. Corpart, D. Charmot, S. Zard, T. Biadatti, and D. Michelet, US Patent No. 6 153 705 (2000).
P. O. Baburkin, P. V. Komarov, A. I. Barabanova, P. G. Khalatur, and A. R. Khokhlov, Dokl. Phys. Chem. 470, 129 (2016).
Light Scattering from Polymer Solutions, Ed. by M. B. Huglin (Academic Press, London; New York, 1972).
M. Beija, J. D. Marty, and M. Destarac, Chem. Commun. 47, 2826 (2011).
R. Devasia, R. Borsali, S. Lecommandoux, R. L. Bindu, N. Mougin, and Y. Gnanou, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.) 46, 448 (2005).
A. Gregory and M. H. Stenzel, Prog. Polym. Sci. 37, 38 (2012).
C. Peng, K. Huang, M. Han, W. Meng, Y. Xiong, and W. Xu, Polym. Adv. Technol. 24, 1089 (2013).
Y. Xia, N. A. D. Burke. and H. D. H. Stöver, Macromolecules 39, 2275 (2006).
S. Furyk, Y. Zhang, D. Ortiz-Acosta, P. S. Cremer, and D. E. Bergbreiter, J. Polym. Sci., Part A: Polym. Chem. 44, 1492 (2006).
L. Shao, M. Hu, L. Chen, L. Xu, and Y. Bi, React. Funct. Polym. 72, 407 (2012).
P. Kujawa, H. Watanabe, F. Tanaka, and F. M. Winnik, Eur. Phys. J. E: Soft Matter Biol. Phys. 17, 129 (2005).
H. Ren, X. P. Qiu, Y. Shi, P. Yang, and F. M. Winnik, Macromolecules 53, 5105 (2020).
M. Wu, H. Zhang, and H. Liu, Polym. Bull. 76, 825 (2019).
Y. E. Kirsh, Water Soluble Poly-N-vinylamides: Synthesis and Physicochemical Properties (Wiley, Chichester, 1998).
I. M. Henderson, P. G. Adams, G. A. Montaño, and W. F. Paxton, J. Polym. Sci., Part B: Polym. Phys. 52, 507 (2014).
J. Yan, W. Ji, E. Chen, Z. Li, and D. Liang, Macromolecules 41, 4908 (2008).
J. Zhao, G. Zhang, and S. Pispas, J. Polym. Sci., Part A: Polym. Chem. 47, 4099 (2009).
Q. Wang, H. Tang, and P. Wu, J. Polym. Sci., Part B: Polym. Phys. 54, 385 (2016).
R. Motokawa, K. Morishita, S. Koizumi, T. Nakahira, and M. Annaka, Macromolecules 38, 5748 (2005).
K. Kubota, S. Fujishige, and I. Ando, J. Phys. Chem. 94, 5154 (1990).
C. Wu and S. Zhou, Macromolecules 28, 5388 (1995).
C. Wu and S. Zhou, Macromolecules 28, 8381 (1995).
X. Wang, X. Qiu, and C. Wu, Macromolecules 31, 2972 (1998).
A. C. W. Lau and C. Wu, Macromolecules 32, 581 (1999).
A. Kermagoret, C. A. Fustin, M. Bourguignon, C. Detrembleur, C. Jérôme, and A. Debuigne, Polym. Chem. 4, 2575 (2013).
A. Kermagoret, K. Mathieu, J. M. Thomassin, C. A. Fustin, R. Duchêne, C. Jérôme, C. Detrembleur, and A. Debuigne, Polym. Chem. 5, 6534 (2014).
J. Chuang, A. Y. Grosberg, and T. Tanaka, J. Chem. Phys. 112, 6434 (2000).
A. S. Dubovik, E. E. Makhaeva, V. Y. Grinberg, and A. R. Khokhlov, Macromol. Chem. Phys. 206, 915 (2005).
S. Sun and P. Wu, J. Phys. Chem. B 115, 11609 (2011).
A. Laukkanen, L. Valtola, F. M. Winnik, and H. Tenhu, Macromolecules 37, 2268 (2004).
Funding
This work was financially supported by the Russian Science Foundation (project no. 14-13-00544). NMR studies and elemental analysis were performed with the financial support from the Ministry of Science and Higher Education of the Russian Federation using the equipment of the Center for Molecular Composition Studies of the A.N. Nesmeyanov Institute of Organoelement Compounds of the Russian Academy of Sciences.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflict of interest.
Additional information
The original online version of this article was revised due to a retrospective Open Access order.
Supplementary Information
Rights and permissions
Open Access. This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Barabanova, A.I., Blagodatskikh, I.V., Vyshivannaya, O.V. et al. Synthesis, Thermoresponsive Behavior, and Catalytic Properties of Amphiphilic Diblock Copolymers of N-Vinylcaprolactam and N-Vinylimidazole. Polym. Sci. Ser. A 63, 382–399 (2021). https://doi.org/10.1134/S0965545X21040027
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0965545X21040027