
Abstract
A porous aromatic framework, namely PAF-30, was structurally modified by the introduction of complexing groups based on dipyridylamine, dipicolylamine, and acetylacetone. The materials synthesized in this manner were used as supports of molybdenum catalysts for epoxidation: PAF-30-dpa-Mo, PAF-30-dpcl-Mo, and PAF-30-AA-Mo. All the materials were examined by various analytic methods, such as IR spectroscopy, low-temperature nitrogen adsorption/desorption, X-ray photoelectron spectroscopy, elemental analysis, and transmission electron microscopy. The catalytic activity was tested in epoxidation of cyclohexene, 1-hexene, 1-octene, and styrene. The reusability of the catalysts was assessed using the case of cyclohexene epoxidation.

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Epoxidation of olefins is of great importance in the basic and fine chemicals industry. Using this process, a wide range of chemicals applicable for the synthesis of polymers, dyes, plasticizers, surfactants, and pharmaceuticals can be produced [1, 2]. Epoxidation catalysts include various salts and complexes of transition metals in the highest valence state that have a low redox potential and high Lewis acidity, such as Mo(VI), W(VI), V(V), and Ti(IV). Among these, soluble Mo(VI) complexes are the most active [3–5]. Mo(VI) naphthenate has been used industrially by Halcon in the synthesis of propylene oxide from propylene [6].
However, implementation of homogeneous catalysis faces a number of challenges, in particular those relating to catalyst removal from reaction mixtures, regeneration, and performance stability. Of late, it has become increasingly relevant to develop stable and active heterogeneous molybdenum catalysts for epoxidation. In addition to their easy removal and long life, immobilization of metal complexes on supports ensures uniform distribution of active sites on the surface, thus preventing the catalyst from deactivation otherwise caused by the formation of oxodimeric, peroxodimeric, and other polymeric species [7].
A number of prior studies have been focused on developing methods for heterogenization of catalysts on various organic and inorganic supports, in particular zeolites [8, 9], mesoporous silica gels [10–12], metal–organic frameworks (MOFs) [13–15], and polymers [16–21]. The properties of heterogeneous catalysts strongly depend on the characteristics of the support, and primarily on its interactions with reactants and metal complexes, as well as the porosity, chemical structure, and type of support. For example, hydrophobic supports such as activated carbons, polystyrenes, and some polymer types are more favorable for the adsorption of non-polar organic substrates (e.g., hydrocarbons), than for the adsorption of polar compounds (e.g., oxidation products of these hydrocarbons). In contrast, hydrophilic supports more readily promote the adsorption of polar compounds. In this context, porous aromatic frameworks (PAFs) can be considered promising precursors for the creation and investigation of new heterogeneous catalysts. This is a novel type of polymeric support that consists of strongly covalently bonded aromatic moieties interconnected into rigid frameworks. This structure is responsible for the high properties of PAFs such as stability under temperatures up to 350–400°C, resistance to strong acids and alkalis, oxidation resistance when exposed to hydrogen peroxide and atmospheric oxygen, and resistance to dissolution and swelling in organic solvents.
In our previous works, we synthesized and studied a PAF-30-Mo catalyst based on molybdenum nanoparticles immobilized in the pores of a PAF-30t aromatic framework [22]. This catalyst exhibited high activity and selectivity in the formation of epoxides during cyclohexene oxidation by tert-butyl hydroperoxide. However, the catalyst was found to lose its activity due to metal leaching. To improve the catalyst stability, one promising technique is functionalization of the support with groups that generate complexes with the metal, thus preventing it from leaching.
The purpose of the present study was to modify PAFs with 2,2′-dipyridylamine, di(2-picolyl)amine, and acetylacetone groups; to load them with molybdenum using MoO2(acac)2 (dioxomolybdenum acetylacetonate) as a metal source; and to investigate the structural properties, activity, and stability of the resultant epoxidation catalysts.
EXPERIMENTAL
The following reagents were used: 1,2-dichloroethane (CP grade, Ekos-1, Russia); tert-butyl hydroperoxide (70% aqueous solution, ABCR); cyclohexene (99%, Aldrich); toluene (EP grade, ChimMed, Russia); bis(acetylacetonato)dioxomolybdenum(VI) (99%, ABCR); 2,2′-dipyridylamine (98%, ABCR); di(2-picolyl)amine (97%, Sigma-Aldrich); styrene (≥99%, Aldrich); 1-octene (98%, Aldrich); acetylacetone (≥99%, Sigma-Aldrich); sodium carbonate (high-purity grade, Reakhim, Russia); potassium iodide (CP grade, Reakhim); phosphorus pentoxide (≥99%, ChimMed); hydrochloric acid (EP grade, Sigma Tec, Russia); ethanol (AR grade, IRea 2000, Russia); 1,4-dioxane (CP grade, RusHim, Russia); paraformaldehyde (95%, Sigma-Aldrich); glacial acetic acid (99.8%, RusHim); acetone (CP grade, Ekos-1); and tetrahydrofuran (THF, AR grade, Component-Reaktiv, Russia).
PAF-30 was synthesized according to the procedures described in [23].
The methods used to modify the PAF-30 are provided below.
Synthesis of PAF-30–CH2Cl. The PAF-30 was chloromethylated according to the procedure described in [24]. Paraformaldehyde (5 g) and hydrochloric acid (100 mL) were placed in a 250 mL flask equipped with a magnetic stir bar and a reflux condenser. When all the paraformaldehyde was dissolved, 20 g of phosphorus pentoxide and 30 mL of glacial acetic acid were carefully added to the mixture. Next, 1 g of PAF-30 was placed in the flask and stirred at 90°C for 72 h. The product was filtered, then washed with water (3×50 mL) and ethanol (3×50 mL). The resultant powder was dried in vacuo at 60°C for 24 h. The final PAF-30–CH2Cl weighed 0.908 g.
Synthesis of PAF-30-dpa and PAF-30-dpcl. The PAF-30–CH2Cl was functionalized with nitrogen-containing ligands according to the procedure described in [25]. PAF-30–CH2Cl (200 mg) in 1,4-dioxane (50 mL) was placed in a 100 mL flask equipped with a magnetic stir bar and a reflux condenser. A 20-fold excess of 2,2′-dipyridylamine (dpa) (600 mg) or di(2-picolyl)amine (dpcl) (640 μL) and catalytic amounts of potassium iodide (10 mg) were added to the suspension obtained. The mixture was stirred at 90°C for 72 h. The resultant materials (PAF-30-dpa or PAF-30-dpcl) were filtered, washed with 1,4-dioxane (3×50 mL), water (3×50 mL), and THF (3×50 mL), then dried in vacuo at 60°C for 24 h. The final PAF-30-dpa and PAF-30-dpcl weighed 184 and 176 mg, respectively.
Synthesis of PAF-30-AA. The PAF-30–CH2Cl was modified with acetylacetone according to the procedure described in [26]. In a 100 mL flask equipped with a magnetic stir bar and a reflux condenser, a solution of acetylacetone (17 µL) in acetone (25 mL) was prepared. The mixture was then cooled to 0°C, and, under stirring, 23 mg of sodium carbonate was added batchwise. The contents were stirred at 0°C for 15 minutes. Next, 200 mg of the PAF-30–CH2Cl and catalytic amounts of potassium iodide (10 mg) were added. The suspension was stirred at 50°C for 72 h. The resultant material was filtered, then washed with water (3×50 mL) and methanol (3×50 mL). Next, it was dried in vacuo at 60°C for 24 h. The final PAF-30-AA weighed 192 mg.
Synthesis of PAF-30-dpa-Mo, PAF-30-dpcl-Mo, and PAF-30-AA-Mo. Molybdenum was immobilized on the synthesized materials according to the procedure described in [27]. In a 50 mL round-bottom flask equipped with a magnetic stir bar and a reflux condenser, 100 mg of a support (PAF-30-dpa, PAF-30-dpcl, or PAF-30-AA) was placed, and 240 mg of MoO2(acac)2 in 25 mL of toluene was added. The mixture was heated under stirring to 90°C and held for 96 h. The product was filtered, then washed with toluene (3×50 mL) and methanol (3×50 mL). Next, it was dried in vacuo at 60°C for 24 h. The PAF-30-dpa-Mo, PAF-30-dpcl-Mo, and PAF-30-AA-Mo catalysts weighed 98, 89, and 95 mg, respectively.
INSTRUMENTS AND METHODS
Inductively coupled plasma atomic emission spectroscopy (ICP-AES). The molybdenum content in the synthesized catalysts was measured by ICP-AES using a SHIMADZU ICPE-9000 instrument.
Low-temperature nitrogen adsorption/desorption. The textural properties of the samples were measured on a Gemini VII 2390 (V1.02t) analyzer. Prior to testing, the samples were degassed at 120°C for 8 h. The specific surface area was evaluated using a Brunauer–Emmett–Teller (BET) model in the relative partial pressure (P/P0) range of 0.05–0.2. The total pore volume was calculated at P/P0 = 0.94.
Infrared (IR) spectroscopy. IR spectra were recorded in the range of 4000–500 cm–1 using a Thermo Scientific Nicolet IR200 instrument with a Multireflection HATR attenuation total reflection accessory (with a 45° ZnSe crystal for various wavelength ranges at a resolution of 4 nm).
The support compositions were determined by elemental analysis on a Thermo Flash 2000 CHNS analyzer.
Transmission electron microscopy (TEM). Micrographs were recorded on a JEOL JEM2100F/Cs/GIF instrument at a resolution of 0.19 nm, with an electron beam up to 200 kV. The micrographs were processed using ImageJ software.
X-ray photoelectron spectroscopy (XPS). The catalyst surface composition was determined by XPS using a PHI5500 Versa Probe II instrument with monochromated AlKα radiation (hν = 1486.6 eV) for excitation, with an anode voltage of 14 kV and a power of 50 W. During the measurements, the residual gas pressure in the test chamber was 5 × 10–8 to 7 × 10–8 Pa.
Catalytic Tests
A magnetic stir bar, 5 mg of a catalyst, 4 mL of dichloroethane, 0.2 mL of a substrate, and 0.2 mL of toluene (as an internal standard) were placed in a two-necked glass reactor equipped with a heating jacket. The reactor was connected to a thermostat and equipped with a reflux condenser fitted with a septum and a needle on the top. The tert-butyl hydroperoxide solution (0.4 mL) was rapidly injected into the mixture through the second neck, after which the septum was inserted, and stirring was started. Epoxidation was carried out at 80°C, with samples being taken by a microsyringe through the septum. The samples were up to 10 μL in volume.
The reaction products were analyzed on a Hewlett-Packard 6890 gas chromatograph (GC) equipped with a flame ionization detector and a HP-1 column (50 m× 0.32 mm×1.05 μm, 100% dimethylsiloxane phase, helium as a carrier gas). The chromatograms were recorded and processed using the HP ChemStation Rev. A. 06. 01 (403) software package. The product concentrations were derived from the ratio of the corresponding peak area to the internal standard peak area.
RESULTS AND DISCUSSION
The PAF-30 was modified with chelating groups in two steps: chloromethyl groups were introduced first, and the resultant PAF-30–CH2Cl was treated with 2,2′-dipyridylamine, di(2-picolyl)amine, or acetylacetone.
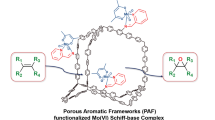
The PAF-30-dpa, PAF-30-dpcl, and PAF-30-AA materials were then impregnated with a MoO2(acac)2 solution in toluene at 90°C to synthesize the target molybdenum catalysts (Fig. 1).

Synthesis of PAF-30-dpa-Mo, PAF-30-dpcl-Mo, and PAF-30-AA-Mo.
The analytical data (Table 1) show that PAF-30-dpa and PAF-30-dpcl contain 1.18 and 2.05 wt % (or 843 and 1464 μmol/g) of nitrogen, respectively, thus confirming the successful immobilization of amines. The higher content of functional groups in PAF-30-dpcl may result from the higher reactivity of nitrogen atoms in dipicolylamine than that in dipyridylamine due to the conjugation of the nitrogen electrons with aromatic rings in dipyridylamine molecules.
The textural properties of the supports were examined by low-temperature nitrogen adsorption/desorption (Table 1). The functionalization of PAF-30 reduced both the surface area and pore volume. The textural properties of the supports were found to depend on the size and concentration of the functional groups: higher concentrations and volumes of the functional groups corresponded to smaller surface areas and smaller available pore volumes.
Molybdenum was introduced into the pores of the synthesized supports by their impregnation with the MoO2(acac)2 toluene solution. To characterize the catalyst structures, IR spectroscopy, XPS, TEM, and ICP-AES methods were employed. The metal content in the catalysts (9–12 wt %, as shown in Table 1, or 940– 1250 μmol/g) exceeded the concentration of functional groups in the precursor supports by a factor of about 2.5–3.5. This may indicate either immobilization of MoO2(acac)2 without chemical binding (i.e., adsorption of the complex by the support), or formation of multinuclear complexes or nanoparticles of MoO2(acac)2 inside the support pores.
The IR spectra of the catalysts display new bands when compared to the initial PAF-30 (Fig. 2). Some of them correspond to bonds in ligands (e.g., the 1530, 1470, and 1150 cm–1 peaks in PAF-30-dpa and PAF-30-dpcl [28]). In all cases, the low intensity of these bands correlates well with the elemental analysis and nitrogen adsorption data. The new peaks at 914 and 957 cm–1 are attributed to symmetric and asymmetric vibrations in the O=Mo=O moiety [29]. Furthermore, there are peaks in the 700– 800 cm–1 range, attributed to Mo–O–Mo vibrations [30]; this points to the formation of molybdenum oxide clusters or nanoparticles inside the support pores. It is worth noting that agglomeration of metal complexes and salts in the pores of aromatic frameworks during impregnation has also been reported for the synthesis of palladium catalysts such as PAF70-Pd and PP-P-Pd also report [31, 32].

IR spectra of PAF-30-dpa-Mo, PAF-30-dpcl-Mo, and PAF-30-AA-Mo.
The TEM micrographs display small (1.5–2.5 nm) nanoparticles uniformly distributed inside the pores (Fig. 3). To determine the valence state of the metal, the catalysts were subjected to XPS examination (Table 2). The data clearly show that, in all catalysts, molybdenum was in the highest oxidation state, i.e. Mo(VI) [33], with the binding energies being lower than those for the MoO2(acac)2 complex [34]. This serves as evidence that the ligands in the PAFs promoted diffusion of MoO2(acac)2 and uniform distribution of the complex in the support. However, because of the decomposition and agglomeration of the complex during the molybdenum loading, a major portion of molybdenum was present as nanoparticles uniformly distributed inside the PAF-30 pores.

TEM micrographs: (a, c) PAF-30-dpcl-Mo; (b) PAF-30-dpa-Mo; and (d) PAF-30-AA-Mo.
The catalysts were tested in epoxidation of olefins (Table 3). The highest activity was observed in the case of cyclohexene epoxidation: over three hours of reaction, the substrate conversion reached above 90%, with epoxide selectivity being at least 90%. The conversion of the other substrates, however, was not as rapid. For example, the styrene conversion after 3 h was as low as 15–17%; moreover, benzaldehyde was formed as a byproduct in significant amounts. In the cases of linear olefins, 1-hexene, and 1-octene, extremely vigorous isomerization of olefins was observed, even despite their slightly higher conversion (up to 22–29%). This process was most probably caused by the weak Lewis acidity of molybdenum oxide nanoparticles [35].
Our previous study revealed rapid deactivation of a PAF-30-Mo catalyst caused by metal leaching from the catalyst pores during epoxidation of cyclohexene [22]. In contrast, the catalysts synthesized in the present work withstood several reuse cycles without significant loss of activity (Fig. 4). This suggests that introduction of functional groups into the support enhances the catalyst stability.

Reuse of PAF-30-dpa-Mo, PAF-30-dpcl-Mo, and PAF-30-AA-Mo in cyclohexene epoxidation. Reaction conditions: 2 mmol substrate; substrate/oxidant ratio = 1 : 1.5; 2 mmol toluene; 5 mg catalyst; 4 mL 1,2-dichloroethane; 80°C; 3 h.
As the catalysts lost some of their initial activity, apparently the metal was partially leached from the support in this study as well. Therefore, an important problem was to assess the epoxidation activity of the metal leached into the solution. For the purpose of this assessment, cyclohexene was repeatedly epoxidated, where the catalyst was filtered off after 1 h, so that the reaction was continued under catalyst-free conditions (Fig. 5). The plot clearly shows that the catalytic process ceased.

Kinetics of cyclohexene epoxidation and catalyst removal experiment: (a) PAF-30-dpa-Mo; (b) PAF-30-dpcl-Mo; and (c) PAF-30-AA-Mo. Reaction conditions: 2 mmol substrate; substrate/oxidant ratio = 1 : 1.5; 2 mmol toluene; 5 mg catalyst; 4 mL 1,2-dichloroethane; 80°C; 3 h.
Thus, we see that the epoxidation occurred on molybdenum’s active sites inside the PAF pores, and that the contribution of the leached metal to the subsequent olefin oxidation was negligible.
CONCLUSIONS
The activity of molybdenum catalysts based on porous aromatic frameworks (PAFs) modified with complexing ligands was investigated in epoxidation of olefins. Although introducing ligands promotes uniform distribution of the metal in the support, molybdenum complexes decompose during impregnation to form molybdenum oxide nanoparticles inside the PAF pores. Moreover, these nanoparticles favor side reactions during epoxidation, such as isomerization of linear α-olefins. Nonetheless, structural modification of PAFs improves the stability of the catalysts compared to their unmodified counterparts. The highest activity was exhibited in epoxidation of cyclohexene: at 80°C, cyclohexene epoxide was produced in high yield (above 84%) over 3 h.
AUTHOR CONTRIBUTION
V.A. Yarchak: synthesis of samples, catalytic test, processing of experimental data.
L.A. Kulikov: test methodology, processing of experimental data.
A.L. Maximov and E.A. Karakhanov: conceptualization.
All co-authors: discussion of results.
Change history
16 February 2023
Corrected PDF Copyright
19 April 2023
An Erratum to this paper has been published: https://doi.org/10.1134/S0965544123010097
REFERENCES
Oyama, T.S., Mechanisms in Homogeneous and Heterogeneous Epoxidation Catalysis, Amsterdam: Elsevier, 2008, 1 ed. https://doi.org/10.1016/B978-0-444-53188-9.X0001-6
Shen, Y., Jiang, P., Wai, P.T., Gu, Q., and Zhang, W., Catalysts, 2019, vol. 9, no. 1, pp. 31–57. https://doi.org/10.3390/catal9010031
Masters, C., Homogeneous Transition-Metal Catalysis—A Gentle Art, London: Chapman and Hall, 1981.
Esnaashari, F., Moghadam, M., Mirkhani, V., Tangestaninejad, S., Mohammadpoor-Baltork, I., Khosoropour, A.R., Zakeri, M., and Hushmandrad, S., Polyhedron, 2012, vol. 48, no. 1, pp. 212–220. https://doi.org/10.1016/j.poly.2012.08.084
Li, T., Zhang, W., Chen, W., Miras, H.N., and Song, Y.F., ChemCatChem., 2018, vol. 10, no. 1, pp. 188–197. https://doi.org/10.1002/cctc.201701056
Nijhuis, T.A., Makkee, M., Moulijn, J.A., and Weckhuysen, B.M., Ind. Eng. Chem. Res., 2006, vol. 45, no. 10, pp. 3447–3459. https://doi.org/10.1021/ie0513090
Bezaatpour, A., Khatami, S., and Amiri, M., RSC Adv., 2016, vol. 6, no. 33, pp. 27452–27459. https://doi.org/10.1039/C5RA27751E
Dai, P.S.E. and Lunsford, J.H., J. Catal., 1980, vol. 64, no. 1, pp. 184–199. https://doi.org/10.1016/0021-9517(80)90491-1
Eghbali, P., Şahin, E., and Masteri-Farahani, M., J. Porous Materials, 2017, vol. 24, no. 1, pp. 39–44. https://doi.org/10.1007/s10934-016-0234-8
Shen, Y., Jiang, P., Zhang, J., Bian, G., Zhang, P., Dong, Y., and Zhang, W., Mol. Catal., 2017, vol. 433, pp. 212–223. https://doi.org/10.1016/j.mcat.2016.12.011
Jia, M., Seifert, A., and Thiel, W.R., J. Catal., 2004, vol. 221, no. 2, pp. 319–324. https://doi.org/10.1016/j.jcat.2003.07.009
Shen, Y., Jiang, P., Wang, Y., Bian, G., Wai, P.T., and Dong, Y., J. Solid State Chem., 2018, vol. 264, pp. 156–164. https://doi.org/10.1016/j.jssc.2018.05.005
Noh, H., Cui, Y., Peters, A.W., Pahls, D.R., Ortuno, M.A., Vermeulen, N.A., Cramer, C.J., Gagliardi, L., Hupp, J.T., and Farha, O.K., J. Am. Chem. Soc., 2016, vol. 138, no. 44, pp. 14720–14726. https://doi.org/10.1021/jacs.6b08898
Tang, J., Dong, W., Wang, G., Yao, Y., Cai, L., Liu, Y., Zhao, X., Xu, J., and Tan, L., RSC Adv., 2014, vol. 4, no. 81, pp. 42977–42982. https://doi.org/10.1039/c4ra07133f
Hlatshwayo, X.S., Xaba, M.S., Ndolomingo, M.J., Bingwa, N., and Meijboom, R., Catalysts, 2021, vol. 11, no. 6, pp. 673–685. https://doi.org/10.3390/catal11060673
Tangestaninejad, S., Habibi, M.H., Mirkhani, V., Moghadam, M., and Grivani, G., Inorg. Chem. Commun., 2006, vol. 9, no. 6, pp. 575–578. https://doi.org/10.1016/j.inoche.2006.03.001
Sherrington, D.C. and Simpson, S., React. Polymer., 1993, vol. 19, nos. 1–2, pp. 13–25. https://doi.org/10.1016/0923-1137(93)90007-3
Miller, M.M. and Sherrington, D.C., J. Catal., 1995, vol. 152, no. 2, pp. 377–383. https://doi.org/10.1006/jcat.1995.1092
Gao, B., Men, J., and Zhang, Y., Metal-Org. Nano-Metal Chem., 2015, vol. 45, no. 6, pp. 821–827. https://doi.org/10.1080/15533174.2013.843556
Mbeleck, R., Ambroziak, K., Saha, B., and Sherrington, D.C., React. Funct. Polym., 2007, vol. 67, no. 12, pp. 1448–1457. https://doi.org/10.1016/j.reactfunctpolym.2007.07.024
Chang, Y., Lv, Y., Lu, F., Zha, F., and Lei, Z., J. Mol. Catal. A: Chem., 2010, vol. 320, nos. 1–2, pp. 56–61. https://doi.org/10.1016/j.molcata.2010.01.003
Kulikov, L.A., Yarchak, V.A., Zolotukhina, A.V., Maksimov, A.L., and Karakhanov, E.A., Petrol. Chem., 2020, vol. 60, no. 9, pp. 1087–1093. https://doi.org/10.1134/S0965544120090169
Yuan, Y., Sun, F., Ren, H., Jing, X., Wang, W., Ma, H., Zhao, H., and Zhu, G., J. Mater. Chem., 2011, vol. 21, no. 35, pp. 13498–13502. https://doi.org/10.1039/c1jm11998b
Kulikov, L., Kalinina, M., Makeeva, D., Maximov, A., Kardasheva, Y., Terenina, M., and Karakhanov, E., Catalysts, 2020, vol. 10, no. 10, pp. 1–17. https://doi.org/10.3390/catal10101106
Kratz, M.R. and Hendricker, D.G., Polymer (Guildf)., 1986, vol. 27, no. 10, pp. 1641–1643. https://doi.org/10.1016/0032-3861(86)90117-5
Thorat, K.G., Kamble, P., Ray, A.K., and Sekar, N., Phys. Chem. Chem. Phys., 2015, vol. 17, no. 26, pp. 17221–17236. https://doi.org/10.1039/c5cp01741f
Tangestaninejad, S., Moghadam, M., Mirkhani, V., Mohammadpoor-Baltork, I., and Ghani, K., J. Iran. Chem. Soc., 2008, vol. 5, no. S1, pp. 71–79. https://doi.org/10.1007/bf03246492
Mirzaee, M., Bahramian, B., Gholizadeh, J., Feizi, A., and Gholami, R., Chem. Eng. J., 2017, vol. 308, pp. 160–168. https://doi.org/10.1016/j.cej.2016.09.055
Tan, Z., Qian, D., Zhang, W., Li, L., Ding, Y., Xu, Q., Wang, F., and Li, Y., J. Mater. Chem. A: Mater., 2012, vol. 1, no. 3, pp. 657–664. https://doi.org/10.1039/C2TA00325B
Tahmasebi, N. and Khalildashti, M., Korean J. Chem. Eng., 2020, vol. 37, no. 3, pp. 448–455. https://doi.org/10.1007/s11814-019-0469-6
Jing, L.P., Sun, J.S., Sun, F., Chen, P., and Zhu, G., Chem. Sci., 2018, vol. 9, no. 14, pp. 3523–3530. https://doi.org/10.1039/C8SC00510A
Zhang, Q., Yang, Y., and Zhang, S., Chem. Eur. J., 2013, vol., 19, no. 30, pp. 10024–10029. https://doi.org/10.1002/chem.201300334
Hong, M., Yao, M.Y., and Pan, H., RSC Adv., 2015, vol. 5, no. 111, pp. 91558–91563. https://doi.org/10.1039/c5ra14581c
Zhang, Z., Liu, B., Lv, K., Sun, J., and Deng, K., Green Chem., 2014, vol. 16, no. 5, pp. 2762–2770. https://doi.org/10.1039/C4GC00062E
Shen, K., Liu, X., Lu, G., Miao, Y., Guo, Y., Wang, Y., and Guo, Y., J. Mol. Catal. A: Chem., 2013, vol. 373, pp. 78–84. https://doi.org/10.1016/J.MOLCATA.2013.02.020
Funding
The study was performed with financial support from the Russian Science Foundation (project no. 22-79-10044).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
A.L. Maximov, a co-author, is the Chief Editor at the Neftekhimiya (Petroleum Chemistry) Journal. The other co-authors declare no conflict of interest requiring disclosure in this article.
Rights and permissions
Open Access. This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yarchak, V.A., Kulikov, L.A., Maximov, A.L. et al. Epoxidation of Olefins in the Presence of Molybdenum Catalysts based on Porous Aromatic Frameworks. Pet. Chem. 63, 327–335 (2023). https://doi.org/10.1134/S0965544123010012
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0965544123010012