
Abstract
The feasibility of biphenyl hydrogenation with syngas for hydrogen purification and binding with the aim of its transportation was demonstrated. Specific features of the hydrogenation of biphenyl as a promising organic hydrogen carrier using unsupported Ni–Mo sulfide catalysts were studied. In particular, the influence of temperature, reaction time, presence of water in the system, and Н2/СО gas mixture composition on the substrate conversion and selectivity with respect to products was examined. The highest conversion and the maximal hydrogen uptake are reached at 380°С in 6–8 h. The dispersed catalysts are active in biphenyl hydrogenation at the CO concentration in the Н2/СО gas mixture of up to 50 vol %, and H2O can act in this case as an in situ hydrogen source owing to the occurrence of the water-gas shift reaction.

Similar content being viewed by others
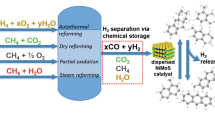

Avoid common mistakes on your manuscript.
The use of hydrogen as an energy carrier requires the development of approaches to producing low-carbon-footprint hydrogen and to making its purification and transportation more efficient [1, 2]. Traditional methods for producing pure hydrogen include several steps. The first step is steam reforming of hydrocarbons, carbon dioxide reforming of methane, or autothermal methane reforming. Then, CO is removed from the gaseous mixture of hydrogen, carbon monoxide, and water vapor by involving it into the water-gas shift reaction with the formation of CO2 and its separation on adsorption and membrane installations [3, 4]. The production of low-carbon-footprint hydrogen requires not only separation but also utilization of CO2, both by disposal or use in oil extraction [5] and by use as chemical feedstock. The latter processes, which include dry reforming of methane [6], dehydrogenation of alkanes [7], and high-temperature and low-temperature electrolysis of СО2 to СО or, in the presence of water, to syngas [8, 9], as a rule, are power-consuming and in many cases lead to the formation of СО, which should be subsequently converted in traditional chemical processes. In this case, the syngas separation into H2 and CO can be an alternative. It is desirable that such separation be combined with hydrogen binding for the subsequent storage and transportation. In particular, this approach can be implemented using liquid organic hydrogen carriers [10–14]. The most cost-saving process implementation should involve hydrogenation of liquid organic hydrogen carriers directly with syngas produced by steam reforming; in this case, hydrogen becomes bound with the liquid carrier and can be readily separated in this form, whereas the remaining СО-containing gases can be involved in further processing. Such approach has certain advantages over other methods such as hydrogen liquefaction, physical adsorption, etc. [12–16]. In this case, there is no need for additional steps of treatment of the vapor–gas mixture to remove СО by its involvement in the water-gas shift reaction; in addition, the use of pressure swing adsorption, liquefaction, and other power-consuming hydrogen separation methods can be abandoned. Liquid organic carriers are, as a rule, stable at ambient temperature and pressure and ensure considerably higher gravimetric hydrogen content compared to liquid hydrogen or metal hydrides [17].
Aromatic compounds form one of promising classes of liquid organic hydrogen carriers. Hydrogen is incorporated in them in the course of hydrogenation and can be released in the course of dehydrogenation. The gravimetric hydrogen content in some cases exceeds 7 wt %. In particular, biphenyl shows much promise as an organic carrier because of its stability and high hydrogen storage density (7.3 wt %), appreciably exceeding the values for other compounds considered as liquid organic hydrogen carriers (carbazole 6.7, toluene 6.2, phenyltoluene 6.7 wt %) [18].
Hydrogenation of aromatic hydrocarbons, in particular, biphenyl, is traditionally performed in a pure hydrogen atmosphere using catalysts based on noble metals, e.g., Ru/Al2O3, Pt/Al2O3, Pd-Rh/Al2O3, and Pd/MoO3 [19–23]. Such catalysts are sensitive to the presence of even minor amounts of СО, which is a strong catalytic poison for them. This fact does not allow these systems to be used for hydrogen binding by hydrogenation of aromatic compounds directly from the hydrogen-containing gas formed by steam, oxidative, or carbon dioxide reforming of methane.
As shown previously, catalysts based on Ni–Mo sulfides are active both in hydrogenation of aromatic compounds [24–27] and in the water-gas shift reaction. Such systems are resistant to СО, which allows the hydrogenation to be performed in the СО–water system using hydrogen formed in situ by the reaction of CO with H2O. Both supported catalysts [28] and dispersions of nanosized sulfides of the corresponding metals [29, 30] can be used in the process. The advantages of the unsupported dispersed catalysts are high specific surface area, high content of the active components, and resistance to catalytic poisons. In addition, the unsupported catalytic systems can be prepared in situ by high-temperature decomposition of oil-soluble metal salts in the presence of a sulfidizing agent precursor (elemental sulfur) and remain stable in the presence of large amounts of water [29–32].
This study deals with the specific features of chemical binding of hydrogen by hydrogenation of biphenyl as one of promising organic hydrogen carriers in a syngas atmosphere using unsupported Ni–Mo sulfide catalysts with in situ formation of the active component from oil-soluble precursors.
EXPERIMENTAL
The following chemicals were used as starting compounds: molybdenum hexacarbonyl Mo(CO)6 (98%, catalog no. 199958, Sigma–Aldrich), nickel naphthenate C22H14NiO4 (Ni 5–12%, catalog no. AA39455A1, Alfa Aesar), elemental sulfur (chemically pure grade, Khimreaktiv, Russia), biphenyl (99%, catalog no. W312908, Sigma–Aldrich), and heptane (98.5%, catalog no. 8.22332.1006, Khimmed, Russia).
Catalytic experiments were performed in a 50 cm3 temperature-controlled steel autoclave. The constant temperature of the reaction mixture was maintained with an electric resistance furnace equipped with a thermocouple and a temperature controller. A 10 wt % solution of biphenyl in heptane was used as a model feed. The active component precursors were molybdenum hexacarbonyl (0.08 wt % in terms of metal) and sodium naphthenate, and the sulfiding agent precursor was elemental sulfur (0.5 wt %). The amounts of the starting compounds and reagents were calculated so as to ensure the Mo/Ni molar ratio in the catalytic formulation of 3/1 and the biphenyl/Mо molar ratio of 65/1. The experiments were performed as follows. The autoclave was charged with 13 mg of Mo(CO)6, 14 mg of C22H14NiO4, 24 mg of S, 500 mg of biphenyl, the required amount of the solvent (heptane), and, when performing the process under the water-gas shift conditions, 500 μL of distilled water. The total volume of the reaction mixture was 5 mL. The autoclave was sealed and filled with hydrogen or syngas (volume ratio Н2/СО = 1–3) to a pressure of 6 MPa at 25°С. The model feed was hydrogenated in the temperature interval 320–380°С with vigorous stirring of the reaction mixture for the required time (from 2 to 8 h). After the reaction completion, the reactor was cooled to room temperature and opened.
The liquid reaction products were analyzed with a Crystallux-4000 gas–liquid chromatograph (Meta-Chrom, Russia) equipped with a flame ionization detector and a 0.25 mm × 50 m Petrocol TM capillary column (Supelco) with polydimethylsiloxane stationary liquid phase; analyses were performed under the conditions of programmed heating. The chromatograms were processed using NetChrom software. The biphenyl conversion (%), selectivity (%) with respect to products (cyclohexylbenzene and dicyclohexyl), and hydrogen uptake relative (%) to the theoretical value were calculated by the following formulas:
where SP is total peak area of products; SBP, biphenyl peak area; SCHB, cyclohexylbenzene peak area; and SDCH, dicyclohexyl peak area.
To determine the physicochemical characteristics, the Ni–Mo sulfide catalyst after performing the reaction (380°С, 6 h, H2/CO = 3/1) was separated from liquid reaction products by centrifugation, washed, and dried. The catalyst surface structure was examined by transmission electron microscopy (TEM) with a JEM-2100 electron microscope (Jeol). A catalyst sample was dispersed in ethanol in an ultrasonic bath and then applied onto a copper grid with Lacey Formvar supporting grid (Ted Pella). X-ray diffraction analysis of the samples was performed with a Rigaku Rotaflex D/max RC device (Rigaku). The X-ray diffraction patterns were recorded using CuKα radiation (λ = 1.5418 Å) in the angle range 2θ = 1.5–70 with a 0.05 step at a rate of 2 deg min–1. The gravimetric content of elements in the samples was determined by X-ray fluorescence analysis with a Thermo Scientific ARL QUANT’X device (Thermo Fisher) in a vacuum. The results were processed using UniQuant® standardless method.
RESULTS AND DISCUSSION
The biphenyl dehydrogenation occurs with the successive formation of dicyclohexylbenzene and then dicyclohexyl. The rate-limiting step of hydrogenation is cyclohexylbenzene hydrogenation, because the previous step, biphenyl hydrogenation to cyclohexylbenzene, occurs at a higher rate [33].

Unsupported catalysts based on transition metal sulfides, which can be prepared directly in the reaction medium, exhibit high activity in hydrogenation of aromatic compounds. The formation of the active catalyst component in situ suggests the presence of an induction period in which the precursors (nickel naphthenate, molybdenum hexacarbonyl) decompose and transform into sulfides. Therefore, we compared the activity of the Ni–Mо sulfide catalyst formed in situ in the course of the reaction and that of its analog prepared in advance by the same procedure and under the same conditions (380°С, hydrogen pressure 6 MPa, t = 6 h) but without substrate.
Experiments on evaluation of the activity of the systems in biphenyl hydrogenation at elevated hydrogen pressure and different temperatures have shown that the biphenyl conversion with the catalyst prepared in advance is 15–20% higher than that with the catalyst prepared in situ because of an induction period observed in the latter case (Fig. 1). With increasing temperature, the conversion and extent of hydrogenation of biphenyl increase. At 380°С, the biphenyl conversion was 65% (products: dicyclohexyl 9, cyclohexylbenzene 56%) with the catalyst prepared in situ and 79% (products: dicyclohexyl 19, cyclohexylbenzene 60%) with the catalyst prepared in advance.

Temperature dependence of the biphenyl conversion and hydrogen uptake (relative to the theoretically possible value) with the Ni–Mо sulfide catalyst prepared in advance and with its analog formed in situ in the course of the reaction. Reaction conditions: ω(Mo) = 0.08 wt %, Mo/Ni molar ratio 3/1, biphenyl/Mо molar ratio 65/1, p(H2) = 60 MPa (at 25°C), t = 6 h.
To obtain pure hydrogen, it is necessary to remove CO and CO2 from the gas mixture after steam or carbon dioxide reforming of hydrocarbons, which requires major capital investments. A significant advantage of sulfide catalysts compared to those based on noble metals is their resistance to CO. It is important to study the activity of catalysts based on transition metal sulfides in hydrogenation processes in H2 and СO atmosphere. Therefore, we performed experiments to evaluate how the СO concentration and Н2/СО ratio in syngas affect the biphenyl conversion and relative hydrogen uptake in the course of hydrogenation at 380°С under the conditions when the active component of Ni–Mo sulfide catalysts was formed in the reaction medium in situ (Fig. 2a). The biphenyl conversion under the syngas and hydrogen pressure was 30 and 65%, respectively. A decrease in the biphenyl conversion in going to syngas may be caused by a decrease in the hydrogen partial pressure in the system due to an increase in the СО fraction in the gas mixture. For example, at the Н2/СО volume ratio of 3, the hydrogen partial pressure is 4.5, and at Н2/СО = 1, 3 MPa. In this case, the biphenyl conversion decreases from 30 to 18%, and the hydrogen uptake, from 17 to 10%. The major reaction product is cyclohexylbenzene; the selectivity of its formation reaches 85–90%.

Plots of the biphenyl conversion and relative hydrogen uptake vs. gas mixture composition for the reaction performed (a) in a hydrocarbon medium and (b) under the water-gas shift conditions. Reaction conditions: ω(Mo) = 0.08 wt %, Mo/Ni molar ratio 3/1, biphenyl/Mо molar ratio 65/1, T = 380°С, p = 6 MPa (at 25°C), t = 6 h; (b) ω(H2O) = 10 wt %.
Transition metal sulfides exhibit activity not only in hydrogenation but also in the water-gas shift reaction, which allows in situ production of additional hydrogen in the presence of water. When performing the biphenyl hydrogenation with the addition of 10 wt % water in a syngas atmosphere, the influence of the gas mixture composition becomes less pronounced. For example, at a lower hydrogen concentration, 50 vol % (Н2/СО ratio 1/1), the biphenyl conversion is comparable to that in the system with Н2/СО = 3/1 (Fig. 2b). In both cases, the relative hydrogen uptake is 7%. Probably, the observed trend is due to the occurrence of the water-gas shift reaction ensuring the hydrogen formation in situ and an increase in its partial pressure.
As the temperature is increased from 320 to 380°С, the biphenyl conversion increases (Fig. 3a). For example, in 6 h the substrate conversion is 4% at 320°C (selectivity: 100% cyclohexylbenzene) and 30% at 380°С (selectivity: cyclohexylbenzene 90, dicyclohexyl 10%). However, further increase in the temperature is undesirable, because dehydrogenation starts to prevail (at 410°С, the conversion decreases to 9%).

Plots of the biphenyl conversion and relative hydrogen uptake vs. (a) temperature and (b) time of the reaction performed in a syngas atmosphere using Ni–Mo sulfide catalysts formed in situ. Reaction conditions: ω(Mo) = 0.08 wt %, Mo/Ni molar ratio 3/1, biphenyl/Mо molar ratio 65/1, p = 6 MPa (at 25°C), H2/CO volume ratio 3/1; (a) t = 6 h; (b) T = 380°С.
An increase in the hydrogenation time leads to an increase in the biphenyl conversion (Fig. 3b). For example, the conversion slightly exceeds 5% in 2 h and reaches 44% in 8 h. Low biphenyl conversion at short times is due to the induction period required for the formation of the catalyst active phase. An increase both in hydrogenation time and in temperature leads to an increase in the relative hydrogen uptake.
Energy-dispersive X-ray fluorescence analysis shows that the obtained sample of the Ni–Mo sulfide catalyst has the following composition (wt %): S 33.4, Mo 50.3, and Ni 16.3.
The phase composition of the catalyst was studied by X-ray diffraction analysis. The X-ray diffraction pattern is difficult to interpret because of the low sample crystallinity (Fig. 4). Furthermore, the reflection at 2θ ≈ 14.4°, corresponding to MoS2, is slightly shifted toward 2θ ≈ 14°. This may be caused by the fact that the coplanarity of the MoS2 planes is disturbed by intercalation of Ni atoms and solvent molecules. The major reflections correspond to the phases of MoS2 [2θ ≈ 14.3° (hkl = 002), 33.0° (hkl = 100), 39.2° (hkl = 103), and 58.4° (hkl = 110)] and double sulfide NixMoSy [2θ ≈ 13.5° (hkl = 101), 20.2° (hkl = 012), 43.8° (hkl = 311), and 58.2° (hkl = 413)]. In addition, relatively weak reflections corresponding to NiS are observed 2θ ≈ 30.0° (hkl = 101) and 34.4° (hkl = 300).

X-ray diffraction pattern of the Ni–Mo sulfide catalyst.
The layered structure of MoS2 with three to seven sulfide layers is clearly seen in the TEM images. The interplanar spacing is 6.2 Å, which corresponds to the basal (002) plane of the MoS2 crystallite (Fig. 5). Thus, the procedure for preparing unsupported sulfides, based on decomposition–sulfidation of metal precursors in situ directly in the reaction medium in a syngas atmosphere, allows preparation of nanosized catalysts with high content of the active component.

TEM image of the Ni–Mo sulfide catalyst.
Thus, with unsupported Ni–Mo sulfide catalysts, biphenyl hydrogenation can be performed using Н2/СО mixtures of different compositions in the temperature interval 320–380°C, which allows this organic hydrogen carrier to be used for binding and storage of hydrogen without preliminary purification and syngas separation. The biphenyl conversion increases with a decrease in the CO fraction. CO can also be involved in the water-gas shift reaction with the formation of “additional” hydrogen in situ. In the case of adding water, the biphenyl conversion at a lower hydrogen concentration, 50 vol % (Н2/СО ratio 1/1), is comparable to that in the system with 75 vol % hydrogen concentration, which may be due to the occurrence of the water-gas shift reaction ensuring in situ hydrogen formation and an increase in its partial pressure.
AUTHOR CONTRIBUTION
A.L. Maximov, E.A. Karakhanov, and A.V. Vutolkina: formulation of objectives and tasks of the research, development of experimental procedures, and suggestion of physicochemical methods for catalyst investigation. I.G. Baigildin: catalytic experiments, interpretation of the results of chromatographic analysis of product mixtures, and X-ray diffraction study of the phase composition of the catalylst. A.V. Vutolkina: analysis of the results obtained by transmission electron microscopy and X-ray fluorescence analysis and preparation of graphic materials for the paper.
REFERENCES
Filippov, S.P. and Yaroslavtsev, A.B., Russ. Chem. Rev., 2021, vol. 90, no. 6, pp. 627–643. https://doi.org/10.1070/RCR5014
Yakubson, K.I., Russ. J. Appl. Chem., 2020, vol. 93, no. 12, pp. 1775–1795. https://doi.org/10.1134/S1070427220120010
Jorschick, H., Vogl, M., Preuster, P., Bösmann, A., and Wasserscheid, P., Int. J. Hydrogen Energy, 2019, vol. 44, pp. 31172–31182. https://doi.org/10.1016/j.ijhydene.2019.10.018
Bazhenov, S.D., Novitskii, E.G., Vasilevskii, V.P., Grushevenko, E.A., Volkov, A.V., and Bienko, A.A., Russ. J. Appl. Chem., 2019, vol. 92, no. 8, pp. 1045–1063. https://doi.org/10.1134/S1070427219080019
Fakher, S. and Imqam, A., Fuel, 2020, vol. 265. ID 116944. https://doi.org/10.1016/j.fuel.2019.116944
Usman, M., Wan Daud, W.M.A., and Abbas, H., Renew. Sust. Energ. Rev., 2015, vol. 45, pp. 710–744. https://doi.org/10.1016/j.rser.2015.02.026
Gomez, E., Yan, B., Kattel, S., and Chen, J., Nat. Rev. Chem., 2019, vol. 3, pp. 638–649. https://doi.org/10.1038/s41570-019-0128-9
Rabinowitz, J.A. and Kanan, M.W., Nat. Commun., 2020, vol. 11. ID 5231. https://doi.org/10.1038/s41467-020-19135-8
Song, Y., Zhang, X., Xie, K., Wang, G., and Bao, X., Adv. Mater., 2019, vol. 31, no. 50. ID 1902033. https://doi.org/10.1002/adma.201902033
Reuß, M., Grube, T., Robinius, M., Preuster, P., Wasserscheid, P., and Stolten, D., Appl. Energy, 2017, vol. 200, pp. 290–302. https://doi.org/10.1016/j.apenergy.2017.05.050
Teichmann, D., Arlt, W., and Wasserscheid, P., Int. J. Hydrogen Energy., 2012, vol. 37, pp. 18118–18132. https://doi.org/10.1016/j.ijhydene.2012.08.066
Makaryan, I.A. and Sedov, I.V., Petrol. Chem., 2021, vol. 61, no. 9, pp. 977–988. https://doi.org/10.1134/S0965544121090085
Makaryan, I.A. and Sedov, I.V., Russ. J. Appl. Chem., 2021, vol. 94, no. 8, pp. 1011–1021 https://doi.org/10.1134/S1070427221080012
Kustov, L.M., Kalenchuk, A.N., and Bogdan, V.I., Russ. Chem. Rev., 2020, vol. 89, no. 9, pp. 897–916. https://doi.org/10.1070/RCR4940
Preuster, P., Papp, C., and Wasserscheid, P., Acc. Chem. Res., 2017, vol. 50, pp. 74–85. https://doi.org/10.1021/acs.accounts.6b00474
Orimo, S., Nakamori, Y., Eliseo, J., Züttel, A., and Jensen, C., Chem. Rev., 2007, vol. 107, pp. 4111–4132. https://doi.org/10.1021/cr0501846
Zhu, Q.-L. and Xu, Q., Energy Environ. Sci., 2015, vol. 8, pp. 478–512. https://doi.org/10.1039/C4EE03690E
Kalenchuk, A.N., Koklin, A.E., Bogdan, V.I., and Kustov, L.M., Russ. Chem. Bull., 2017, vol. 66, pp. 1208–1212. https://doi.org/10.1007/s11172-017-1874-9
Li, L.L., Yang, M., Dong, Y., Mei, P., and Cheng, H.S., Int. J. Hydrogen Energy, 2016, vol. 41, pp. 16129–16134. https://doi.org/10.1016/j.ijhydene.2016.04.240
Auer, F., Blaumeiser, D., Bauer, T., Bösmann, A., Szesni, N., Libuda, J., and Wasserscheid, P., Catal. Sci. Technol., 2019, vol. 9, pp. 3537–3547. https://doi.org/10.1039/C9CY00817A
Brückner, N., Obesser, K., Bösmann, A., Teichmann, D., Arlt, W., Dungs, J., and Wasserscheid, P., ChemSusChem., 2014, vol. 7, pp. 229–235. https://doi.org/10.1002/cssc.201300426
Xue, W., Liu, X., Mao, B., Liu, H., Qiu, M., Yang, C., Chen, X., and Sun, Y., Chem. Eng. J., 2021, vol. 421. ID 127781. https://doi.org/10.1016/j.cej.2020.127781
Fei, S., Han, B., Li, L., Mei, P., Zhu, T., Yang, M., and Cheng, H., Int. J. Hydrogen Energy., 2017, vol. 42, pp. 25942–25950. https://doi.org/10.1016/j.ijhydene.2017.08.204
Knyazeva, M.I., Panyukova, D.I., Kuchinskaya, T.S., Kulikov, A.B., and Maximov, A.L., Petrol. Chem., 2019, vol. 59, no. 12, pp. 1285–1292. https://doi.org/10.1134/S0965544119120065
Su, X., An, P., Gao, J.,Wang, R., Zhang, Y., Li, X., Zhao, Y., Liu, Y., Ma, X., and Sun, M., Chin. J. Chem. Eng., 2020, vol. 28, pp. 2566–2576. https://doi.org/10.1016/j.cjche.2020.05.010
Naseri, H., Mazloom, G., and Akbari, A., Microp. Mesop. Mater., 2021, vol. 325. ID 111341. https://doi.org/10.1016/j.micromeso.2021.111341
Sizova, I.A., Serdyukov, S.I., and Maksimov, A.L., Petrol. Chem., 2015, vol. 55, no. 4, pp. 470–480. https://doi.org/10.1134/S0965544115060110
Vutolkina, A.V., Glotov, A.P., Zanina, A.V., Makhmutov, D.F., Maximov, A.L., Egazar’yants, S.V., and Karakhanov, E.A., Catal. Today, 2019, vol. 329, pp. 156–166. https://doi.org/10.1016/j.cattod.2018.11.030
Vutolkina, A., Glotov, A., Baygildin, I., Talanova, M., Maximov, A., and Karakhanov, E., Pure Appl. Chem., 2020, vol. 92, pp. 949–966. https://doi.org/10.1515/pac-2019-1115
Vutolkina, A., Baygildin, I., Glotov, A., Cherednichenko, K., Maximov, A., and Karakhanov, E., Appl. Catal. B, 2021, vol. 282. ID 119616. https://doi.org/10.1016/j.apcatb.2020.119616
Knyazeva, M.I., Panyukova, D.I., and Maximov, A.L., Petrol. Chem., 2019, vol. 59, no. 5, pp. 504–510. https://doi.org/10.1134/S0965544119050049
Zakharyan, E.M., Onishchenko, M.I., and Maksimov, A.L., Petrol. Chem., 2018, vol. 58, no. 1, pp. 22–31. https://doi.org/10.1134/S0965544118010164
Li, L., Rong, Z., Du, W., and Ma, S., ChemCatChem., 2009, vol. 1, pp. 369–371 https://doi.org/10.1002/cctc.200900141
Funding
The study was financially supported by the Russian Science Foundation (project no. 21-79-10140, https://rscf.ru/project/21-79-10140/) in the part of preparing and studying the catalysts and as part of State Plan for the Topchiev Institute of Petrochemical Synthesis, Russian Academy of Sciences.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
A.L. Maximov is the Editor-in-Chief, and E.A. Karakhanov, a member of the Editorial Board of Nanogeterogennyi Kataliz journal. The other authors declare no conflict of interest requiring disclosure in this article.
Rights and permissions
Open Access. This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Baigildin, I.G., Karakhanov, E.A., Maximov, A.L. et al. Biphenyl Hydrogenation with Syngas for Hydrogen Purification and Transportation: Performance of Dispersed Catalytic Systems Based on Transition Metal Sulfides. Pet. Chem. 61, 1131–1137 (2021). https://doi.org/10.1134/S0965544121100078
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0965544121100078