
Abstract
Polynuclear bismuth(III) iodide complexes with 1-ethyl-4-dimethylaminopyridinium (1-EtDMAP)4[Bi8I28] (1) and (1-EtDMAP)BiI4 (2) have been obtained by the reactions of bismuth(III) iodide with an organic iodide salt cations in organic solvents and characterized by X-ray diffraction. The optical properties and thermal stability of the obtained compounds have been studied.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Interest in the chemistry of halide complexes of p‑elements is largely due to the variety of their structural types [1]. For example, halogen bismuthate anions can have molecular structure with a nuclearity from 1 to 8 [2–11]. A large number of complexes with polymer anions are also known, usually one-dimensional [12–19]; examples of two-dimensional anions are extremely rare [20]. On the other hand, halogenated metals [21–23] can possess a number of physicochemical properties that are of interest from the point of view of materials science. These include, in particular, thermochromism [24–30] and photochromism [31, 32]. The most actively developing areas include the study of solar cells and photodetectors based on iodometallates, especially iodoplumbates(II) [33–36]. The latter circumstance predetermines the increased interest in iodide complexes. However, as it was repeatedly noted earlier [1], there are still no known approaches that allow one to obtain compounds of this class with a pre-planned structure. Screening using a wide range of organic cations as salt precursors remains the most rational way to search for halogenated metalates that are promising from the point of view of materials science.
In this work, we obtained and structurally characterized two new bismuth iodide complexes with 1-ethyl-4-dimethylaminopyridinium cation (-EtDMAP), namely (1-EtDMAP)4[Bi8I28] (1) and (1-EtDMAP)BiI4 (2). Their structure and preparation conditions, as well as optical properties and thermal stability are discussed.
EXPERIMENTAL
Compounds 1 and 2 were synthesized in air. Reagents of the chemically pure grade (Russian State Standard) were obtained from commercial sources and used without further purification. 1-EtDMAP iodide salt was obtained by the reaction of 4-dimethylaminopyridine and ethyl iodide (1 : 1) in CH3CN (reflux, 18 h, >90% yield); the purity was verified by 1H NMR spectroscopy and elemental analysis.
Synthesis of (1-EtDMAP)4[Bi8I28] (1). Weighed portions BiI3 (100 mg, 0.17 mmol) and 1-EtDMAP-I (24 mg, 0.8 mmol) were dissolved in CH3CN (15 mL) when heating to 80°C for 1 h. After dissolution, the mixture was slowly cooled to room temperature and the solvent was evaporated. After partial evaporation of the solvent at room temperature, dark red crystals were obtained. Yield, 49%.
For C36H60N8Bi8I28 anal. calcd. (%): C, 7.4; H, 1.0; N, 1.9.
Found (%): C, 7.7; H, 1.0; N, 2.0.
Synthesis of (1-EtDMAP)BiI4 (2). Weighed portions of BiI3 (59 mg, 0.1 mmol) and 1-EtDMAP-I (28 mg, 0.1 mmol) were dissolved in 3 mL of a mixture of CH3CN/acetone (1 : 1) solvents when heating to 80°C for 1 h. After dissolution, the mixture was slowly cooled to room temperature and kept for a day. Dark red crystals were obtained. Yield, 52%.
For C9H15N2BiI4 anal. calcd. (%): C, 12.5; H, 1.7; N, 3.2.
Found (%): C, 12.7; H, 1.5; N, 3.0.
X-ray diffraction. Diffraction data for single crystals of compounds 1 and 2 were obtained at 130 K on an Agilent Xcalibur automatic diffractometer equipped with an AtlasS2 two-coordinate detector (graphite monochromator, λ(MoKα) = 0.71073 Å, ω‑scan). Integration, absorption corrections, and unit cell parameters were determined using the CrysAlisPro software package. The crystal structures were solved using the SHELXT program and refined by full-matrix least squares in the anisotropic (except for hydrogen atoms) approximation using the SHELXL program [37]. The positions of hydrogen atoms were calculated geometrically and refined according to the “riding” model. Crystallographic data and details of diffraction experiments are given in Table 1. Complete tables of interatomic distances and bond angles, atomic coordinates, and atomic displacement parameters have been deposited with the Cambridge Crystallographic Data Center (CCDC nos. 2068994, 2068995).
X-ray powder diffraction studies of polycrystals was carried out on a Shimadzu XRD-7000 diffractometer (CuKα radiation, Ni-filter, OneSight linear detector, 2θ angle range 5°–50°, 2θ step 0.0143°, accumulation at a point 2 s). Samples for the study were prepared as follows: polycrystals were ground in an agate mortar in the presence of heptane, the resulting suspension was applied to the polished side of a standard quartz cell. After drying of heptane, the sample was a thin even layer (~100 μm thick).
Thermogravimetric analysis was carried out on a TG 209 F1 Iris thermal balance (Germany). The measurements were carried out in a flow of helium in the temperature range 30–450°C at a heating rate of 10 K/min in open aluminum crucibles.
Optical properties. The diffuse reflectance spectra of the powders were recorded using a spectrophotometric system consisting of a Kolibri-2 spectrometer (VMC Optoelektronika, Russia), an Avantes FCR-7UVIR400-2-ME-HT reflection/backscattering probe, and an AvaLight-DHS deuterium-tungsten lamp (Avantes, Netherlands). The spectra were recorded in the wavelength range of 300–1000 nm at room temperature relative to the standard of 100% reflection which was powder barium sulfate BaSO4.
RESULTS AND DISCUSSION
As noted earlier [1], the factors affecting the composition and structure of metal halides include the nature of the cation, the salt of which is used in the synthesis, as well as the ratio of reagents and the nature of the solvent [38]. The role of the latter factor has been studied scarcely, although individual examples demonstrating its importance have been described [39].
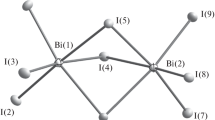
When the reaction was carried out in an acetonitrile solution at a reagent ratio of 2 : 1 (BiI3 : CatI), iodobismuthate 1 with a discrete octanuclear anion was obtained (Fig. 1). Complex compounds with an anion of a similar structure are already known [40, 41], although they are few in number. In this anion, eight bismuth atoms are located on two parallel straight lines and are linked by μ2- and μ3-bridging iodide ligands. The lengths of the Bi–Iterm, Bi–μ2-I, and Bi–μ3-I bonds in 1 are in the ranges 2.848–2.904, 2.931–3.457, and 3.059–3.455 Å, respectively.

Structure of anion [α-Bi8I28]4– in compound 1.
When carrying out the reaction in CH3CN with a different ratio of reagents (1 : 1), we failed to obtain single crystals suitable for X-ray diffraction; however, they were obtained using a mixture of acetone and acetonitrile. According to the results of X-ray diffraction, the anion in 2 has a polymer structure (Fig. 2). This structural type (type E according to our proposed classification [1]) occurs quite often [42, 43]. In the structure of this anion, the {BiI6} octahedra are linked by two pairs of μ2-bridging ligands (Fig. 2) and form an endless polymer chain. The lengths of the Bi–Iterm and Bi–μ2-I bonds in 2 fall in the ranges 2.942–2.919 and 3.038–3.282 Å, respectively. Packings in crystals 1 and 2 are shown in Figs. 3 and 4, respectively.

Structure of anion [BiI4]– in compound 2.

Crystal packing in the structure of 1.

Crystal packing in the structure of 2.
According to X-ray powder diffraction data, samples 1 and 2 are single-phase, which made it possible to study their thermal stability and optical properties. According to TGA data (Fig. 5), both compounds decompose at temperatures >300°C, but 1 demonstrates higher stability. Diffuse reflectance spectra are shown in Fig. 6. They have a clear absorption edge and are similar to each other. To determine the band gap, these spectra were recalculated into absorption spectra using the Kubelka–Munk formula. The band gap was calculated from the absorption spectra in the Tauz coordinates and amounted to 1.92 and 1.93 eV for 1 and 2, respectively, which is comparable with the data [44, 45].

Thermal analysis data for compounds 1 (black) and 2 (red).

Diffuse reflectance spectra of compounds 1 (blue) and 2 (red).
REFERENCES
S. A. Adonin, M. N. Sokolov, and V. P. Fedin, Coord. Chem. Rev. 312, 1 (2016). https://doi.org/10.1016/J.CCR.2015.10.010
H. Eickmeier, B. Jaschinski, A. Hepp, et al., Z. Naturforsch, Sect. B 54, 305 (1999).
M. A. Tershansy, A. M. Goforth, M. D. Smith, et al., Acta Crystallogr., Sect. E 62, M3269 (2006). https://doi.org/10.1107/S1600536806043960
V. V. Sharutin, I. V. Egorova, M. V. Levchuk, et al., Russ. J. Coord. Chem. 28, 613 (2002). https://doi.org/10.1023/A:1020082731096
V. V. Sharutin, I. V. Yegorova, N. N. Klepikov, et al., Russ. J. Inorg. Chem. 54, 52 (2009). https://doi.org/10.1134/S0036023609010124
A. M. Goforth, M. A. Tershansy, M. D. Smith, et al., Acta Crystallogr., Sect. C 62, M381 (2006). https://doi.org/10.1107/S0108270106025972
A. M. Goforth, L. R. Peterson, M. D. Smith, et al., J. Solid State Chem. 178, 3529 (2005). https://doi.org/10.1016/j.jssc.2005.09.010
K. Y. Monakhov, C. Gourlaouen, R. Pattacini, et al., Inorg. Chem. 51, 1562 (2012). https://doi.org/10.1021/ic201859c
N. A. Yelovik, T. A. Shestimerova, M. A. Bykov, et al., Russ. Chem. Bull. 66, 1196 (2017). https://doi.org/10.1007/s11172-017-1872-y
T. A. Shestimerova, A. V. Mironov, M. A. Bykov, et al., Cryst. Growth Des. 20, 87 (2020). https://doi.org/10.1021/acs.cgd.9b00636
P. A. Buikin, A. Y. Rudenko, A. B. Ilyukhin, et al., Russ. J. Coord. Chem. 46, 111 (2020). https://doi.org/10.1134/S1070328420020049
A. N. Usoltsev, M. Elshobaki, S. A. Adonin, et al., J. Mater. Chem. A 7, 5957 (2019). https://doi.org/10.1039/C8TA09204D
S. Pandey, A. P. Andrews, and A. Venugopal, Dalton Trans. 1, 8705 (2016). https://doi.org/10.1039/c6dt01032f
G. Xu, G.-C. Guo, M.-S. Wang, et al., Angew. Chem., Int. Ed. 46, 3249 (2007). https://doi.org/10.1002/anie.200700122
J. Heine, Dalton Trans. 10069 (2015). https://doi.org/10.1039/c5dt00813a
T. A. Shestimerova, N. A. Yelavik, A. V. Mironov, et al., Inorg. Chem. 57, 4077 (2018). https://doi.org/10.1021/acs.inorgchem.8b00265
T. A. Shestimerova, N. A. Golubev, N. A. Yelavik, et al., Cryst. Growth Des. 18, 2572 (2018). https://doi.org/10.1021/acs.cgd.8b00179
T. A. Shestimerova, A. V. Mironov, M. A. Bykov, et al., Molecules 25, 2765 (2020).
N. A. Yelovik, A. V. Mironov, M. A. Bykov, et al., Inorg. Chem. 55, 4132 (2016). https://doi.org/10.1021/acs.inorgchem.5b02729
D. B. Mitzi, Inorg. Chem. 39, 6107 (2000). https://doi.org/10.1021/ic000794i
O. V. Rudnitskaya, E. K. Kultyshkina, E. V. Dobrokhotova, et al., J. Struct. Chem. 60, 1086 (2019). https://doi.org/10.1134/S0022476619070096
V. V. Sharutin, O. K. Sharutina, and E. V. Lobanova, Russ. J. Inorg. Chem. 65, 870 (2020). https://doi.org/10.1134/S0036023620060200
V. V. Sharutin, O. K. Sharutina, and V. S. Senchurin, Russ. J. Inorg. Chem. 65, 1712 (2020). https://doi.org/10.1134/S0036023620110170
V. R. Shayapov, A. N. Usoltsev, S. A. Adonin, et al., New J. Chem. 43, 3927 (2019). https://doi.org/10.1039/C9NJ00320G
A. Gagor, M. Weclawik, B. Bondzior, et al., CrystEngComm 17, 3286 (2015). https://doi.org/10.1039/C5CE00046G
D.-H. Wang, L.-M. Zhao, X.-Y. Lin, et al., Inorg. Chem. Front. 5, 1162 (2018). https://doi.org/10.1039/C7QI00755H
B. V. Bukvetskii, T. V. Sedakova, and A. G. Mirochnik, Russ. J. Coord. Chem. 38, 106 (2012). https://doi.org/10.1134/S1070328412020017
B. V. Bukvetskii, T. V. Sedakova, and A. G. Mirochnik, Russ. J. Inorg. Chem. 56, 213 (2011). https://doi.org/10.1134/S0036023611020045
B. V. Bukvetskii, T. V. Sedakova, and A. G. Mirochnik, Russ. J. Coord. Chem. 36, 651 (2010). https://doi.org/10.1134/S1070328410090034
P. Wang, Z. R. Chen, and H. H. Li, J. Clust. Sci. 31, 943 (2020). https://doi.org/10.1007/s10876-019-01699-1
R.-G. Lin, G. Xu, G. Lu, et al., Inorg. Chem. 53, 5538 (2014). https://doi.org/10.1021/ic5002144
J.-J. Shen, X.-X. Li, T.-L. Yu, et al., Inorg. Chem. 55, 8271 (2016). https://doi.org/10.1021/acs.inorgchem.6b01599
A. A. Petrov, I. P. Sokolova, N. A. Belich, et al., J. Phys. Chem. C 121, 20739 (2017). https://doi.org/10.1021/acs.jpcc.7b08468
S. A. Fateev, A. A. Petrov, V. N. Khrustalev, et al., Chem. Mater. 30, 5237 (2018). https://doi.org/10.1021/acs.chemmater.8b01906
N. A. Belich, A. S. Tychinina, V. V. Kuznetsov, et al., Mendeleev Commun. 28, 487 (2018). https://doi.org/10.1016/j.mencom.2018.09.011
E. I. Marchenko, S. A. Fateev, A. A. Petrov, et al., J. Phys. Chem. C 123, 26036 (2019). https://doi.org/10.1021/acs.jpcc.9b08995
G. M. Sheldrick, Acta Crystallogr., Sect. C 71, 3 (2015). https://doi.org/10.1107/S2053229614024218
A. N. Usoltsev, N. A. Korobeynikov, A. S. Novikov, et al., Inorg. Chem. 59, 17320 (2020). https://doi.org/10.1021/acs.inorgchem.0c02599
I. A. Ahmed, R. Blachnik, and H. Reuter, Z. Anorg. Allg. Chem. 627, 2057 (2001). https://doi.org/10.1002/1521-3749(200109)627:9<2057::AID-ZAAC2057>3.0.CO;2-7
V. V. Sharutin, I. V. Egorova, N. N. Klepikov, et al., Russ. J. Inorg. Chem. 54, 239 (2009). https://doi.org/10.1134/S0036023609020120
H. Krautscheid, Z. Anorg. Allg. Chem. 621, 2049 (1995). https://doi.org/10.1002/zaac.19956211212
W. Bi, N. Leblanc, N. Mercier, et al., Chem. Mater. 21, 4099 (2009). https://doi.org/10.1021/cm9016003
N. Leblanc, N. Mercier, M. Allain, et al., J. Solid State Chem. 195, 140 (2012). https://doi.org/10.1016/j.jssc.2012.03.020
Z.-P. Zhang, Q.-Y. Feng, Q.-L. Wang, et al., J. Clust. Sci. 29, 367 (2018). https://doi.org/10.1007/s10876-018-1339-9
Z. P. Zhang, Q. Y. Feng, Y. L. Wei, et al., J. Clust. Sci. 29, 725 (2018). https://doi.org/10.1007/s10876-018-1397-z
Funding
This work was supported by the Russian Science Foundation (project no. 18-73-10040).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflicts of interest.
Additional information
Translated by V. Avdeeva
Rights and permissions
Open Access. This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Usol’tsev, A.N., Shentseva, I.A., Shayapov, V.R. et al. Bismuth(III) Iodide Complexes with 1-Ethyl-4-Dimethylaminopyridinium: Structure, Thermal Stability, and Optical Properties. Russ. J. Inorg. Chem. 66, 1482–1487 (2021). https://doi.org/10.1134/S0036023621100193
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0036023621100193