
Abstract
Bacteriophages—viruses that infect bacterial cells—are the most abundant biological entities on Earth. The use of phages in fundamental research and industry requires tools for precise manipulation of their genomes. Yet, compared to bacterial genome engineering, modification of phage genomes is challenging because of the lack of selective markers and thus requires laborious screenings of recombinant/mutated phage variants. The development of the CRISPR-Cas technologies allowed to solve this issue by the implementation of negative selection that eliminates the parental phage genomes. In this manuscript, we summarize current methods of phage genome engineering and their coupling with CRISPR-Cas technologies. We also provide examples of our successful application of these methods for introduction of specific insertions, deletions, and point mutations in the genomes of model Escherichia coli lytic phages T7, T5, and T3.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Generation of mutants lies at the core of the modern biology reductionist approach and represents the first stage in any study of biological function of DNA sequence of interest [1, 2]. While early works relied on non-targeted mutagenesis induced by X-rays or chemical compounds, the development of genetic engineering allowed to precisely mutate specific genes. In particular, in bacteria, the recombineering approach, which is based on the homologous recombination mechanism, is suitable for the introduction of desired mutations from plasmid or oligonucleotide templates [3–5]. Screening of recombinants is usually carried out with the use of positive selection markers, such as antibiotic resistance genes, that are introduced in the modified genome during recombination.
Standard recombineering can be used to engineer genomes of temperate phages, while they reside in the bacterial host as stable lysogens or episomes [6–8]. However, phages with lytic life cycle could not be subjected to positive selection with markers selecting for bacterial growth. Further, rapid DNA replication typical for lytic phages limits the time-window for efficient recombination, which should occur at the early stages of infection before additional copies of viral genome are synthesized. Phage genomes are compact and do not tolerate the introduction of large insertions (which may be necessary, for example, when constructing gene fusions) that affect the packaging of DNA in the virions. To overcome this, non-essential regions of phage genome can be identified and deleted before useful cargo DNA segments can be introduced [9, 10]. However, though large-scale screens have been carried out only for a few model phages [11–13], it is expected that a large proportion of viral genes are essential. Since many phages have genes organized in long operons, one can expect polar effects upon introduction of changes in upstream genes [14]. To overcome this, mutations introduced in phage genomes need to be scarless, which may require complicated additional steps [3, 7, 8].
Why would one need to edit a genome of a lytic phage? Besides obvious tasks of studying functions of viral genes, genome editing opens up opportunities for the development and fine-tuning of phages for various practical applications. With the increasing dangers of the antibiotic-resistance crisis, phages once again are gaining attention as potential antimicrobial agents [15, 16]. Though standardized clinical trials carried so far demonstrated limited efficiency of phage therapies, they proved the safety of such treatments and highlighted problems associated with large-scale phage applications [17–20]. A few cases of successful applications of phages for personalized treatments of Acinetobacter baumannii, Pseudomonas aeruginosa, and Mycobacterium abscessus infections were reported [21–23]. An ideal phage for therapeutic purposes should possess a series of features, such as the lack of gene products toxic to humans, efficient recognition of a specific bacterial pathogen and killing of its different forms (including biofilms), scalable production, and long-term stability. These features can be enhanced or engineered using genome editing [24, 25]. As a long-term prospect, one can envision synthetic modular phages constructed with desired properties. Removal of non-essential genes solves the problem of most toxin genes, which appear to be dispensable for infection, and releases space in the genome for the introduction of required cargo genes or labels, such as fluorescent proteins. Simultaneous phage/antimicrobials treatment proved to be more efficient [26, 27] and phages can be designed to express bacteria-killing cargo such as colicins during infection [28, 29]. In a similar manner, phages can be engineered to express enzymes degrading extracellular matrix, capsular polymers or bacterial cell wall, which improves the efficiency of biofilms treatment [30, 31]. The key step defining the host range of the phage and efficiency of its infection is the recognition of a receptor on the surface of the cell. Resistant bacteria often arise due to mutations in receptor genes [32, 33]. Genetic engineering has been applied to modulate receptor binding proteins of phages to extend their host range and similar principles could help overcome evolved bacterial resistance [34–38]. Besides therapeutic applications, phage genome engineering is relevant for construction of phage-based bacterial biosensors and development of vaccines [10, 39–41].
CRISPR-Cas systems (from Clustered Regularly Interspaced Palindromic Repeats and CRISPR-associated proteins) provide RNA-based adaptive immunity to prokaryotes by targeting specific DNA (or sometimes RNA) sites [42, 43]. In most CRISPR-Cas systems, annealing of a segment of guide RNA (gRNA) bound by Cas proteins to a complementary DNA target (protospacer) leads to endonucleolytic cleavage of the latter. Since gRNA can be easily programmed to target any desired DNA region, CRISPR–Cas systems were employed for the development of a plethora of gene editing tools [44, 45]. Class II CRISPR–Cas systems rely on a single Cas protein (such as Cas9, Cas12 or Cas13 in Types II, V and VI, respectively) for target recognition and cleavage and thus became systems of choice for most applications [46–48]. The basic principle of CRISPR-induced gene disruption relies on the fact that dsDNA breaks introduced by the action of Cas proteins activate host DNA repair mechanisms [49] such as NHEJ (non-homologous end joining) or HDR (homology-directed repair). Error-prone repair of a double-stranded break introduced in the targeted sequence eliminates the gRNA recognition site, while non-mutated DNA molecules are subjected to additional attack as long as the effector Cas protein and gRNA are present (Fig. 1). Screening of surviving clones allows identification of the loss-of-function variants carrying In-Dels in the gene of interest. A more versatile approach involves homologous recombination of DNA containing cleaved target sequence with a template containing a sequence with desired change (Fig. 1), which allows one to introduce any type of mutation (insertion, deletion, or single nucleotide substitution) in a scarless way. In this case, the Cas-gRNA effector plays a dual role of generating hotspots for recombination at required locations and eliminating non-edited sequences.

CRISPR–Cas guided mutagenesis. A Cas–gRNA complex recognizes target DNA (protospacer) with correct Protospacer Adjacent Motif (PAM) and introduces a double-stranded break that can be either repaired through NHEJ or through homologous recombination.
The negative selection coupled with recombination mediated by Cas-gRNA constitutes a powerful approach for phage genome engineering significantly simplifying screening for desired mutants. However, phages evolved multiple mechanisms to inhibit immunity systems of their hosts, which may complicate genome editing and CRISPR targeting. Examples include, among others, DNA modifications, such as glycosylation of cytosines, which decrease the efficiency of Cas9 cleavage [50, 51]; production of anti-CRISPR or DNA-protecting proteins [52, 53]; creation of pseudo-nucleus structures that physically block access of host proteins to viral DNA and inhibit DNA-targeting CRISPR systems [54, 55]. Genome editing for such phages might require specific strategies like the use of RNA-targeting Cas effectors [56].
Below we summarize current methods available for genome editing of lytic phages.
METHODS FOR GENETIC ENGINEERING OF PHAGE GENOMES
In vitro phage “rebooting” strategies. The most direct way of phage genomes manipulation is in vitro, using purified or synthesized DNA material. For obvious reasons, such methods are limited by the phage genome size. A small ssDNA phage φX174 with its ~5.6 kB genome was the first to be assembled from scratch using synthetic oligonucleotides [57]. Development of in vitro DNA assembly methods, like Gibson Assembly or Golden Gate Cloning, allowed approaching the construction of large DNA molecules from multiple fragments which is suitable for the whole-genome assembly of medium sized (tens of thousands of base pairs) phages (Fig. 2) [35, 58–60]. In vitro assembly allows one to generate all types of mutations without the introduction of selection markers and does not require plaques screening (reviewed in [25, 61]). It can also be used for generation of mutant phage libraries. For example, error-prone amplification of genomic DNA region encoding receptor binding protein followed by in vitro assembly with the rest of the genome allowed to generate a series of Listeria phage PSA mutants with extended host range [35].

In vitro phage genome assembly and reactivation workflow.
Direct transformation of reconstructed whole viral genomes into host cell could be problematic and thus different variants of in vivo assembly/amplification could be deployed. In the work of Ando et al., E. coli, Salmonella, Pseudomonas, and Klebsiella phages were assembled into Yeast Artificial Chromosomes (YACs) after transformation of PCR products into yeast cells. Assembled YACs were next transformed in E. coli resulting in production of infectious phage, an approach that was called “rebooting” or “reactivation of the synthetic genome” [62, 63]. Rebooting of phages infecting Gram-positive bacteria requires transformation of DNA fragments into cell-wall deficient L-form cells. This method was shown to be applicable for the Listeria, Bacillus, and Staphylococcus phages [64]. Phage particles can also be produced in cell-free transcription-translation coupled systems (TXTL) programmed with phage genomic DNA [65, 66]. Infectious particles production for phages as large as T4, was demonstrated in TXTL systems, providing apparently the most straightforward approach for construction of modified phages directly from the in vitro-assembled genomes [67, 68] (Fig. 2).
In vivo recombination-based methods. In vivo methods rely on homologous recombination (HDR) and the allelic exchange between DNA molecules carrying flanking homologous regions and different central regions (Fig. 3). While some phages fully depend on the host recombination functions [69], others encode their own recombination proteins that outperform the host RecA-mediated pathway and are more tolerant to mismatches [70–73]. Thus, the mere presence of a template with desired mutation and homology regions in a cell infected with a phage can be sufficient for production of recombinant phage progeny. Such a template could be a plasmid, a PCR product, or even a single-stranded oligonucleotide of sufficient length electroporated into the infected cell [74, 75]. To enhance the efficiency of recombination, overexpression of recombination-stimulating proteins, such as Exo, Beta, Gam components of the phage λ Red system or RecET-like proteins in infected cells has been reported [3, 76]. The λ Red recombination system is by far the most popular choice for recombineering [3, 5, 7]. Exo is a 5'→3' exonuclease that generates free single-stranded 3'-ends, which interact with the ssDNA-binding protein Beta and recombination most likely occurs when the ssDNA intermediates anneal to the lagging DNA strand during replication. The Gam protein protects the DNA template from degradation by inhibiting host RecBCD machinery [77]. Only the Beta protein is required for recombineering with ssDNA and different viral ssDNA annealing proteins were shown to enhance recombination efficiency in vivo [73]. As an alternative, ssDNA can be generated in the cell using retron elements. Retrons are RNA–DNA hybrids synthesized by reverse transcriptase and can be introduced on plasmids providing a source of programmable ssDNA [78]. Retron-mediated mutagenesis has been applied to modify E. coli phage T5 [53]. One of the most efficient methods of phage recombineering—BRED (Bacteriophage Recombineering of Electroporated DNA) exploits simultaneous electroporation of purified phage genome and recombination template into host cells overexpressing recombination-stimulating proteins (Fig. 3). It was successfully used for editing of phages infecting Mycobacteria, Escherichia, Salmonella, and Klebsiella [74, 79–82]. A simplified protocol—BRIP (Bacteriophage Recombineering with Infectious Particles)—separates the electroporation and phage infection steps, which is less efficient but allows one to avoid the need to purifying phage genomes [75, 83]. It should be noted that these methods are limited to bacteria for which efficient transformation protocols are available.

Methods of in vivo phage genome editing through recombination. Recombination can be carried with plasmid template, ssDNA oligos electroporated into the cell, or ssDNA in a form of plasmid-encoded retorn elements. Expression of the λ Red system enhances efficiency of recombination.
Classical methods for the selection of recombinants. As a result of in vivo recombination, a mixed progeny of the wild-type and mutated phage variants will arise, and identification of the latter can be challenging. The simplest but laborious approach involves screening of individual plaques through PCR with mutation-specific primers. Single-nucleotide substitutions could be detected with the MAMA-PCR when the 3'-terminal base of the primer anneals to the mutated position but not to the wild-type sequence [53, 84]. Amplification-dilution screening could be applied to enhance the efficiency of recombinants selection: in this approach a phage lysate carrying possible mutants is serially diluted and mixed with host bacterial culture in a 96‑well plates; PCR screening of phages reproduced in each well allows to determine wells with highest concentration of mutants, which increases the chances of selecting desired rare variants in the following single plaques screening [53]. Phage plaques carrying desired mutations can also be identified through hybridization with fluorescently- or radioactively-labelled oligonucleotide probes. Even point substitutions can be identified by gradually increasing the annealing temperature to remove non-specific binding [85]. When efficiency of recombination is low, the positive selection methods help visually discriminate wild-type and mutant variants or prevent reproduction of non-mutated phages. When tolerated by the phage, introduced mutations could be coupled with genes encoding fluorescent proteins or LacZ markers, which allows identification of recombinant plaques under a fluorescent microscope or on X-gal supplemented agar plates [53, 83, 86]. Sometimes proteins dispensable for host survival could be essential for the phage propagation, for example, E. coli TrxA (thioredoxin) is required for the activity of phage T7 DNA polymerase [87, 88]. This allows one to use the trxA gene as a positive selection marker when the phage is propagated on a ΔtrxA E. coli host [89–91]. A similar trxA insertion strategy was applied for selection of non-T7-like phages from the Felixounovirus genus [92].
CRISPR–Cas-based phage genome editing and selection. The Cas9 nuclease from the Streptococcus pyogenes (SpCas9) was used in different Gram-positive and Gram-negative bacteria species, such as E. coli, Streptococcus thermophilus, Lactococcus lactis, Mycobacterium smegmatis, B. subtilis and became a preferable tool for phage genome editing [13, 51, 83, 93–95]. To avoid cleavage of CRISPR loci, which contains spacers complementary to gRNA, many CRISPR–Cas systems rely on a specific short sequence—PAM (Protospacer Adjacent Motif, NGG in the case of SpCas9). PAM should be located at a flank of DNA site complementary to gRNA to allow it’s recognition and cleavage; PAM is absent in gRNA-encoding loci [96]. Thus, a gRNA design should begin with the screening for PAM sequences in the phage genome region of interest. Several different gRNAs targeting the same gene are usually used since their efficiency of target recognition/cleavage can vary by an order of magnitude or more. Many tools are available for prediction of gRNA editing efficiency [97–99]. Expression of Cas9 along with a gRNA targeting the wild-type genomic sequence provides means of negative selection strong enough to completely eliminate the parental phage. Viruses can escape CRISPR interference through accumulation of mutations in the PAM or protospacer region that either arise randomly or accumulate, for example, as a result of error-prone NHEJ (Fig. 1). A recent study investigated a large set of B. subtilis phage Goe1 escaper mutants, using gRNAs designed to target each gene of its 18 kBp genome, which allowed to determine viral genes essentiality and tolerance to mutations [13].
CRISPR editing is much more efficient when cells are provided with a template for homologous recombination and with recombineering proteins [82, 83]. To introduce deletions, insertions or substitutions, cells are transformed with a template carrying the desired sequence flanked by 100–200 bp 5' and 3' homology regions identical to the wild-type viral sequences (Fig. 4a). A gRNA is designed in a way that recombination leads to the loss of the targeted protospacer in phage genome; point mutations are placed in the PAM or protospacer positions that when altered prevent the recognition by the Cas9–gRNA complex. Thus, recombinant phages will evade CRISPR restriction, while wild-type, non-recombinant variant will be eliminated. The first use of CRISPR-assisted recombination involved gene deletions in E. coli T7 phage with the native I–E CRISPR–Cas system, which resulted in ~40% efficiency (in terms of the amount of screened plaques containing the desired mutation) [100], and gene deletions or point mutations in S. thermophilus phage 2972 with the use of SpCas9, which provided efficiency close to 100% [93]. Other works relying on I–E or SpCas9 for phage genome editing also reported efficiencies in the range of 50‒100% [94, 101, 102]. When combined with BRED or BRIP methods, SpCas9 counter-selection also significantly improved the efficiency of editing, which approached 100% for majority of deletions introduced in various M. smegmatis phages [83]. The Type V Cas12a effector was successfully applied to edit the E. coli phage T4 genome [10], while Type III system proved to be efficient for engineering staphylococcal phages [102]. A promising approach was recently proposed that relies on an RNA-targeting Type VI Cas13a system [56, 103, 104]. Cas13 does not require PAM for cleavage and allows to target any site in the genome, provided it is transcribed. LbCas13a was employed to enhance selection of edited T4 carrying single codon mutations [104]. Moreover, RNA targeting may help to overcome problems associated with editing of phages bearing modified DNA or those that rely on specialized compartments such as pseudonuclei to separate host defence proteins from phage DNA. In another approach, a gene encoding an anti-CRISPR protein (acrVIA1) was introduced into the genome of P. aeruginosa jumbo phage ΦKZ as a positive selection marker [56]. Introduction of acr gene inhibits Cas13-directed RNA cleavage and protospacers complementary to gRNAs preserved in the recombinant phage, which allows one to edit various loci in the viral genome with a single gRNA, once a relevant recombination template is provided (Fig. 4b).

CRISPR-Cas assisted phage genome editing and selection of phage mutants. (a) Negative selection with Cas9–wild-type phage is eliminated in a result of gRNA–Cas9 cleavage of target DNA, while recombinant phages carrying specific insertions, deletions, or substitutions destroying PAM/protospacer region evade cleavage. (b) An example of positive selection with the acr marker gene that inhibits cleavage by RNA-targeting Cas13. Similar principle can be applied to DNA-targeting Cas effectors.
All systems described above need to be carefully adjusted and optimized for specific applications, phages, and their hosts. Below we describe the results of our own experience of combining SpCas9 negative-selection with λ Red recombineering to introduce insertions, deletions, or point mutations into the genomes of model coliphages T7, T5 and T3.
MATERIALS AND METHODS
SpCas9 gRNAs and their editing efficiencies are summarized in Table 1; constructed plasmids and primers used in this work are listed in Tables S1 and S2 (see Supplementary Information).
Modification of a plasmid encoding λ Red system and guide RNA for phage genome editing. pKDsgRNA-ack plasmid was used to deliver guide RNAs and the λ Red system into bacteria [105]. The plasmid contains the λ Red system under control of arabinose-inducible promoter and gRNA under control of Ptet. The leaky expression of gRNA from Ptet was sufficient and aTc induction was not provided to avoid toxicity associated with gRNA overexpression [105]. The default gRNA sequence of pKDsgRNA-ack was replaced with sequences designed to introduce mutations in the T7, T3, or T5 genomes (Table 1). The gRNA sequences were chosen to contain an appropriately positioned NGG PAM downstream of the targeted complementary site. For introduction of point substitutions, mutations were designed to disrupt the PAM sequence of the targeted protospacer. To replace the gRNA sequence we performed Circular Polymerase Extension Cloning (CPEC) [106] with Q5 DNA Polymerase (New England Biolabs) and primers encoding required gRNA; the vectors next were assembled from linear PCR products using the KLD reaction mixture (New England Biolabs).
Creation of a recombination templates. To replace a region of phage genome with a sequence containing a desired mutation, plasmid templates were used for recombination. The plasmids were created based on the pBAD/His B vector [107], which is compatible with pKDsgRNA-ack and pCas9 [108]. For insertion of a EYFP tag in the T7 genome, the resulting plasmid (Supplementary Information, Table S1) was designed to carry 3' and 5' 155 bp homology regions, flanking the insertion site, while the middle part carried an in-frame fusion of gp10B with EYFP coding sequences. To generate the T3 0.3 gene deletion, a plasmid carrying a fragment of fused 260 bp flanking regions bordering the 0.3 gene in the phage was constructed. All plasmids were prepared using Gibson assembly with HiFi Master Mix (New England Biolabs) [59] from 2 or 3 PCR products amplified with Phusion DNA polymerase (New England Biolabs). To introduce point mutations into the T7 or T5 genomes, a ~600 bp region of interest was first cloned into pBAD/His B vector using Gibson Assembly and desired mutations were introduced using the Q5 Quick Mutagenesis Kit (New England Biolabs).
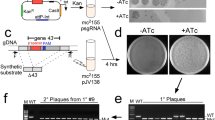
Modification of the phage genome. The pipeline for the phage genome editing and selection of recombinants is provided in the Fig. 5. To perform phage genome editing, three plasmids (pCas9, pBad with appropriate recombination insert, and pKDsgRNA-ack) were introduced in E. coli DH5α (for T7 editing), BW25113 (for T7 and T5 editing) or BL-21 (for T3 editing) [109]. Transformed cells were plated on Petri dishes containing LB 1.2% agar medium (Amresco) with the addition of ampicillin (100 μg/mL), chloramphenicol (50 μg/mL) and spectinomycin (100 μg/ml) and incubated at +37°С. Overnight cultures were obtained from single colonies in liquid LB medium with the addition of appropriate antibiotics at +37°С and moderate shaking. Initial estimation on the efficiency of SpCas9 targeting with specific gRNAs in the absence of recombination template was carried in 96-well plates: mid-log liquid cultures were infected with the phage in a range of Multiplicities of Infection (MOI) and optical density was monitored using EnSpire Multimode Plate Reader (Perkin Elmer). To carry recombination, log-phase cultures were 100-fold diluted in fresh LB medium supplemented with antibiotics and grown till OD600 = 0.3 when the arabinose was added to 0.2%. ~1 hour after λ Red induction cultures were infected with phages at different MOIs and left overnight. Lysates were plated on 0.4% top-agar containing susceptible E. coli BW25113 (or BL-21) cells and plaques developed at 37°С overnight.

A pipeline for the phage genome editing using SpCas9 counter-selection. HA—Homology Arm.
Phage plaques screening. Individual plaques were screened for the presence of desired mutations using PCR with primers specific for the mutated sequence (Supplementary Information, Table S2). For the screening of point substitutions, 3' end of the primer was designed to anneal to the mutated nucleotide and the annealing temperature was sometimes raised up to 72°С to discriminate specific annealing to mutated sequence and non-specific annealing to the wild-type sequene. PCR screens were carried with TaqHS DNA-Polymerase ScreenMix (Evrogen, Russia). All mutations were confirmed by Sanger sequencing using additional pairs of primers annealing ~150 bp away from the introduced mutation site.
To confirm the presence of the fusion gp10B::EYFP protein in the T7 capsid, Western blotting of proteins in the modified phage lysate was performed. The gp10B::EYFP protein was visualized using mouse monoclonal antibodies to EYFP (Living Colors® JL-8, Clontech) in 1:1000 dilution. Samples of phage lysate in serial dilutions were run on a 10% SDS-PAGE gel and then transferred to a nitrocellulose membrane (Amersham, USA). Anti-Mouse Peroxidase antibodies produced in rabbits (Sigma 9044) were used as secondary antibodies. A stained nitrocellulose membrane was visualized using SuperSignal West Pico Chemiluminescent Substrate Kit (Thermo Scientific, USA) in Chemidoc (BioRad, USA).
RESULTS
Introduction of point mutations and fluorescent labelling of the T7 phage. To check the validity of recombineering-assisted SpCas9 modification/selection approach we infected E. coli cultures lacking the pBAD recombination template or carrying three plasmids—pBAD, a plasmid expressing SpCas9, and the pKDsgRNA-ack derivative carrying λ Red system and gRNA_1 targeting the early phage RNA polymerase gene (Fig. 5)—with T7 at different MOIs. Cells were infected with or without λ Red system induction. Our expectation was that at a certain MOI Cas9 targeting will completely inhibit the production of phage in the absence of recombination template, while at conditions of λ Red induction and in the presence of recombination template on pBAD, modified phage progeny will appear and lyse the culture. Indeed, at MOIs below 10–4 the culture remained completely protected in the absence of λ Red induction and without the pBAD recombination template but lyzed when λ Red was induced in the presence of the recombination template plasmid (Fig. 6). The obtained phage lysate was plated on a lawn of E. coli BW25113 and single plaques were screened for the presence of desired mutation. It should be noted, that some gRNAs did not produce the phenotype shown in Fig. 6, however, even in conditions when Cas9 did not protect bacteria from lysis, or when infected cultures with induced and uninduced λ Red system grew apparently in the same way, we were able to select recombinants in a single screening experiment (Table 1).

Growth curves of indicated bacterial cultures infected with phage T7 at MOI = 10–5. To induce λ Red system expression from the pKD vector cultures were supplemented with 0.2% arabinose 30 min prior to infection. Phage was added at t = 0. Each growth curve represents the mean values and standard deviations obtained from three technical replicates.
In our previous work with the Type I BREX defence system from E. coli HS [91, 110] we detected phage T7 escaper mutants that partially overcame the host defence. Genomic sequencing revealed that these mutants lost a BREX methylation site GGTAAG closest to the left (early) end of the phage genome. To confirm this observation and investigate the importance of mutual orientation of BREX sites for efficient defence, we constructed a series of T7 mutants carrying insertions, deletions, or inversions of BREX sites located in the early part of the genome (Table 1, gRNA_1,2,3). BREX sites modifications in recombination templates were designed in such a way that introduced changes disrupted the closely located PAM region in the targeted wild-type phage sequence, to make mutated variants insensitive to SpCas9 cleavage. The same gRNA was used to introduce additional BREX sites in the forward or reverse orientation using different pBAD recombination templates. Overall, 33 mutant phages were generated from parental T7 strains with different genetic backgrounds using a set of 3 distinct gRNAs. PCR screening of lysates obtained at various MOIs confirmed the appearance of desired mutants and, for most cases, the presence of mutations was detected in 50‒100% of single plaques subsequently tested, demonstrating the efficiency of the λ Red assisted SpCa9 phage genome editing (Table 1, Fig. 7a). Using the same approach, we constructed a fluorescently-labelled T7 strain with a C-terminal fusion of the capsid protein gp10B with EYFP [111, 112]. PCR of randomly selected plaques readily confirmed the presence of desired product (Fig. 7b). The fusion was further validated by separating proteins from recombinant T7 lysates by SDS-PAGE followed by Western blot with EYFP-specific antibodies, which confirmed the presence of the gp10B-EYFP fusion protein (Fig. 7c). As a drawback of the method, we note high toxicity of the pKDsgRNA-ack for E. coli. This is apparently unrelated to encoded gRNAs and the efficiency of transformation of the starting plasmid was low even in highly competent bacterial cells— which significantly complicated construction of derivatives encoding required gRNA sequences.

(a) An example of MAMA-PCR with individual plaques and primers specific to mutated BREX site edited with gRNA_2. The product of 339 bp should appear only when a primer anneals to the mutated BREX site. M—DNA ladder, Gene Ruler 100 bp (Thermo Scientific). (b) Confirmation of the EYFP-encoding sequence insertion at the 3' end of T7 gene 10B. The fusion sequence should produce a 1137 bp PCR product. M—DNA ladder, Gene Ruler 1 kb+ (Thermo Scientific). (c) A Western blot of proteins in lysates of engineered T7 gp10B-EYFP produced from 5 randomly picked individual plaques was carried out using EYFP-specific antibodies. The molecular weight of the stained band corresponds to the gp10B-EYFP fusion protein.
Introduction of a short insertion into the First Strand Transfer (FST) region of the T5 genome. Phage T5 injects its genome in two stages: after translocation of the ~10 kBp FST region the injection temporarily stops. The rest of the genome (Second Step Transfer region—SST) enters the cell after an unknown signal, presumably generated by a product of a phage gene introduced during FST, triggers the process [113, 114]. Phage T5 is resistant to many host defences including Type I/II/III Restriction–Modification systems and Types I and II CRISPR–Cas systems when their recognition sites are located in the SST region, but not in the FST region [53, 115, 116]. This suggests that one of the FST gene products could be a broad range anti-restriction protein [53, 116]. To reproduce the restriction-sensitivity phenotype, we employed SpCas9 to introduce an EcoRV recognition site (GATATC) in the non-coding region of the T5 FST. Although the designed gRNA_5 did not protect E. coli from lysis by T5 in liquid culture, screening of individual plaques allowed us to readily recover desired EcoRV-sensitive T5 mutants with ~90% efficiency (Table 1).
Deletion of phage T3 0.3 gene and estimation of the editing efficiency. The 0.3 gene of phage T3 encodes an anti-restriction protein S-adenosyl-methionine (SAM) lyase that inhibits Type I R–M systems by lowering the intracellular concentration of SAM, a co-factor required for restriction [117–119]. To study phenotypes associated with the loss of SAM lyase function, we created a 0.3 gene deletion T3 strain. The pBAD recombineering template carried fused 3' and 5' homology regions upstream and downstream of the 0.3 ORF to completely and scarlessly remove the gene. The deletion of the 459 bp 0.3 sequence allows facile distinction of mutated and parental genomes by PCR. Thus, comparison of efficiencies of recombination at different conditions became possible. Expression of SpCas9 along with gRNA_6 against the 0.3 gene provided ~2 orders of magnitude defence against T3. We have monitored T3 liquid culture infections in a range of MOIs (1 to 10–7) using strains with the recombination template pBAD only (1) or carrying the recombination template and λ Red system in the presence (3) or in the absence (2) of λ Red induction. Cultures additionally carrying pCas9 were also infected to provide counter-selection in the presence (5) or in the absence (4) of λ Red induction. As can be seen from Fig. 8a, only at conditions of negative selection (culures 4 and 5) a PCR amplicon indicative of recombinant T3Δ0.3 became detectable in the lysates of infected cultures. Thus, production of recombinants did not depend on λ Red induction. At these conditions, no amplicon corresponding to wild-type phage was detected, suggesting it’s complete elimination by SpCas9. A PCR screen of 10 individual plaques obtained from culture 4 and 5 lysates confirmed the 100% editing efficiency of the phage genome (Fig. 8b). The difference in λ Red proteins requirement observed in experiments involving T3 and T7 (Fig. 6 vs. Fig. 8) is likely due to differences in the efficiency of negative selection by gRNA used.

(a) Estimation of T3 editing efficiency in different conditions. Results of the PCR reaction with primers specific for the T3 0.3 gene carried after overnight infection of indicated culture with phage T3 in a range of MOI from 1 to 10–7. First 5 rows represent results of the PCR with non-infected culture, followed by 2 rows of PCR products obtained from control gDNA of T3 wild-type (864 bp) or T3 Δ0.3 (406 bp). M—DNA ladder GeneRuler 1 kb + (Thermo Scientific). (b) Screning of individual plaques obtained from lysates of cultures 4 and 5. PCR confirms 100% editing efficiency in conditions tested.
Overall, the results of experiments presented above demonstrate that mutations of all possible types, i.e., point substitutions, insertions, or deletions can be introduced with high efficiency into specific locations of E. coli phages T7, T5, and T3 using recombineering-assisted SpCas9 genome editing and counter-selection. There is still much to be learned from these model phages and new avenues of inquiry into the infection process/interactions with the host are opened up by the facile editing of their genomes.
REFERENCES
Sarkar S. 1998. Genetics and Reductionism. Cambridge Univ. Press.
Williams E.G., Auwerx J. 2015. The convergence of systems and reductionist approaches in complex trait analysis. Cell. 162, 23–32.
Datsenko K.A., Wanner B.L. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97, 6640–6645.
Baba T., Ara T., Hasegawa M., Takai Y., Okumura Y., Baba M., Datsenko K.A., Tomita M., Wanner B.L., Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2, 8–2006.
Thomason L.C., Sawitzke J.A., Li X., Costantino N., Court D.L. 2014. Recombineering: genetic engineering in bacteria using homologous recombination. Curr. Protoc. Mol. Biol. 106, 1–16.
Thomason L.C., Oppenheim A.B., Court D.L. 2009. Modifying bacteriophage λ with recombineering. In Bacteriophages. Springer, pp. 239–251.
Marinelli L.J., Hatfull G.F., Piuri M. 2012. Recombineering: a powerful tool for modification of bacteriophage genomes. Bacteriophage. 2, 5–14.
Piya D., Vara L., Russell W.K., Young R., Gill J.J. 2017. The multicomponent antirestriction system of phage P1 is linked to capsid morphogenesis. Mol. Microbiol. 105, 399–412.
Pires D.P., Monteiro R., Mil-Homens D., Fialho A., Lu T.K., Azeredo J. 2021. Designing P. aeruginosa synthetic phages with reduced genomes. Sci. Rep. 11, 1–10.
Zhu J., Ananthaswamy N., Jain S., Batra H., Tang W.-C., Lewry D.A., Richards M.L., David S.A., Kilgore P.B., Sha J. 2021. A universal bacteriophage T4 nanoparticle platform to design multiplex SARS-CoV-2 vaccine candidates by CRISPR engineering. Sci. Adv. 7, eabh1547.
Dedrick R.M., Marinelli L.J., Newton G.L., Pogliano K., Pogliano J., Hatfull G.F. 2013. Functional requirements for bacteriophage growth: gene essentiality and expression in mycobacteriophage Giles. Mol. Microbiol. 88, 577–589.
Thomas J.A., Benítez Quintana A.D., Bosch M.A., Coll De Peña A., Aguilera E., Coulibaly A., Wu W., Osier M.V., Hudson A.O., Weintraub S.T. 2016. Identification of essential genes in the Salmonella phage SPN3US reveals novel insights into giant phage head structure and assembly. J. Virol. 90, 10284–10298.
Kohm K., Basu S., Nawaz M.M., Hertel R. 2021. Chances and limitations when uncovering essential and non-essential genes of Bacillus subtilis phages with CRISPR–Cas9. Environ. Microbiol. Rep. 13, 934–944.
Ciampi M.S. 2006. Rho-dependent terminators and transcription termination. Microbiology. 152, 2515–2528.
Pirnay J.-P., Ferry T., Resch G. 2022. Recent progress toward the implementation of phage therapy in Western medicine. FEMS Microbiol. Rev. 46, fuab040.
Hatfull G.F., Dedrick R.M., Schooley R.T. 2022. Phage therapy for antibiotic-resistant bacterial infections, Annu. Rev. Med. 73, 197–211.
Jault P., Leclerc T., Jennes S., Pirnay J.P., Que Y.-A., Resch G., Rousseau A.F., Ravat F., Carsin H., Le Floch R. 2019. Efficacy and tolerability of a cocktail of bacteriophages to treat burn wounds infected by Pseudomonas aeruginosa (PhagoBurn): a randomised, controlled, double-blind phase 1/2 trial. Lancet Infect. Dis. 19, 35–45.
Sarker S.A., Sultana S., Reuteler G., Moine D., Descombes P., Charton F., Bourdin G., McCallin S., Ngom-Bru C., Neville T. 2016. Oral phage therapy of acute bacterial diarrhea with two coliphage preparations: a randomized trial in children from Bangladesh. EBioMedicine. 4, 124–137.
Petrovic Fabijan A., Lin R.C.Y., Ho J., Maddocks S., Ben Zakour N.L., Iredell J.R. 2020. Safety of bacteriophage therapy in severe Staphylococcus aureus infection. Nat. Microbiol. 5, 465–472.
Furfaro L.L., Payne M.S., Chang B.J. 2018. Bacteriophage therapy: clinical trials and regulatory hurdles. Front. Cell. Infect. Microbiol. 8, 376. https://doi.org/10.3389/fcimb.2018.00376
Schooley R.T., Biswas B., Gill J.J., Hernandez-Morales A., Lancaster J., Lessor L., Barr J.J., Reed S.L., Rohwer F., Benler S. 2017. Development and use of personalized bacteriophage-based therapeutic cocktails to treat a patient with a disseminated resistant Acinetobacter baumannii infection. Antimicrob. Agents Chemother. 61, e00954-17.
Chan B.K., Turner P.E., Kim S., Mojibian H.R., Elefteriades J.A., Narayan D. 2018. Phage treatment of an aortic graft infected with Pseudomonas aeruginosa. Evol. Med. Public Heal. 2018, 60–66.
Dedrick R.M., Guerrero-Bustamante C.A., Garlena R.A., Russell D.A., Ford K., Harris K., Gilmour K.C., Soothill J., Jacobs-Sera D., Schooley R.T. 2019. Engineered bacteriophages for treatment of a patient with a disseminated drug-resistant Mycobacterium abscessus. Nat. Med. 25, 730–733.
Lenneman B.R., Fernbach J., Loessner M.J., Lu T.K., Kilcher S. 2021. Enhancing phage therapy through synthetic biology and genome engineering. Curr. Opin. Biotechnol. 68, 151–159.
Kilcher S., Loessner M.J. 2019. Engineering bacteriophages as versatile biologics. Trends Microbiol. 27, 355–367.
Ryan E.M., Alkawareek M.Y., Donnelly R.F., Gil-more B.F. 2012. Synergistic phage-antibiotic combinations for the control of Escherichia coli biofilms in vitro. FEMS Immunol. Med. Microbiol. 65, 395–398.
Segall A.M., Roach D.R., Strathdee S.A. 2019. Stronger together? Perspectives on phage-antibiotic synergy in clinical applications of phage therapy. Curr. Opin. Microbiol. 51, 46–50.
Mills S., Ross R.P., Hill C. 2017. Bacteriocins and bacteriophage; a narrow-minded approach to food and gut microbiology. FEMS Microbiol. Rev. 41, S129–S153.
Du J., Meile S., Baggenstos J., Jaeggi T., Piffaretti P., Hunold L., Matter C.I., Leitner L., Kessler T.M., Loessner M.J. 2022. Enhancing bacteriophage therapeutics through in situ production and release of heterologous antimicrobial effectors. bioRxiv.
Lu T.K., Collins J.J. 2007. Dispersing biofilms with engineered enzymatic bacteriophage. Proc. Natl. Acad. Sci. U. S. A. 104, 11197–11202.
São-José C. 2018. Engineering of phage-derived lytic enzymes: improving their potential as antimicrobials. Antibiotics. 7, 29.
Hesse S., Rajaure M., Wall E., Johnson J., Bliskovsky V., Gottesman S., Adhya S. 2020. Phage resistance in multidrug-resistant Klebsiella pneumoniae ST258 evolves via diverse mutations that culminate in impaired adsorption. MBio. 11, e02530-19.
Azam A.H., Tanji Y. 2019. Bacteriophage-host arm race: an update on the mechanism of phage resistance in bacteria and revenge of the phage with the perspective for phage therapy. Appl. Microbiol. Biotechnol. 103, 2121–2131.
Altamirano F.L.G., Barr J.J. 2021. Unlocking the next generation of phage therapy: the key is in the receptors. Curr. Opin. Biotechnol. 68, 115–123.
Dunne M., Rupf B., Tala M., Qabrati X., Ernst P., Shen Y., Sumrall E., Heeb L., Plückthun A., Loessner M.J. 2019. Reprogramming bacteriophage host range through structure-guided design of chimeric receptor binding proteins. Cell Rep. 29, 1336–1350.
Yehl K., Lemire S., Yang A.C., Ando H., Mimee M., Torres M.D.T., de la Fuente-Nunez C., Lu T.K. 2019. Engineering phage host-range and suppressing bacterial resistance through phage tail fiber mutagenesis. Cell. 179, 459–469.
Dunne M., Prokhorov N.S., Loessner M.J., Leiman P.G. 2021. Reprogramming bacteriophage host range: design principles and strategies for engineering receptor binding proteins. Curr. Opin. Biotechnol. 68, 272–281.
Yosef I., Goren M.G., Globus R., Molshanski-Mor S., Qimron U. 2017. Extending the host range of bacteriophage particles for DNA transduction. Mol. Cell. 66, 721–728.
Farooq U., Yang Q., Ullah M.W., Wang S. 2018. Bacterial biosensing: Recent advances in phage-based bioassays and biosensors. Biosens. Bioelectron. 118, 204–216.
Aliakbar Ahovan Z., Hashemi A., De Plano L.M., Gholipourmalekabadi M., Seifalian A. 2020. Bacteriophage based biosensors: trends, outcomes and challenges. Nanomaterials. 10, 501.
Bao Q., Li X., Han G., Zhu Y., Mao C., Yang M. 2019. Phage-based vaccines. Adv. Drug Deliv. Rev. 145, 40–56.
Nussenzweig P.M., Marraffini L.A. 2020. Molecular mechanisms of CRISPR–Cas Immunity in bacteria. Annu. Rev. Genet. 54, 93‒120. https://doi.org/10.1146/annurev-genet-022120-112523
Hille F., Richter H., Wong S.P., Bratovič M., Ressel S., Charpentier E. 2018. The biology of CRISPR–Cas: backward and forward. Cell. 172, 1239–1259.
Liu G., Lin Q., Jin S., Gao C. 2021. The CRISPR–Cas toolbox and gene editing technologies. Mol. Cell. 82 (2), 333‒347. https://doi.org/10.1016/j.molcel.2021.12.002
Knott G.J., Doudna J.A. 2018. CRISPR-Cas guides the future of genetic engineering. Science. 361, 866–869.
Hsu P.D., Lander E.S., Zhang F. 2014. Development and applications of CRISPR–Cas9 for genome engineering. Cell. 157, 1262–1278.
Zetsche B., Heidenreich M., Mohanraju P., Fedorova I., Kneppers J., DeGennaro E.M., Winblad N., Choudhury S.R., Abudayyeh O.O., Gootenberg J.S. 2017. Multiplex gene editing by CRISPR–Cpf1 using a single crRNA array. Nat. Biotechnol. 35, 31–34.
Abudayyeh O.O., Gootenberg J.S., Konermann S., Joung J., Slaymaker I.M., Cox D.B.T., Shmakov S., Makarova K.S., Semenova E., Minakhin L. 2016. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science. 353 (6299), aaf5573. https://doi.org/10.1126/science.aaf5573
Jang H.-K., Song B., Hwang G.-H., Bae S. 2020. Current trends in gene recovery mediated by the CRISPR–Cas system. Exp. Mol. Med. 52, 1016–1027.
Bryson A.L., Hwang Y., Sherrill-Mix S., Wu G.D., Lewis J.D., Black L., Clark T.A., Bushman F.D. 2015. Covalent modification of bacteriophage T4 DNA inhibits CRISPR–Cas9. MBio. 6, e00648-15.
Tao P., Wu X., Tang W.-C., Zhu J., Rao V. 2017. Engineering of bacteriophage T4 genome using CRISPR–Cas9. ACS Synth. Biol. 6, 1952–1961.
Pawluk A., Davidson A.R., Maxwell K.L. 2018. Anti-CRISPR: discovery, mechanism and function. Nat. Rev. Microbiol. 16, 12–17.
Ramirez-Chamorro L., Boulanger P., Rossier O. 2021. Strategies for bacteriophage T5 mutagenesis: expanding the toolbox for phage genome engineering. Front. Microbiol. 12, 667332. https://doi.org/10.3389/fmicb.2021.667332
Mendoza S.D., Nieweglowska E.S., Govindarajan S., Leon L.M., Berry J.D., Tiwari A., Chaikeeratisak V., Pogliano J., Agard D.A., Bondy-Denomy J. 2020. A bacteriophage nucleus-like compartment shields DNA from CRISPR nucleases. Nature. 577, 244–248.
Malone L.M., Warring S.L., Jackson S.A., Warnecke C., Gardner P.P., Gumy L.F., Fineran P.C. 2020. A jumbo phage that forms a nucleus-like structure evades CRISPR–Cas DNA targeting but is vulnerable to type III RNA-based immunityю Nat. Microbiol. 5, 48–55.
Guan J., Bosch A.O., Mendoza S.D., Karambelkar S., Berry J., Bondy-Denomy J. 2022. RNA targeting with CRISPR–Cas13a facilitates bacteriophage genome engineering. bioRxiv. https://doi.org/10.1101/2022.02.14.480438
Smith H.O., Hutchison C.A., Pfannkoch C., Venter J.C. 2003. Generating a synthetic genome by whole genome assembly: φX174 bacteriophage from synthetic oligonucleotides. Proc. Natl. Acad. Sci. U. S. A. 100, 15440–15445.
Gibson D.G., Benders G.A., Andrews-Pfannkoch C., Denisova E.A., Baden-Tillson H., Zaveri J., Stockwell T.B., Brownley A., Thomas D.W., Algire M.A. 2008. Complete chemical synthesis, assembly, and cloning of a Mycoplasma genitalium genome. Science. 319, 1215–1220.
Gibson D.G., Young L., Chuang R.-Y., Venter J.C., Hutchison C.A., Smith H.O. 2009. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods. 6, 343–345.
Engler C., Gruetzner R., Kandzia R., Marillonnet S. 2009. Golden gate shuffling: a one-pot DNA shuffling method based on type IIs restriction enzymes. PLoS One. 4, e5553.
Pires D.P., Cleto S., Sillankorva S., Azeredo J., Lu T.K. 2016. Genetically engineered phages: a review of advances over the last decade. Microbiol. Mol. Biol. Rev. 80, 523–543.
Ando H., Lemire S., Pires D.P., Lu T.K. 2015. Engineering modular viral scaffolds for targeted bacterial population editing. Cell Syst. 1, 187–196.
Latka A., Lemire S., Grimon D., Dams D., Maciejewska B., Lu T., Drulis-Kawa Z., Briers Y. 2021. Engineering the modular receptor-binding proteins of Klebsiella phages switches their capsule serotype specificity. MBio. 12, e00455-21.
Kilcher S., Studer P., Muessner C., Klumpp J., Loessner M.J. 2018. Cross-genus rebooting of custom-made, synthetic bacteriophage genomes in L-form bacteria. Proc. Natl. Acad. Sci. U. S. A. 115, 567–572.
Bundy B.C., Franciszkowicz M.J., Swartz J.R. 2008. Escherichia coli-based cell-free synthesis of virus-like particles. Biotechnol. Bioeng. 100, 28–37.
Garenne D., Thompson S., Brisson A., Khakimzhan A., Noireaux V. 2021. The all-E. coli TXTL toolbox 3.0: new capabilities of a cell-free synthetic biology platform. Synth. Biol. 6, ysab017.
Shin J., Jardine P., Noireaux V. 2012. Genome replication, synthesis, and assembly of the bacteriophage T7 in a single cell-free reaction. ACS Synth. Biol. 1, 408–413.
Rustad M., Eastlund A., Jardine P., Noireaux V. 2018. Cell-free TXTL synthesis of infectious bacteriophage T4 in a single test tube reaction. Synth. Biol. 3, ysy002.
Bobay L.-M., Touchon M., Rocha E.P.C. 2013. Manipulating or superseding host recombination functions: a dilemma that shapes phage evolvability. PLoS Genet. 9, e1003825.
Muniyappa K., Radding C.M. 1986. The homologous recombination system of phage lambda. Pairing activities of beta protein. J. Biol. Chem. 261, 7472–7478.
Murphy K.C. 2012. Phage recombinases and their applications. Adv. Virus Res. 83, 367–414.
Brewster J.L., Tolun G. 2020. Half a century of bacteriophage lambda recombinase: in vitro studies of lambda exonuclease and Red-beta annealase. IUBMB Life. 72, 1622–1633.
Filsinger G.T., Wannier T.M., Pedersen F.B., Lutz I.D., Zhang J., Stork D.A., Debnath A., Gozzi K., Kuchwara H., Volf V. 2021. Characterizing the portability of phage-encoded homologous recombination proteins. Nat. Chem. Biol. 17, 394–402.
Marinelli L.J., Piuri M., Swigoňová Z., Balachandran A., Oldfield L.M., van Kessel J.C., Hatfull G.F. 2008. BRED: a simple and powerful tool for constructing mutant and recombinant bacteriophage genomes. PLoS One. 3, e3957.
Oppenheim A.B., Rattray A.J., Bubunenko M., Thomason L.C., Court D.L. 2004. In vivo recombineering of bacteriophage λ by PCR fragments and single-strand oligonucleotides. Virology. 319, 185–189.
van Kessel J.C., Hatfull G.F. 2008. Mycobacterial recombineering. In Chromosomal Mutagenesis. Springer, pp. 203–215.
Mosberg J.A., Lajoie M.J., Church G.M. 2010. Lambda red recombineering in Escherichia coli occurs through a fully single-stranded intermediate. Genetics. 186, 791–799.
Lopez S.C., Crawford K.D., Lear S.K., Bhattarai-Kline S., Shipman S.L. 2022. Precise genome editing across kingdoms of life using retron-derived DNA. Nat. Chem. Biol. 18, 199–206.
Goldfarb T., Sberro H., Weinstock E., Cohen O., Doron S., Charpak-Amikam Y., Afik S., Ofir G., Sorek R. 2015. BREX is a novel phage resistance system widespread in microbial genomes. EMBO J. 34, 169–183.
Shin H., Lee J.-H., Yoon H., Kang D.-H., Ryu S. 2014. Genomic investigation of lysogen formation and host lysis systems of the Salmonella temperate bacteriophage SPN9CC. Appl. Environ. Microbiol. 80, 374–384.
Pan Y.-J., Lin T.-L., Chen C.-C., Tsai Y.-T., Cheng Y.-H., Chen Y.-Y., Hsieh P.-F., Lin Y.-T., Wang J.-T. 2017. Klebsiella phage ΦK64-1 encodes multiple depolymerases for multiple host capsular types. J. Virol. 91, e02457-16.
Jensen J.D., Parks A.R., Adhya S., Rattray A.J., Court D.L. 2020. λ Recombineering used to engineer the genome of phage T7. Antibiotics. 9, 805.
Wetzel K.S., Guerrero-Bustamante C.A., Dedrick R.M., Ko C.-C., Freeman K.G., Aull H.G., Divens A.M., Rock J.M., Zack K.M., Hatfull G.F. 2021. CRISPY-BRED and CRISPY-BRIP: efficient bacteriophage engineering. Sci. Rep. 11, 1–6.
Cha R.S., Zarbl H., Keohavong P., Thilly W.G. 1992. Mismatch amplification mutation assay (MAMA): application to the cH-ras gene. Genome Res. 2, 14–20.
Klimuk E., Mekler V., Lavysh D., Serebryakova M., Akulenko N., Severinov K. 2020. Novel Escherichia coli RNA polymerase binding protein encoded by bacteriophage T5. Viruses. 12, 807.
Oda M., Morita M., Unno H., Tanji Y. 2004. Rapid detection of Escherichia coli O157: H7 by using green fluorescent protein-labeled PP01 bacteriophage. Appl. Environ. Microbiol. 70, 527–534.
Mark D.F., Richardson C.C. 1976. Escherichia coli thioredoxin: a subunit of bacteriophage T7 DNA polymerase. Proc. Natl. Acad. Sci. U. S. A. 73, 780–784.
Ghosh S., Hamdan S.M., Cook T.E., Richardson C.C. 2008. Interactions of Escherichia coli thioredoxin, the processivity factor, with bacteriophage T7 DNA polymerase and helicase. J. Biol. Chem. 283 (46), 32077‒32084.
Qimron U., Marintcheva B., Tabor S., Richardson C.C. 2006. Genomewide screens for Escherichia coli genes affecting growth of T7 bacteriophage. Proc. Natl. Acad. Sci. U. S. A. 103, 19039–19044.
Tabib-Salazar A., Liu B., Shadrin A., Burchell L., Wang Z., Wang Z., Goren M.G., Yosef I., Qimron U., Severinov K. 2017. Full shut-off of Escherichia coli RNA-polymerase by T7 phage requires a small phage-encoded DNA-binding protein. Nucleic Acids Res. 45, 7697–7707.
Isaev A., Drobiazko A., Sierro N., Gordeeva J., Yosef I., Qimron U., Ivanov N.V., Severinov K. 2020. Phage T7 DNA mimic protein Ocr is a potent inhibitor of BREX defence. Nucleic Acids Res. 48, 5397–5406.
Šimoliūnienė M., Kazlauskas D., Zajančkauskaitė A., Meškys R., Truncaitė L. 2021. Escherichia coli trxA gene as a molecular marker for genome engineering of felixounoviruses. Biochim. Biophys. Acta, Gen. Subj. 1865, 129967.
Martel B., Moineau S. 2014. CRISPR-Cas: an efficient tool for genome engineering of virulent bacteriophages. Nucleic Acids Res. 42, 9504–9513.
Lemay M.-L., Tremblay D.M., Moineau S. 2017. Genome engineering of virulent lactococcal phages using CRISPR–Cas9. ACS Synth. Biol. 6, 1351–1358.
Schilling T., Dietrich S., Hoppert M., Hertel R. 2018. A CRISPR–Cas9-based toolkit for fast and precise in vivo genetic engineering of Bacillus subtilis phages. Viruses. 10, 241.
Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E. 2012. A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science. 337, 816–821.
Wang J., Zhang X., Cheng L., Luo Y. 2020. An overview and metanalysis of machine and deep learning-based CRISPR gRNA design tools. RNA Biol. 17, 13–22.
Xiang X., Corsi G.I., Anthon C., Qu K., Pan X., Liang X., Han P., Dong Z., Liu L., Zhong J. 2021. Enhancing CRISPR–Cas9 gRNA efficiency prediction by data integration and deep learning. Nat. Commun. 12, 1–9.
Kirillov B., Savitskaya E., Panov M., Ogurtsov A.Y., Shabalina S.A., Koonin E.V., Severinov K.V. 2022. Uncertainty-aware and interpretable evaluation of Cas9–gRNA and Cas12a–gRNA specificity for fully matched and partially mismatched targets with Deep Kernel Learning. Nucleic Acids Res. 50, e11.
Kiro R., Shitrit D., Qimron U. 2014. Efficient engineering of a bacteriophage genome using the type IE CRISPR-Cas system. RNA Biol. 11, 42–44.
Box A.M., McGuffie M.J., O’Hara B.J., Seed K.D. 2016. Functional analysis of bacteriophage immunity through a type IE CRISPR-Cas system in Vibrio cholerae and its application in bacteriophage genome engineering. J. Bacteriol. 198, 578–590.
Bari S.M.N., Walker F.C., Cater K., Aslan B., Hatoum-Aslan A. 2017. Strategies for editing virulent staphylococcal phages using CRISPR–Cas10. ACS Synth. Biol. 6, 2316–2325.
Liu L., Li X., Ma J., Li Z., You L., Wang J., Wang M., Zhang X., Wang Y. 2017. The molecular architecture for RNA-guided RNA cleavage by Cas13a. Cell. 170, 714–726.
Adler B.A., Hessler T., Cress B.F., Mutalik V.K., Barrangou R.K., Banfield J., Doudna J.A. 2022. RNA-targeting CRISPR–Cas13 provides broad-spectrum phage immunity. bioRxiv. https://doi.org/10.1101/2022.03.25.485874
Reisch C.R., Prather K.L.J. 2015. The no-SCAR (Scarless Cas9 Assisted Recombineering) system for genome editing in Escherichia coli. Sci. Rep. 5, 1–12.
Quan J., Tian J. 2011. Circular polymerase extension cloning for high-throughput cloning of complex and combinatorial DNA libraries. Nat. Protoc. 6, 242–251.
Guzman L.-M., Belin D., Carson M.J., Beckwith J.O.N. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 177, 4121–4130.
Jiang W., Bikard D., Cox D., Zhang F., Marraffini L.A. 2013. RNA-guided editing of bacterial genomes using CRISPR–Cas systems. Nat. Biotechnol. 31, 233–239.
Inoue H., Nojima H., Okayama H. 1990. High efficiency transformation of Escherichia coli with plasmids. Gene. 96, 23–28.
Gordeeva J., Morozova N., Sierro N., Isaev A., Sinkunas T., Tsvetkova K., Matlashov M., Truncaite L., Morgan R.D., Ivanov N.V., Siksnys V., Zeng L., Severinov K. 2019. BREX system of Escherichia coli distinguishes self from non-self by methylation of a specific DNA site. Nucleic Acids Res. 47, 253–265.
Jusuk I., Vietz C., Raab M., Dammeyer T., Tinnefeld P. 2015. Super-resolution imaging conditions for enhanced yellow fluorescent protein (eYFP) demonstrated on DNA origami nanorulers. Sci. Rep. 5, 1–9.
Znobishcheva E.A., Morozova N.E., Khodorkovskii M.A. 2019. Fluorescent labeling of bacteriophage T7 by CRISPR-Cas9. J. Phys.: Conf. Ser. IOP Publ., 1400. p. 33005.
Davison J. 2015. Pre-early functions of bacteriophage T5 and its relatives. Bacteriophage. 5, e1086500.
Davison J. 2020. Phage T5 two-step injection. bioRxiv. https://doi.org/https://doi.org/10.1101/866236
Davison J., Brunel F. 1979. Restriction insensitivity in bacteriophage T5 I. Genetic characterization of mutants sensitive to EcoRI restriction. J. Virol. 29, 11.
Strotskaya A., Savitskaya E., Metlitskaya A., Morozova N., Datsenko K.A., Semenova E., Severinov K. 2017. The action of Escherichia coli CRISPR–Cas system on lytic bacteriophages with different lifestyles and development strategies. Nucleic Acids Res. 45, 1946–1957.
Studier F.W., Movva N.R. 1976. SAMase gene of bacteriophage T3 is responsible for overcoming host restriction. J. Virol. 19, 136.
Spoerel N., Herrlich P., Bickle T.A. 1979. A novel bacteriophage defence mechanism: the anti-restriction protein. Nature. 278, 30–34.
Guo X., Söderholm A., Isaksen G.V., Warsi O., Eckhard U., Trigüis S., Gogoll A., Jerlström-Hultqvist J., Åqvist J., Andersson D.I. 2021. Structure and mechanism of a phage-encoded SAM lyase revises catalytic function of enzyme family. Elife. 10, e61818.
FUNDING
The study was supported by grants from Russian Foundation for Basic Research (Ko_A_21-54-10001), Russian Science Foundation (Grant 22-14-00004 and Grant 22-74-00126) and the Ministry of Science and Higher Education of the Russian Federation (075-10-2021-114).
Author information
Authors and Affiliations
Contributions
NM, KS and AI conceived the study; AI, NM and EZ carried T7 mutagenesis; AI and AA carried T3 mutagenesis; AI and EZ carried T5 mutagenesis; AI prepared the manuscript; KS edited the text; all authors read and approved the final version.
Corresponding authors
Ethics declarations
COMPLIANCE WITH ETHICAL STANDARDS
Authors declare no competing interests.
This article does not contain any research involving humans or animals as subjects of research.
ADDITIONAL INFORMATION
Illustrations were prepared using BioRender.com under the paid subscription.
Supplementary Information
Rights and permissions
Open Access. This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Isaev, A., Andriianov, A., Znobishcheva, E. et al. Editing of Phage Genomes—Recombineering-assisted SpCas9 Modification of Model Coliphages T7, T5, and T3. Mol Biol 56, 801–815 (2022). https://doi.org/10.1134/S0026893322060073
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0026893322060073