
Abstract—
Biodiversity of the bacterial communities in the digestive system of Crenomytilus grayanus inhabiting the coastal Sea of Japan waters with chronic anthropogenic pollution was investigated using metabarcoding. Apart from marine bacteria, the taxa typical under contamination with oil (Rhodobacteraceae, Corynebacteriaceae), heavy metals (Asinibacterium), and unprocessed municipal waste (Cloacibacterium, Globicatella) were revealed in the microbiota. A collection of 411 cultured heterotrophic bacterial strains isolated in the course of this study was characterized taxonomically. The intestinal microbiome of the studied mollusks was shown to have a unique composition, depending on their habitat. Ability of bacterial strains isolated from the C. grayanus digestive system to degrade various nutrient substrates (sugars, amino acids, and polysaccharides) and xenobiotics (oil hydrocarbons, bisphenol A, and atrazine) was studied. Most isolates degraded a broad range of organic substrates; 13% (54 strains) oxidized oil hydrocarbons; 1% (4 strains) oxidized bisphenol A; and 0.5% (2 strains) degraded atrazine. The possible role of the microbiome C. grayanus microbiome in symbiotic digestion and in detoxication of the mollusk is discussed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Microbiocenoses occupying a specific ecological niche within the host organism are characterized by a complex hierarchical structure, established interspecies relations, and multistage metabolic processes. Thy carry out a number of functions useful to the host: increase its resistance to colonization by pathogenic microorganisms, participate in food digestion, detoxication, and maintenance of the water, salt, and pH homeostasis, synthesize amino acids, organic acids, vitamins, and other metabolites, and may act as a nutrient source under stress (Harris, 1993).
Mussels are the key component of marine ecosystems and one of the promising objects for commercial reproduction of bivalves. Considering the important role of indigenous microflora in the preservation of the hydrobiont health, numerous publications concerning the mussels-associated microbiota are available. Microbial diversity has been described for such traditional subjects of aquaculture as Mytilus galloprovincialis (Cavallo et al., 2009; Santisi et al., 2015), Mytilus coruscus (Li et al., 2018), Mytilus trossulus (Beleneva et al., 2003), and Mytilus edulis (Bezgachina and Kozitskiy, 2010; Motiei, 2014). In bivalves, the composition of microbiota was found to depend on a number of factors, including hydrobiont species and genetic characteristics, developmental stage, type of organ or tissue, physicochemical characteristics of the habitat, season, temperature, dissolved oxygen concentration in the water, nutrients, presence of pollutants, etc. (Paillard et al., 2022). However, members of the following taxa are most commonly found in the bacterial communities of various mussel species: Verrucomicrobia, Actinobacteria, Fusobacteria, Aeromonas, Pseudomonas, Alcaligenes, Moraxella, Acinetobacter, Flavobacterium, Cytophaga, Spongiobacter, Shewanella, Vibrio, Escherichia, Chromobacterium, Photobacterium, Desulfovibrio, Rhodococcus, Microbacterium, and Micrococcus (Beleneva et al., 2003; Cavallo et al., 2009; Bezgachina and Kozitskiy, 2010; Motiei, 2014; Santisi et al., 2015; Li et al., 2018).
As for Crenomytilus grayanus (Dunker, 1853), the microbiota of its digestive system is poorly studied, although this is on of the most common bivalve species in the coastal northern part of the Sea of Japan and a traditional commercial object. Previous studies dealt with investigation of the effect of the microflora of the water and sediments on Crenomytilus grayanus microflora (Bogatyrenko et al., 2018), enzymatic activity of molluck-associated microorganisms (Ivanova et al., 1992), and seasonal dynamics of abundance of heterotrophic bacteria in C. grayanus tissues (Beleneva and Zhukova, 2009).
The goal of the present work was to investigate the biodiversity and metabolic properties of bacterial communities of the digestive system of C. grayanus from the coastal areas of the northern Sea of Japan.
MATERIALS AND METHODS
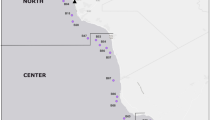
Sampling area. Four coastal areas of the northern part of the Sea of Japan were investigated (Fig. 1, Table 1).

Sampling area.
The Ajax Bay is located at the northeast of the Russky Island, in the southern part of the Eastern Bosphorus Strait. It is located at the coast of a large city of Vladivostok and experiences chronic pollution with hydrocarbons, DDT (4,4'-dichlorodiphenyltrichloroethane), phenol, heavy metals, and municipal garbage (Moshchenko and Shaikhlislamova, 2010).
The Stark Strait separates the islands Russky and Popov, which both fall under Vladivostok administration. It experiences significant recreational load in summer. The area is polluted with unprocessed wastewater from tourist facilities, as well as with oil hydrocarbons and heavy metals (Kozhenkova and Khristoforova, 2009).
The Vostok Bay is located at the northeast of the Peter the Great Bay, near the Primorsky krai large ports, including the Koz’mino oil-loading terminal. The area is polluted with phenols and oil hydrocarbons, with their concentrations exceeding the maximum permissible ones 10–30 and 2–12 times, respectively, depending on the season (Zhuravel’ et al., 2012).
The Matrosskaya Bay is located at the north of the Primorsky krai. It was chosen as a reference area with the minimal anthropogenic load. No large settlements and industrial objects are located in its vicinity.
In August 2019, 10 samples of adult C. grayanus were collected at each of the studied areas from the depth of 6–10 m. In the laboratory, the animals were cleaned from external contamination and dissected. Samples of the gastrointestinal tract were aseptically removed, homogenized, and frozen prior to subsequent research.
Metabarcoding. Total DNA (in three replicates) was isolated from the homogenate using the GeneJET Genomic DNA Purification Kit (Thermo Scientific, United States) according to the manufacturer’s protocol. DNA concentration and purity were analyzed using NanoDrop 2000 (Thermo Scientific, United States).
Amplification of the V3−V4 region of the 16S rRNA genes was carried out on CFX-96 (Bio-Rad, United States) using the Phusion Hot Start II DNA Polymerase kit (Thermo Fisher Scientific, United States), 0.2 mM dNTP (Life Technologies, United States), and the primers 343F (5'-CTCCTACGGRRSGCAGCAG-3') and 806R (5'-GGACTACNVGGGTWTCTAAT-3') containing adaptive sequences (Illumina), a linker, and a barcode.
Amplification products of the required length were excised from the gel, and DNA was extracted using the MinElute Gel Extraction Kit (Qiagen, Germany). Sequencing was carried out in the Genomika Joint Use Center, Institute of Chemical Biology and Fundamental Medicine, Siberian Branch, Russian Academy of Sciences) on a MiSeq sequencer (Illumina, United States), using the Reagent Kit v3 (2 × 300) (Illumina, United States).
The reads were processed using the R programming environment and the DADA2 package (https://github.com/benjjneb/dada2). After construction of the table of sequences, chimera removal, and clusterization, taxonomic classification was carried out using the SILVA v. 138 database with 80% bootstrap support. The sequences were normalized, aligned, and grouped into the operational taxonomic units (OTUs) with 97% identity (Mikhailov et al., 2019).
The data obtained in the present work were deposited to NCBI, project PRJNA892878.
Analysis of cultured bacterial forms. Intestinal samples were plated on the universal medium for marine microorganisms (MMM) containing the following (g/L): CaCO3, 1; MgSO4, 1; K2HPO4, 0.2; glucose, 0.2; peptone enzymatic, dry, for bacteriology, 5; agar bacteriological, 15; yeast extract 5; artificial seawater, 500 mL; and distilled water, 500 mL; pH 7.8–8.1. Artificial seawater contained the following (g/L): NaCl, 27.5; MgCl2, 5; MgSO4·7H2O, 2; CaCl2, 0.5; KCl, 1; and FeSO4, 0.001, 500 mL (Youchimizu, 1976). The plates were incubated at 22°C for 5 days, and pure bacterial cultures were isolated.
Bacterial isolates were identified based on analysis of the 16S rRNA gene sequences using the primer pair 27F (5'–AGAGTTTGATCATGGCTCAG–3') and 1350R (5'–GACGGGCGGTGTGTACAAG–3') (Lane et al., 1985). Sanger sequencing was carried out on an ABI PRISM 3130 Genetic Analyzer (Applied Biosystems, United States).
Metabolic Activity of Bacterial Isolates
Growth on natural organic substrates. Ability of the strains to degrade sugars (lactose, glucose, sucrose, arabinose, mannose, xylose, dulcitol, sorbitol, and mannitol) was tested in Hiss media with the BP indicator (aurin and aniline blue 1 : 1) (Biotekhnovatsiya, Russia). Ability to degrade amino acids (arginine, tryptophan, phenylalanine, ornithine, alanine, and glycine) was tested using the relevant commercial kits (Saint Petersburg Pasteur Institute, Russia).
Ability to degrade starch, chitin, chitosan, fucoidan, sodium alginate, collagen, and chondroitin sulfate was determined by plating 24-h cultures on the modified Voroshiliva−Dianova (VD) medium containing the following (g/L distilled water): NaCl, 10; NH4NO3, 1; K2HPO4, 1; KH2PO4, 1; MgSO4, 0.2; CaCl2, 0.02; FeCl2, 2 drops of saturated solution; agar, 15.0 (Kim, 2022). The relevant substrate (1%) was used as the sole carbon source. Inoculated plates of the medium containing no organic substrate were used as the controls. The plates were incubated for up to 10 days at 22°C. Bacterial growth in the presence of a substrate and its absence in the control were recorded. To reveal amylolytic activity, Lugol solution was added to the starch medium, with clear zones around the colonies indicating the presence of amylases. All experiments were carried out in triplicate.
Growth in the presence of xenobiotics. To determine the ability of the isolates to utilize oil hydrocarbons, they were plated on the modified VD mineral medium with 1% crude oil, and growth was recorded after incubation for 14 days at 22°C.
To determine the ability to utilize naphthalene, benzene, toluene, and xylene, suspensions were prepared using the cells of 24-h cultures grown in the medium with oil (OD600 = 1). Bushnell-Haas medium containing the following (g/L): MgSO4·7H2О, 0.2; CaC12·2H2O, 0.002; KH2PO, 1.0; K2HPО4, 1.0; NH4-NO3, 1.0; and FeCl3, 0.05; pH 7.0 was dispensed into the wells of a multiwell plate (1.5 mL) and inoculated with 25 µL of the bacterial suspension. As the sole carbon source, 5 µL of one of the substrates (hexadecane, naphthalene, benzene, toluene, or xylene) and 1.5 µg 2,6-dichlorophenolindophenol (DCPIP) were added to the wells. In the course of microbial hydrocarbon oxidation, electrons are transported to electron acceptors. Since DCPIP is an electron acceptor, it is possible to estimate the ability of microorganisms to utilize a hydrocarbon substrate by DCPIP changing its color from blue (oxidized) to colorless (reduced). The plates were incubated at 22°C, and discoloration or lightening of the color of the Bushnell−Haas medium was monitored for 24 h (Hanson et al., 1993).
To determine the ability to degrade bisphenol A, the strains were plated on the modified VD mineral medium with 0.1% bisphenol A and incubated for 14 days at 22°C. Suspensions of bacterial strains growing on this medium were prepared (OD600 = 1), and 1 mL of the suspension was used to inoculate 50 mL of liquid VD medium with 0.1% bisphenol A. Sterile VD medium with 0.1% bisphenol A was used as the control. After incubation for 5 days at 22°C, the samples were centrifuged for 3 min at 1500 rpm. The supernatant was collected into clean test tubes, supplemented with 0.5 mL of the Folin−Ciocalteu reagent, and incubated for 5 min. Bisphenol A degradation was determined using a Genesys 10S UV-VIS spectrophotometer (Thermo Scientific, United States) at 765 nm. The phosphotungstic and phosphomolybdic acids present in the Folin−Ciocalteu reagent are reduced by phenolic compounds, forming a blue-colored complex, with its color intensity proportional to the concentration of phenolic compounds (Ingale et al., 2021).
Ability of all isolates to degrade atrazine was determined by assessing their dehydrogenase activity. During redox reactions of microbial decomposition of the xenobiotic by dehydrogenases, the colorless salt 2,3,5-triphenyltetrazolium chloride (TTC) is reduced to red triphenyl formazan. The medium E-8 used for the purpose contained the following (g/L): KH2PO4, 0.7; (NH4)2HPO4, 1.5; MgSO4, 0.8; NaCl, 0.5; agar, 20.0; pH 6.6–6.7. It was supplemented with atrazine (0.6 mg/L) and 1% 2,3,5-triphenyltetrazolium chloride and dispensed into sterile petri dishes (20 mL per plate). The plates with 20 mL of E-8 agar medium with TTC and without atrazine were used as the control. Overnight bacterial cultures were inoculated on the experimental and control media. After incubation for 12 days at 22°C, diameter of the red zone around the inoculated material was measured (RF Patent 2051961).
Statistical processing of the data was carried out in the R environment. The Shannon and Simpson indices were calculated according to the results of metabarcoding. The data on diversity of cultured bacteria were used to construct the Venn diagram and to calculate the Jaccard similarity coefficients. Ability to degrade organic substrates, including xenobiotics, depending on the sampling site, was assessed using the Shapiro−Wilk criterion testing the normal distribution hypothesis, single-factor dispersion analysis (ANOVA), and the Scheffe a posteriori test.
RESULTS AND DISCUSSION
Metabarcoding. A total of 271 453 nucleotide sequences and 58 bacterial OTUs were obtained in the course of analysis of the samples from C. grayanus digestive system (Table 2). The values of the Shannon and Simpson diversity indices were relatively high for the C. grayanus microbiomes from all habitats, except for the Stark Strait (Table 2). Dispersion analysis (ANOVA) did not reveal significant differences between the microbiome samples (p > 0.05).
While members of 7 phyla were found in the studied gastrointestinal tract samples, only Proteobacteria were present in the mollusks from all regions. Representation of this phylum varied significantly from sample to sample (Table 3).
At the family level, the structure off the bacterial community of the Ajax Bay mollusks was represented by the following taxa: unclassified Pseudomonadales_uncultured (18%), Rhodobacteraceae (14.8%), Sphingomonadaceae (10.9%), Pseudomonadaceae (9%), Comamonadaceae (7.7%), Propionibacteriaceae (6.9%), Corynebacteriaceae (6.3%), and unclassified Chlamydiales_uncultured (5.8%).
In the microbiome of the Stark Strait mollusks, the families Bacillaceae (57.3%) and Paenibacillaceae (19%) predominated. Other taxa were less abundant: Chitinophagaceae (9.4%), Comamonadaceae (2.2%), and Sphingomonadaceae (1.8%). This microbiome was characterized by the presence of Weeksellaceae (2.9%) and Pseudoalteromonadaceae (1.7%).
Members of the families Comamonadaceae (33.8%), Chitinophagaceae (10.4%), Sphingomonadaceae (10.3%), Aerococcocaceae (8.2%), Bacillaceae (6.2%), Pseudomonadaceae (6.2%), and Actinomycetaceae (4.2%) were responsible for most of the diversity in the Vostok Bay microbiome. Members of 13 families constituted the remaining 20.7% of the taxonomic diversity.
Predominant families of the Matrosskaya Bay C. grayanus microbiome were Pseudomonadaceae (23.7%), Rhizobiales Incertae Sedis (22.2%), Chitinophagaceae (12.6%), and Sphingomonadaceae (7.2%). The minor part of the community was represented by Moraxellaceae, Actinomycetaceae, Micrococcaceae, Nitrosomonadaceae, Polyangia, Comamonadaceae, Brevibacteriaceae, Caulobacteraceae, Alcaligenaceae, Sphingobacteriales_env.OPS 17, and Nocardiaceae.
At the family level, members of Comamonadaceae and Sphingomonadaceae were common in all samples.
At the genus level, all samples exhibited unique taxonomic composition. In the microbiome of the Ajax Bay mollusks, uncultured genera of the order Pseudomonadales (18.4%), as well as Haematobacter (14.8%) and Sphingomonas (10.9%), predominated. At other sites, predominant genera were Bacillus (57.3%) and Paenibacillus (19%) for the Stark Strait; Alicycliphilus (27.4%), Sediminibacterium (10.4%), Sphingomonas (10.3%) at the Vostok Bay; and Pseudomonas (23.7%), Phreatobacter (22.2%), and Sediminibacterium (12.6%) at the Matrosskaya Bay (Fig. 2).

Bacterial genera revealed in the microbiome of Crenomytilus grayanus digestive system using metabarcoding. The genera with representation over 6% are shown.
According to a number of publications, the microbiota composition of bivalves depends significantly on a number of factors, such as hydrobiont species and its genetic properties, its developmental stage, type of organ or tissue, physico-geographical characteristics of the habitat, season, temperature, concentrations of dissolved oxygen and nutrients, the presence of pollutants, etc. (Paillard et al., 2022). However, in some works the authors revealed the microbiome nucleus, which had a stable composition during long periods or under the impact of varying ambient conditions. Thus, Spirochaetaceae, Vulcaniibacterium, and Delftia were described as the nucleus of the gastrointestinal microbiome of the Pacific oyster Crassostrea gigas (King et al., 2020); Mycoplasmataceae and Spirochaetaceae are the basis of the Ruditapes philippinarum microbiome (Offret et al., 2021); and Peptostreptococcaceae, Pirellulaceae, and Mollicutes form the nucleus of the microbiome of Pinctada margaritifera (Dube et al., 2019).
According to our results, the microbiome of the Stark Strait mussels was characterized by low biodiversity with predominance of the genera Bacillus and Paenibacillus. These microbial groups have a broad range of metabolic capabilities, and Bacillus were used in a number of probiotic preparations for aquaculture due to their ability to synthesize antimicrobial compounds active against most known infectious agents (Lee et al., 2021).
While Bacillus and Paenibacillus were revealed only in the Stark Strait mussels, members of the family Pseudomonadaceae were predominant in the microbiomes of mollusks from other studied sites. Antagonistic relationships between Bacillus and Pseudomonas species occupying the same ecological niches have been reported (Simoes et al., 2008). Investigation of the trepang Apostichopus japonicus revealed that either Bacillus or Pseudomonas predominated in the cultured microflora of the hydrobionts collected in different areas of the Sea of Japan. The enzymatic activity of both groups was similar, probably indicating the same contribution of these microbial groups to the digestive process of the host (Bogatyrenko and Buzoleva, 2016).
Molecular identification revealed in the microbiota of Crenomytilus grayanus from the Sea of Japan coastal waters the taxa indicating anthropogenic pollution. Thus, members of Rhodobacteraceae and Corynebacteriaceae contributed significantly to the Ajax Bay communities. These groups, apart from typical aquatic microorganisms, comprise also active degraders of oil hydrocarbons. Intracellular parasites of the order Chlamydiales were also revealed in the digestive tract of mussels from this site. Such microorganisms are known to attack the cytoplasm of the epithelial cells of the digestive diverticulum in bivalves, causing impaired metabolism and death of these animals (Getchell et al., 2016). The microbiome of the Stark Strait contained members of the genus Asinibacterium, which are typical of the soils contaminated with high heavy metal concentrations (Brzoska et al., 2022).
The mollusks from the Stark Strait and Vostok Bay harbored also the bacteria typical of untreated municipal waste. Thus, members of the genus Cloacibacterium were found in the microbiome of the Stark Strait mollusks, while opportunistic pathogens of the genus Globicatella, which cause meningitis and bacteriemia in humans, occurred in the Vostok Bay microbiome. No taxa indicating anthropogenic load were found in the animals collected in the reference area.
The coastal areas of the northern Sea of Japan experience chronic pollution with heavy metals, oil products, phenols, pesticides, and municipal waste (Barysheva et al., 2019; Moshchenko et al., 2020), which, according to our results, affects the composition of bacterial communities of C. grayanus in the studied region.
Biodiversity of the cultured heterotrophic bacteria. Plating of the samples from C. grayanus digestive system on MMM medium resulted in isolation of 411 heterotrophic bacterial strains belonging to 34 genera: 106 from Ajax Bay, 121 from Stark Strait, 101 from Vostok Bay, and 83 from Matrosskaya Bay (Fig. 3).

Genera of cultured bacteria isolated from the Crenomytilus grayanus digestive system using metabarcoding. The genera with representation over 6% are shown.
Among the Vostok Bay samples, the highest numbers of isolates were obtained for members of the genera Sphingomonas (11), Kocuria (10), Pseudoalteromonas (9), and Paracoccus and Arthrobacter (8 strains each).
From the Matrosskaya Bay samples, the most common isolates belonged to of the genera Pseudomonas (21 strain), Arthrobacter (14), Sphingomonas (13), Actinomyces (9), and Psychrobacter (7). Predominance of Pseudomonas and Psychrobacter, as well as Synechococcus, has been reported for the microbiome of Crassostrea virginica (Pathak et al., 2021).
Intestinal microflora of the Stark Strait mussels was characterized by high abundance of members of the genera Bacillus (25 strains), Paenibacillus (18 strains), and Aerococcus and Paracoccus (10 strains each).
Similar results were obtained in the work on associates of Crenomytilus grayanus from the Troitsa Bay (Peter the Great Gulf, Sea of Japan): 30.6% of the total bacterial diversity of the mollusk stomach was represented by the genus Bacillus (Ivanova et al., 1992). High share of these bacteria may be due to their ability to form spores, providing for their survival in the intestine due to resistance to digestive enzymes and low pH values (Pandiyan et al., 2013). Strains of the genus Paenibacillus were unique for the community of Stark Strait mussels; some members of this genus have been reported to enhance the growth of hydrobionts, improve their immune and antioxidant activity, and provide for resistance to pathogenic vibrios (Amoah et al., 2020).
Predominant isolates from the community of the Ajax Bay mussels belonged to the genera Rhodococcus (13), Pseudomonas (12), Sphingomonas (10), and Corynebacterium (7). Opportunistic pathogens of the genera Escherichia, Klebsiella, Proteus, and Yersinia were found in the samples collected at this area. Detection of these potentially hazardous bacteria indicates the problematic sanitary and epidemiological state of the area caused by chronic contamination with municipal waste and significant recreational load in summer. Although opportunistic pathogens were not dominant in the biocenosis, they shifted the equilibrium to the higher abundance of the microbiota not characteristic of the hydrobiont. Such pathogenic microorganisms as Escherichia coli, Salmonella, and Vibrio parahaemolyticus (Cavallo et al., 2009; Rubiolo et al., 2019), as well as hepatitis A virus and noroviruses (Schrader et al., 2003), have been previously found in other mussel species from the areas with high anthropogenic load. Interestingly, while vibrios are widespread in marine environments and are often revealed in the microflora of bivalves (Moriarty, 1998; Motiei, 2014; Rubiolo et al., 2019), in the present work a single Vibrio strain was isolated only from the Ajax Bay mollusks.
Comparison of the biodiversity of cultured heterotrophic bacteria isolated from C. grayanus of the studied regions by means of the Venn diagram (Fig. 4) the Jaccard coefficient (Кj) revealed that the microbiome of the mollusks of each region was characterized by a specific set of bacterial taxa. The highest values of the similarity coefficient were obtained for the mollusks of the Ajax Bay and Vostok Bay (Кj = 0.46) and for those of the Vostok Bay and Stark Strait (Кj = 0.44), while the lowest values were found for the sets of the Matrosskaya Bay and Stark Strait (Кj = 0.27) and for the Stark Strait and Ajax Bay (Кj = 0.28).

Venn diagram showing the diversity of cultured bacteria isolated from the Crenomytilus grayanus digestive system at the genus level.
The present work showed that the intestinal microbiome of C. grayanus exhibited a unique taxonomic composition, depending on the site where the animals were collected. These results indicate a significant effect of the environment of development of the symbiotic microflora of this bivalves.
At the same time, our analysis revealed that the presence of members of the genera Psychrobacter and Sphingomonas characterized the animals from all studied regions. These groups probably belonged to the autochthonous microflora of the host and could have some useful functions.
Thus, members of the genus Sphingomonas are known to inhibit pathogenic Vibrio anguillarum and may have a positive effect on growth, survival, and biochemical parameters of young fish (Chaudhary et al., 2021). A number of works reported a positive probiotic potential of some Psychrobacter spp. strains for artificial reproduction of a shrimp Exopalaemon carinicauda (Lai et al., 2022), Atlantic cod Gadus morhua (Lazado et al., 2010), Epinephelus coioides (Liu et al., 2021), and Seriola lalandi (Ramírez et al., 2020).
Actinomyces, which were also abundant in the C. grayanus digestive system, are known as producers of a broad spectrum of secondary metabolites, including those with pronounced antimicrobial effects (Cera et al., 2022).
The animals collected at all stations, except the Stark Strait, often contained members of the genus Arthrobacter, which may belong to the indigenous microflora of the studied hydrobionts. Some Arthrobacter species isolated from seawater were shown to possess probiotic properties, e.g., ability to synthesize antibiotics. Arthrobacter strain XE-7 was shown to have a positive effect on the gut microbiota and immunity of Pacific white shrimps and to exhibit antagonistic activity against Vibrio parahaemolyticus (Li et al., 2006). An Arthrobacter davidanieli strain was shown to be efficient for treatment of infections caused in salmonids b Renibacterium salmoninarum and Piscirickettsia salmonis (Salonius et al., 2004).
Numerous Paracoccus strains were isolated from the mollusks collected at all sites, except for Matrosskaya Bay. According to the literature data, these microorganisms are symbionts of insects, corals, and bryozoa and possess antibacterial, algicidal, and fungicidal properties against such organisms as Escherichia coli, Pseudomonas aeruginosa, Bacillus subtilis, Staphylococcus aureus, Prorocentrum donghaiense, and Candida albicans (Leinberger et al., 2021).
Ten Kocuria strains were isolated from the Vostok Bay mussels. According to the literature, this genus contains numerous pathogenic species. However, according to some data, these microorganisms are able to degrade a broad spectrum of xenobiotics and may be used for prevention and treatment of infections caused in trout by Vibrio anguillarum and Vibrio оrdalii (Sharifuzzaman and Austin, 2010).
Alteromonas spp. strains were unique components of the microbiome of the Stark Strait mussels. Alteromonas biofilms were found to be responsible for metamorphosis induction in the larval stages of Mytilus galloprovincialis (Bao et al., 2007).
Unique microbiome composition for each region is probably responsible for the adaptation of the mussels to specific environmental conditions.
Metabolic properties of bacterial isolates. Most of the strains isolated in the present work were found to degrade a broad spectrum of organic substrates (Fig. 5). Analysis did not reveal statistically significant differences in the ability to degrade simple sugars and amino acids between bacteria isolated from the mollusks collected in different regions (Shapiro−Wilk criterion—p > 0.05, ANOVA—F < Fcrit). Thus, in spite of the differences in the species composition, the microbiota of the mussels from different sites have similar enzymatic properties and probably carry out similar functions for the host organism.

Shares of bacterial strains capable of degrading organic substrates.
Utilization of complex organic substrates, which are abundant in marine environments (chitin, chitosan, alginate, fucoidan, collagen, and chondroitin sulfate) was carried out by a lower number of strains (Fig. 5). The share of microorganisms capable of using these compounds as sole carbon sources was reliably higher in the mussels from the Matrosskaya Bay reference site (Shapiro−Wilk criterion p > 0.05, ANOVA— F > Fcrit, the Scheffe test critical value—p < 0.05). It may be suggested that anthropogenic pollution affects both the biodiversity and the functional properties of the symbiotic microflora in bivalves. Anthropogenic load may both decrease the abundance of the autochthonous microbiota capable of degrading complex substrates and affect its activity. Anthropogenic pollution was shown to result in decreased relative abundance of bacteria degrading chitin and its derivatives, cellulose, sodium alginate, and fucoidan in microbial cenoses of the surface sea water and the rate of utilization of these compounds (Kim, 2022). The author linked these results with the adaptation of marine communities at polluted areas to utilization of easily decomposed organic matter, which is constantly supplied by municipal and industrial waste, river flow, and ballast water. Long-term existence under such conditions may result in the loss or decrease of the hydrolytic activity against polysaccharides in the autochthonous microbiota (Kim, 2022).
According to the literature, a number of the Primorsky krai coastal areas experience chronic contamination with various pollutants (Barysheva et al., 2019; Moshchenko et al., 2020). We have therefore analyzed the ability of the isolated strains to grow on media with oil and oil hydrocarbons (hexadecane, benzene, xylene, toluene, and naphthalene), a pesticide (atrazine), and a plasticizer (bisphenol A) as sole carbon sources.
On mineral medium with oil, growth of 54 bacterial strains (13%) from the mollusks of the Ajax Bay, Vostok Bay, and Stark Strait was observed. Most of the active strain belonged to the genera Kocuria, Rhodococcus, Streptomyces, Micrococcus, Pseudomonas, Bacillus, Arthrobacter, and Corynebacterium, which, according to the literature data, are among the most widespread degraders of oil hydrocarbons in general and in the Sea of Japan in particular (Bogatyrenko et al., 2021).
All 54 strains growing on oil (13% of the collection) were able to oxidize hexadecane (Fig. 6). Degradation of aromatic compounds was carried out by a lower number of strains. Thus, xylene was utilized by 46 strains (11.2%); benzene, by 45 (11%); toluene, by 42 (10.2%); and naphthalene, by 35 strains (8.5%) (Fig. 6).

Shares of bacterial strains capable of xenobiotic degradation.
It may be suggested that in nature such microorganisms may be involved in detoxication of the mollusks, protecting them from the deleterious effects of oil hydrocarbons. Such conclusion was made in the work, in which addition of benzopyrene to the young trepang Apostichopus japonicus for 14 days resulted in decreased abundance of autochthonous bacteria in the trepang microbiome (Lactococcus, Bacillus, Lactobacillus, Enterococcus, Leuconostoc, and Weissella) and a sharp increase in the abundance of hydrocarbon-oxidizing microorganisms Lutibacter, Pseudoalteromonas, Polaribacter, Rhodopirellula, and Blastopirellula (Zhao et al., 2019).
Bisphenol A is among the most widespread micropollutants, which may cause metabolic and reproductive disorders in humans. It was recently shown to be dangerous not only to humans, but also to marine organisms. In one work bisphenol A and its analogs were shown to affect the microbiome of Mytilus galloprovincialis larvae (Balbi et al., 2020). Application of pollutants resulted in increased relative abundance of potential pathogens (Vibrio, Arcobacter, and Tenacibaculum) and of bacteria involved in xenobiotic biotransformation (Oleispira and Shewanella).
In the present work, four strains were found to utilize bisphenol A: Bacillus sp. StM5, Bacillus sp. StM11, and Bacillus sp. StM24 from the Stark Strait and Sphingomonas sp. VtM13 from the Vostok Bay. After 5 days, bisphenol degradation by these strains was 52 ± 2.1, 37 ± 1.5, 41 ± 1.9, and 45 ± 2.3%, respectively. Bisphenol A-degrading strains of Bacillus and Sphingomonas have been previously isolated by many researchers from diverse environments (López-Moreno et al., 2021; de Morais Farias and Krepsky, 2022). The key role in bisphenol degradation is played by the cytochrome P450 monooxygenase system, ferredoxin, ferredoxin reductase, and laccase, the enzymes occurring in bacteria of these genera.
Treatment of Crassostrea virginica oysters with atrazine, a common agricultural herbicide, was shown to result in a significant loss of the key species of mutualistic bacteria with subsequent colonization by pathogenic Nocardia (Britt et al., 2020).
In the present work, two bacterial strains isolated from the Vostok Bay mussels were able to degrade atrazine. On day 12 of the experiment, the zones of pollutant oxidation on the plates with Arthrobacter agilis VtM7 and Rhodococcus sp. VtM19 were 30 ± 2.4 and 26 ± 3.1 mm, respectively, which indicated relatively high dehydrogenase activity of these microorganisms. Although rhodococci are known to utilize many xenobiotics, complete degradation of atrazine to CO2 and ammonia has been best described in the literature for Arthrobacter strains (Abd Rani et al., 2022). In Arthrobacter agilis, the atzA/trzN genes are known to encode atrazine chlorohydrolaze, which catalyzes atrazine dechlorination (Mili et al., 2022). The strain isolated in the present work belongs to the species widespread in soil, highly resistant to environmental fluctuations, an degrading numerous pollutants.
Thus, our results may be used for further in-depth assessment of the contribution of the C. grayanus microbiome to the symbiotic digestion and detoxication of this mollusk.
REFERENCES
Amoah, K., Huang, Q.C., Dong, X.H., Tan, B.P., Zhang, S., Chi, S.Y., Yang, Q., Liu, H., and Yang, Y.Z., Paenibacillus polymyxa improves the growth, immune and antioxidant activity, intestinal health, and disease resistance in Litopenaeus vannamei challenged with Vibrio parahaemolyticus, Aquaculture, 2020, vol. 518, p. 734563.
Balbi, T., Vezzulli, L., Lasa, A., Pallavicini, A., and Canesi, L., Insight into the microbial communities associated with first larval stages of Mytilus galloprovincialis: possible interference by estrogenic compounds, Comp. Biochem. Physiol. Part C: Toxicol. Pharmacol., 2020, vol. 237, p. 108833.
Bao, W.Y., Yang, J.L., Satuito, C.G., and Kitamura, H., Larval metamorphosis of the mussel Mytilus galloprovincialis in response to Alteromonas sp. 1: evidence for two chemical cues?, Marine Biol., 2007, vol. 152, pp. 657–666.
Barysheva, V.S., Chernova, E.N., and Patrusheva, O.V., Contamination of the mirine environment of the Vostok Bay, Sea of Japan, by organic compounds (2016−2018), Vestn. Dal’nevost. Otd. Ross. Akad. Nauk, 2019, no. 2 (204), pp. 87–94.
Beleneva, I.A. and Zhukova, N.V., Seasonal dynamics of cell numbers and biodiversity of marine heterotrophic bacteria inhabiting invertebrates and water ecosystems of the Peter the Great Bay, Sea of Japan, Microbiology (Moscow), 2009, vol. 78, pp. 369–375.
Beleneva, I.A., Zhukova, N.V., and Maslennikova, E.F., Comparative study of microbial communities from cultured and natural populations of the mussel Mytilus trossulus in Peter the Great Bay, Microbiology (Moscow), 2003, vol. 72, no. 4, pp. 472–477.
Bezgachina, T.V. and Kozitskiy, A.N., Concerning the sanitary and microbiological tests of mussels from the White Sea, Current Problems of Physiology and Biochemistry of Aquatic Organisms, Coll. Sci. Papers 1st Int. Sem. PhD Workshop, Petrozavodsk, 2010, p. 12.
Bogatyrenko, E.A. and Buzoleva, L.S., Characterization of the gut bacterial community of the Japanese sea cucumber Apostichopus japonicus, Microbiology (Moscow), 2016, vol. 85, no. 1, pp. 116–123.
Bogatyrenko, E.A., Dunkai, T.I., Buzoleva, L.S., and Kim, A.V., Influence of Vladivostok coastal waters pollution on a microflora of mussel Crenomytilus grayanus, IOP Conf. Series: Earth Environ. Sci., 2018, vol. 107, p. e012052.
Bogatyrenko, E.A., Kim, A.V., Dunkai, T.I., Ponomareva, A.L., Es’kova, A.I., Sidorenko, M.L., and Okulov, A.K., Taxonomic diversity of culturable hydrocarbon-oxidizing bacteria in the Sea of Japan, Russ. J. Mar. Biology, 2021, vol. 47, no. 3, pp. 232–239.
Britt, A., Bernini, M., McSweeney, B., Dalapati, S., Duchin, S., Cavanna, K., Santos, N., Donovan, G., O’Byrne, K., Noyes, S., Romero, M., Poonacha, K.N.T., and Scully, T., The effects of atrazine on the microbiome of the eastern oyster: Crassostrea virginica, Sci. Rep., 2020, vol. 10, no. 1, p. 11088.
Brzoska, R.M., Edelmann, R.E., and Bollmann A., Physiological and genomic characterization of two novel Bacteroidota strains Asinibacterium spp. OR43 and OR53, Bacteria, 2022, vol. 1, no. 1, pp. 33–47.
Cavallo, R.A., Acquaviva, M.I., and Stabili, L., Culturable heterotrophic bacteria in seawater and Mytilus galloprovincialis from a Mediterranean area (Northern Ionian Sea–Italy), Environ. Monitor. Assess., 2009, vol. 149, pp. 465—475.
Chaudhary, A., Ahmad, Q.U.A., Akram, A.M., Iqtedar, M., and Qazi, J.I., Effect of Sphingomonas sp., as a probiotic on survival, growth and biochemical constituents of Vibrio anguillarum challenged Labeo rohita fingerlings, Pakistan J. Zool., 2021, vol. 53, no. 6, pp. 2071‒2081. https://doi.org/10.17582/journal.pjz/20181009061048
de Morais Farias, J. and Krepsky, N., Bacterial degradation of bisphenol analogues: an overview, Environ. Sci. Pollut. Res., 2022, vol. 29, рр. 76543–76564.
Dubé, C.E., Ky, C.L., and Planes, S., Microbiome of the black-lipped pearl oyster Pinctada margaritifera, a multi-tissue description with functional profiling, Front. Microbiol., 2019, vol. 10, p. 1548.
Getchell, R.G., Smolowitz, R.M., McGladdery, S.E., and Bower, S.M., Diseases and parasites of scallops, Dev. Aquacult. Fisheries Sci., 2016, vol. 40, pp. 425–467.
Hanson, K.G., Desai, J.D., and Desai, A.J., A rapid and simple screening technique for potential crude oil degrading microorganisms, Biotechnol. Techniques, 1993, vol. 7, no. 10, pp. 745–748.
Harris, J.M., The presence, nature, and role of gut microflora in aquatic invertebrates: a synthesis, Microb. Ecol., 1993, vol. 25, pp. 195—231.
Ingale, S., Patel, K., Sarma, H., and Joshi, S.J., Bacterial biodegradation of bisphenol A (BPA), in Biotechnology for Sustainable Environment, Singapore: Springer, 2021, pp. 95–110. https://doi.org/10.1007/978-981-16-1955-7_4
Ivanova, E., Kiprianova, E., Aminin, D., Mikhailov, V., ana Agafonova, I., Biological activity of microorganisms associated with the Crenomytilus grayanus mussel, Biotechnology & Biotechnological Equipment, 1992, vol. 6, no. 1, pp. 25–30.
Kim, A.V., Effect of anthropogenic pollution on the taxonomic diversity and biological properties of cultivated bacteria of the Primorsky krai marine areas, Extended Abstract Cand. Sci. (Biol.) Dissertation, Vladivostok, 2022.
King, W.L., Siboni, N., Kahlke, T., Dove, M., O’Connor, W., Mahbub, K.R., Jenkins, C., Seymour, J.R., and Labbate, M., Regional and oyster microenvironmental scale heterogeneity in the Pacific oyster bacterial community, FEMS Microbiol. Ecol., 2020, vol. 96, no. 5, p. fiaa054.
Kozhenkova, S.I. and Khristoforova, N.K., Distribution of macrophytic green algae in th Amur Bay, Sea of Japan, Izv. TINRO, 2009, vol. 159, pp. 156–167.
Lai, X., Chen, J., Liang, S., Chen, H., Liu, S., and Gao, H., Effects of the probiotic Psychrobacter sp. B6 on the growth, digestive enzymes, antioxidant capacity, immunity, and resistance of Exopalaemon carinicauda to Aeromonas hydrophila, Probiot. Antimicrobю Proteins, 2022, pp. 1–8. https://doi.org/10.1007/s12602-022-09919-3
Lane, D.J., Pace, B., Olsen, G.J., Stahl, D.A., Sogin, M.L., and Pace, N.R., Rapid determination of 16S ribosomal RNA sequences for phylogenetic analyses, Proc. Natl. Acad. Sci. U. S. A., 1985, vol. 82, no. 20, pp. 6955—6959.
Lazado, C.C., Caipang, C.M.A., Rajan, B., Brinchmann, M.F., and Kiron, V., Characterization of GP21 and GP12: two potential probiotic bacteria isolated from the gastrointestinal tract of Atlantic cod, Probiot. Antimicrob. Proteins, 2010, vol. 2, pp. 126–134.
Lee, C., Kim, S., Shin, Ja., Kim, M.-G., Gunathilaka, B.E., Kim, S.H., Kim, J.E., Ji, S.-C., Han, J.E., and Lee, K.-J., Dietary supplementations of Bacillus probiotic improve digestibility, growth performance, innate immunity, and water ammonia level for Pacific white shrimp, Litopenaeus vannamei, Aquacult. Int., 2021, vol. 29, pp. 2463–2475.
Leinberger, J., Holste, J., Bunk, B., Freese, H.M., Spröer, C., Dlugosch, L., Kück, C., Schulz, S., and Brinkhoff, T., High potential for secondary metabolite production of Paracoccus marcusii CP157, isolated from the crustacean Cancer pagurus, Front. Microbiol., 2021, vol. 12, p. 688754.
Li, J., Tan, B., Mai, K., Ai, Q., Zhang, W., Xu, W., and Ma, H., Comparative study between probiotic bacterium Arthrobacter XE-7 and chloramphenicol on protection of Penaeus chinensis post-larvae from pathogenic vibrios, Aquaculture, 2006, vol. 253, nos. 1–4, pp. 140–147.
Liu, Z.Y., Yang, H.L., Hu, L.H., Yang, W., Ai, C.X., and Sun, Y.Z., Autochthonous probiotics alleviate the adverse effects of dietary histamine in juvenile grouper (Epinephelus coioides), Front. Microbiol., 2021, vol. 12, p. 792718.
Li, Y.F., Yang, N., Liang, X., Yoshida, A., Osatomi, K., Power, D., Batista, F.M., and Yang, J.L., Elevated seawater temperatures decrease microbial diversity in the gut of Mytilus coruscus, Front. Physiol., 2018, vol. 9, p. 839.
López-Moreno, A., Torres-Sánchez, A., Acuña, I., Suárez, A., and Aguilera, M., Representative Bacillus sp. AM1 from gut microbiota harbor versatile molecular pathways for bisphenol A biodegradation, Int. J. Mol. Sci., 2021, vol. 22, no. 9, p. 4952.
Mikhailov, I.S., Zakharova, Y.R., Bukin, Y.S., Galachyants, Y.P., Petrova, D.P., Sakirko, M.V., and Likhoshway, Y.V., Co-occurrence networks among bacteria and microbial eukaryotes of Lake Baikal during a spring phytoplankton bloom, Microb. Ecol., 2019, vol. 77, no. 1, pp. 96–109.
Moriarty, D.J.W., Control of luminous Vibrio species in penaeid aquaculture ponds, Aquaculture, 1998, vol. 164, pp. 351—358.
Moshchenko, A.V. and Shaikhlislamova, L.E., Ecological state of the eastern part of the Eastern Bosphorus Strait (Peter the Great Gulf, Sea of Japan), Izv. TINRO, 2010, vol. 161, pp. 199–211.
Moshchenko, A.V., Belan, T.A., Lishavskaya, T.S., Sevast’yanov, A.V., and Borisov, B.M., Long-term dynamics of the concentrations of priority pollutants and of total chemical pollution of the Vladivostok coastal sea areas (Peter the Great Gulf, Sea of Japan), Izv. TINRO, 2020, vol. 200, no. 2, pp. 377–400.
Motiei, A., Mytilus edulis associated bacteria: diversity and interactions based on bioactive molecules, Doctoral Dissertation., Christian-Albrechts-Universität Kiel, 2014.
Offret, C., Paulino, S., Gauthier, O., Château, K., Bidault, A., Corporeau, C., Miner, P., Petton, B., Pernet, F., Fabioux, C., Paillard, C., and Le Blay, G., The marine intertidal zone shapes oyster and clam digestive bacterial microbiota, FEMS Microbiol. Ecol., 2020, vol. 96, no. 8, p. fiaa078.
Paillard, C, Gueguen, Y., Wegner, K.M., Bass, D., Pallavicini, A., Vezzulli, L., and Arzul, I., Recent advances in bivalve-microbiota interactions for disease prevention in aquaculture, Curr. Opin. Biotechnol., 2022, vol. 73, pp. 225–232.
Pandiyan, P., Balaraman, D., Thirunavukkarasu, R., George, E.G.J., Subaramaniyan, K., Manikkam S., and Sadayappan, B., Probiotics in aquaculture, Drug Invention Today, 2013, vol. 5, no. 1, pp. 55‒59.
Pathak, A., Stothard, P., and Chauhan, A., Comparative genomic analysis of three Pseudomonas species isolated from the eastern oyster (Crassostrea virginica) tissues, mantle fluid, and the overlying estuarine water column, Microorganisms, 2021, vol. 9, no. 3, p. 490.
Ramírez, C., Rojas, R., and Romero, J., Partial evaluation of autochthonous probiotic potential of the gut microbiota of Seriola lalandi, Probiot. Antimicrob. Proteins, 2020, vol. 12, pp. 672–682.
Rubiolo, J.A., Botana, L.M., and Martinez, P., Insights into mussel microbiome, in Microbial Communities in Aquaculture Ecosystems, Cham: Springer Int. Publ., 2019, 1st ed.,pp. 95–120.
Salonius, K., Siderakis, C., MacKinnon, A.M., and Griffiths, S.G., Use of Arthrobacter davidanieli as a live vaccine against Renibacterium salmoninarum and Piscirickettsia salmonis in salmonids, Devel. Biologicals, 2004, vol. 121, pp. 189–197.
Santisi, S., Genovese, M., Bonsignore, M., Fiumara, E., Maricchiolo, G., Mancuso, M., Genovese, L., Giuliano, L., and Cappello, S., Study of bacterial communities in mussel Mytilus galloprovincialis by 16S rDNA, Int. J. Microbiol. Applic., 2015, vol. 2, pp. 18—24.
Schrader, C., Reetz, J., and Süss, J., Food-borne human pathogenic viruses and their molecular detection with special consideration of noroviruses and hepatitis A virus in mussels, Berliner und Munchener Tierarztliche Wochenschrift, 2003, vol. 116, nos. 11−12, pp. 496–505.
Sharifuzzaman, S.M. and Austin, B., Kocuria SM1 controls vibriosis in rainbow trout (Oncorhynchus mykiss, Walbaum), J. Appl. Microbiol., 2010, vol. 108, no. 6, pp. 2162–2170.
Simoes, M., Simões, L.C., Pereira, M.O., and Vieira, M.J., Antagonism between Bacillus cereus and Pseudomonas fluorescens in planktonic systems and in biofilms, Biofouling, 2008, vol. 24, no. 5, pp. 339–349.
Youchimizu, M. and Kimura, T., Study of intestinal microflora of salmonids, Fish Pathol., 1976, vol. 10, pp. 243–259.
Zhao, Y., Liu, H., Wang, Q., Li, B., Zhang, H., and Pi, Y., The effects of benzo [a] pyrene on the composition of gut microbiota and the gut health of the juvenile sea cucumber Apostichopus japonicus Selenka, Fish Shellfish Immunol., 2019, vol. 93, pp. 369–379.
Zhuravel’, E.V., Khristoforova, N.K., Drozdovskaya, N.K., and Tokarchuk, T.N., Assessment of the state of the Vostok Bay water (Peter the Great Gulf, Sea of Japan) by the hydrochemical and microbiological parameters, Izv. Samar. Tsentr. Ross. Akad. Nauk, 2012, vol. 14, no. 1(9), pp. 2325–2329.
Funding
This work was supported by Far Eastern Federal University (Program “PRIORITY 2030”: Ocean Science, project no. 22-05.-1.60-0012).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflicts of interest. All applicable international, national, and/or institutional guidelines for the care and use of animals were followed.
Additional information
Translated by P. Sigalevich
Rights and permissions
Open Access. This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dunkai, T.I., Bogatyrenko, E.A. & Kim, A.V. Biodiversity and Metabolic Properties of Bacterial Communities from the Digestive System of the Bivalve Crenomytilus grayanus. Microbiology 92, 552–563 (2023). https://doi.org/10.1134/S0026261723600982
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0026261723600982