
Abstract
The optimal conditions for C3 oxidative biotransformation of 1.0 g/L pentacyclic triterpenoids oleanolic (OA) and glycyrrhetinic (GA) acids were determined using the resting cells of Rhodococcus rhodochrous IEGM 1360 from the Regional Specialised Collection of Alkanotrophic Microorganisms. Resting cell suspensions (OD600 2.6, pH 8.0, and OD600 2.2, pH 6.0) showed the highest catalytic activity against OA and GA, resulting in the formation of 61 and 100% of their 3-oxo derivatives, respectively. Using phase contrast, atomic force, and confocal laser scanning microscopy, an adaptive response of rhodococci to the effects of OA and GA was revealed. In silico, the apoptotic activity of 3-oxo-OA and antioxidant activity of 3-oxo-GA have been assumed. In vitro, a pronounced antibacterial activity of 3-oxo-OA against Micrococcus luteus, Escherichia coli, Staphylococcus aureus, and Bacillus subtilis was shown. The absence of toxic effects of the above triterpenoids and their 3-oxo derivatives on aquatic objects and plants was demonstrated in silico and in vitro, respectively.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Nowadays, amidst the deficit of effective pharmacological agents for socially significant diseases, an essential task is to synthesize new chemical compounds with potential biological activities, including those derived from plant terpenoids (Calixto, 2019). One of the intensively employed compounds in this area are oleanane type plant pentacyclic triterpenoids, in particular oleanolic (OA, 1, 3β-hyd-roxyolean-12-en-28-oic acid) and glycyrrhetinic (GA, 3, 3β-hydroxy-11-oxoolean-12-en-30-oic acid) acids (Kumar and Dubey, 2019). OA and GA are actively used to obtain semi-synthetic derivatives with pronounced antiviral, antimicrobial, anti-inflammatory, antitumor, and hepatoprotective activities (Capel et al., 2011; Yu et al., 2013; Yan et al., 2018; Alho et al., 2019; Luchnikova et al., 2020). At present, the conversion of these triterpenoids is mainly carried out using methods of chemical synthesis under conditions of extreme acidity and temperature, frequently with expensive catalysts as well as protective groups of the reactive centers of the molecule (Alho et al., 2019). Along with chemical modification, attempts are being made to biologically transform OA and GA. Compared to traditional organic synthesis, the use of enzymes and whole cells of microorganisms to obtain target compounds is technologically promising. This is due to their exceptional chemo- and stereoselectivity, a wide range of metabolizable substrates, minimal side reactions, unnecessary multiple protection and deprotection steps, and importantly, stable activity under extreme environmental conditions. The largest part of the processes of OA and GA biological transformation is catalyzed by filamentous fungi (Capel et al., 2011; Martinez et al., 2013; Gong et al., 2014; Wu et al., 2018), although fungi are technologically impractical and unsafe due to their mycelial growth and the ability to produce mycotoxins. Examples of bacterial transformation of OA and GA are rare and include processes catalyzed by members of the genera Bacillus, Nocardia, and Streptomyces, which exhibit transforming activity at triterpenoid concentrations no more than 0.3 g/L (Ludwig et al., 2015; Xu et al., 2017, 2020). We have previously described a nonpathogenic strain Rhodococcus rhodochrous IEGM 1360 catalyzing the directed transformation of 1.0 g/L OA or GA for 7 days (Luchnikova et al., 2021, 2022).
One of the effective approaches for intensifying the processes of bioconversion of complex hydrophobic compounds is the use of resting cells—bacterial cells in the stationary phase of growth washed from nutrients and resuspended in a buffer solution (Grishko et al., 2013; Ivshina et al., 2015; Nawawi et al., 2016; Ivanova et al., 2022). The absence of growth factors allows regulating the amount of biomass and its physiological state that helps to increase the efficiency and reduce the duration of the target process. At the same time, the use of a buffer solution as a biotransformation medium reduces the risk of bacterial contamination, limits the growth of foreign microflora, and facilitates the process of metabolite isolation. Previously, we have demonstrated successful intensification of the biotransformation processes of plant terpenoids, namely betulin and dehydroabietic acid (Grishko et al., 2013; Ivanova et al., 2022).
The present work was aimed at studying the possibility of employing resting cells of R. rhodochrous IEGM 1360 to enhance the processes of OA and GA biotransformation.
MATERIALS AND METHODS
Culture collection. In this research, Rhodococcus rhodochrous IEGM 1360—an OA and GA biotransforming strain—from the Regional Specialised Collection of Alkanotrophic Microorganisms (acronym IEGM, WFCC #285; USU 73559; www.iegmcol.ru) was used. R. rhodochrous IEGM 1360 was isolated from moss rhizosphere (Tikhaya Bay, Guker Island, Franz Josef Land, Arkhangel’sk region, Russia). The complete genome of the strain has been sequenced and is available in the NCBI database (JAJNCN000000000.1).
Chemicals. High-purity (≥97%) OA (CAS 508-02‑1) and GA (CAS 471-53-4) manufactured by Acros Organics (United States) and TCI (Belgium), respectively, were used in the experiments. Chemical reagents, including acetonitrile, dimethyl sulfoxide (DMSO), methanol, chloroform, ethyl acetate, n-hexane, and isopropanol, were of chemically pure or analytically pure grades (Cryochrome, Russia; Merck, Germany; Sigma-Aldrich, United States). A Millipore Simplicity Personal Ultrapure Water System (Millipore, United States) was used to obtain the ultrapure water.
OA and GA solubility. Solubility of triterpenoids was determined by a micromethod of serial two-fold dilutions using 96-well round-bottomed polystyrene microplates. 100 µL of phosphate-alkaline buffers (pH 5.0, 6.0, 7.0, 8.0, or 9.0) were inoculated into microplate wells. 10 mg of OA or GA dissolved in 100 µL of DMSO were added to the first well of each row and mixed thoroughly. A portion of 100 μL from the resulting mixture was transferred to the next well. The procedure was repeated until a series of two-fold dilutions were formed. The concentration of OA or GA in one row ranged from 0.5 g/L to 0.003907 g/L. Solubility was defined as a concentration at which OD630 of the experimental OA or GA solutions and OD630 of the control buffer were comparable (Multiscan Ascent microplate reader, Thermo Electron Corporation, Finland).
Preparation of resting cell suspensions. Rhodococci were pre-grown in meat-peptone broth (MPB) for 48 h. Bacterial cells of the stationary phase were precipitated by centrifugation (3000 rpm, Hermle Z 200 A, Germany) for 10 min and washed three times with an equivalent volume of the phosphate-alkaline buffer (pH 7.0). The washed cells were resuspended in 25 mL of the Clark−Labs phosphate-alkaline buffers with various 0.1 М KH2PO4 and 0.1 M NaOH ratio (рН 5.0, 6.0, 7.0, 8.0, or 9.0) (Dawson et al., 1986). The optical density (OD600) was adjusted to 2.0, 2.2, 2.4, 2.6, or 2.8 (Lambda EZ201 spectrophotometer, Perkin Elmer, United States). Additionally, the concentration of cells (g/L) was estimated by calculating the weight of dry biomass.
Biotransformation conditions. The experiments were carried out under constant shaking conditions on a Certomat IS orbital shaker (Sartorius, Germany) at 160 rpm and 28°C. OA or GA dissolved in DMSO (1 mg : 10 µL) were used in the concentration of 1.0 g/L. The resting cell suspensions without triterpenoids were used as biotic controls, and the phosphate-alkaline buffers with terpenoids were used as abiotic ones.
Phase-contrast microscopy. Visualization of cells and measurement of their sizes were performed with an Axio Imager M2 optical microscope (Carl Zeiss, Germany) equipped with an Axiocam 506 Color camera in a phase-contrast mode with ×1000 magnification. The volume (V) and surface area (S) of cells were calculated by Equations (1) and (2) (Neumann et al., 2005):
where r is 1/2 of cell width; π is 3.14; h is cell length.
Atomic force and confocal laser scanning microscopy. The morphology of bacterial cells was analyzed using a combined microscopic system, which consists of an Olympus FV1000 laser confocal scanning microscope (CLSM) (Olympus Corporation, Japan) and an Asylum MFP-3D-BIO atomic force microscope (AFM) (Asylum Research, United States). Prior to imaging, a drop (15–20 µL) of the cell suspension was placed on a glass cover slip, mixed with the same volume of Live/Dead® BacLightTM Bacterial Viability Kit (Invitrogen, United States) and allowed to dry at room temperature in darkness for 10–15 min. Then the cover slip was rinsed with deionized water and scanned with CLSM. SYTO9 and propidium iodide were excited by the argon laser (λ = 488 nm) with a 505/525 nm barrier filter and by the He–Ne laser (λ = 543 nm) with a 560/660 nm barrier filter, respectively. Images (size, 0.12 × 0.12 mm; resolution, 1600 × 1600 pixels) were obtained at a rate of 40 nm/pixels. The images were analyzed by the FV10-ASW 3.1 program (Olympus Corporation, Japan). CLSM images were imported into the Igor Pro 6/22A AFM software (Wave Metrics, United States). AFM scanning was carried out in the semi-contact mode in air using an AC240TS silicon cantilever with a resonant frequency of 50–90 kHz and a spring constant of 0.5–4.4 N/m.
Qualitative and quantitative analysis of OA, GA and their derivatives. To isolate residual OA, GA and their derivatives, the post-fermentation media with bacterial cells were acidified with 10% HCl solution and extracted three times with an equal volume of ethyl acetate. The combined organic layers were washed with an aqueous solution of 1% Na2CO3 and then with distilled water to a final рH of 7.0. The ethyl acetate extract was dried over anhydrous Na2SO4. The solvent was removed with a Laborota 4000 rotary evaporator (Heidolph, Germany). The ethyl acetate extracts were qualitatively analyzed by thin layer chromatography (TLC) on Alugram® Xtra SIL G/UV254 plates (Macherey-Nagel, Germany) with the n-hexane–ethyl acetate (1 : 1 or 4 : 1, v/v) mixture as an eluent. The sample spots were visualized by spraying the plates with a solution of 15% H2SO4 followed by heating at 100–120°C for 2–3 min.
Qualitative analysis of the extracts and assessment of the dynamics of GA transformation product formation were performed by gas chromatography-mass spectrometry (GC-MS) using an Agilent 6890N/5975B chromatograph (Agilent Technologies, United States) equipped with a HP-5ms UI column (30 m × 0.25 mm, 0.25 µm) and operating in an electron impact ionization mode (70 eV). Helium was used as a carrier gas. The evaporator temperature was 300°С. The column temperature was programmed from 100 to 300°C in increments of 30°C/min; the retention time was 18.5–23.0 min. The sample was injected in a volume of 0.1‒0.2 µL with a flow split of 1 : 9–1 : 39. Mass spectra were recorded in the range of 35‒535 m/z at a rate of 1.5 scans/s. The samples were initially treated with (trimethylsilyl)diazomethane (Sigma-Aldrich, United States). The obtained mass spectra were compared with the known mass spectra from the NIST08 MS Library.
The formation dynamics of OA transformation product was assessed by high performance liquid chromatography (HPLC) on a Kromasil 100-5-C18 reversed phase column (С18; particle size, 5 µm; pore size, 100 Å; 250 mm × 4.6 mm, Eka Chemicals AB, Sweden). The eluent was a mixture of acetonitrile and deionized water in a percentage ratio of 80 : 20 (v/v). The flow rate was 1 mL/min, the temperature of the column was 40°С, and the injected sample volume was 20 μL. Samples were previously dissolved in isopropanol (Cryochrome, Russia). Concentrations of OA and its derivative were calculated according to Equation (3) compiled from the dependence curve of the OA analytical standard concentration on the peak area.
where x is peak area, mAU; y is concentration, %.
Isolation and identification of OA and GA derivatives. The primary identification of the derivatives was performed by comparing the mass spectra of methyl esters of the obtained compounds (Fig. S1) with the mass spectra of methyl esters of the known compounds from the NIST08 Mass Spectral Library (GC-MS (m/z): 3-oxo-OA, 468.3 (М+); 3-oxo-GA, 482.4 (М+)). Mass spectra were considered identified if the mass spectrum of the studied substance matched with the library one with a similarity coefficient exceeding 90%. The structure of the derivatives was confirmed by NMR spectroscopy. 1H, 13C, and DEPT NMR spectra were recorded on a Bruker AVANCE II spectrometer (Bruker BioSpin GmbH, Germany) at 400 and 100 MHz, respectively (Figs. S2, S3). CDCl3 was used as a solvent. Optical rotation was measured on a Perkin Elmer 341 polarimeter (Perkin Elmer, United States) at 589 nm for solutions of the derivatives in CHCl3. The melting point was recorded using an OptiMelt MPA100 automated melting point system (Stanford Research Systems, United States) with a heating rate of 1°C/min.
The extract (190.5 mg) obtained during OA biotransformation by resting cells of R. rhodochrous IEGM 1360 was separated using a flash chromatograph (Buchi, Switzerland) on a Sepacore Silica 40g cartridge (26.7 × 127 mm) at a substance to sorbent ratio of 1 : 30 (w/w). Using 100% chloroform as an eluent, 114.4 mg of compound 2 and 40.2 mg of residual OA were successively obtained. Recrystallization of compound 2 was carried out in the isopropanol–chloroform system (3 : 1, v/v).
3-Oxoolean-12-en-28-oic acid (2). White powder, mp 202.6°C (lit.: 167−169°C (Maldonado et al., 2015)), Rf 0.45 (n-hexane–ethyl acetate 1 : 1, v/v), \(\left[ \alpha \right]_{D}^{{20}}\) = +38.8° (с 0.5, CHCl3) (lit.: \(\left[ \alpha \right]_{D}^{{25}}\) = +73.6° (c 0.26, CHCl3) (Ma et al., 2002); \({{\left[ \alpha \right]}_{D}}\) = +93.5° (c 0.23, CHCl3) (Maldonado et al., 2015)). 1Н NMR (400 MHz, CDCl3, δ, ppm, J/Hz): 5.30 (1H, H-12); 2.85 (1H, dd, J = 4.0, 16.0 Hz), 2.53 and 2.35 (2H, 2m), 1.14; 1.08; 1.04; 1.02; 0.93; 0.90; 0.81 (each 3Н, 7s, 7CH3). 13С NMR (100 MHz, CDCl3, δ, ppm): 217.47 (C-3); 183.19 (C-28); 143.64 (C-13); 122.42 (C-12); 55.36; 47.40; 46.91; 46.58; 45.86; 41.78; 41.12; 39.32; 39.12; 36.81; 34.11; 33.83; 33.01; 32.42; 32.21; 30.65; 27.71; 26.48; 25.80; 23.54; 23.50; 22.96; 21.42; 19.57; 16.99; 14.99.
The extract (180.9 mg) obtained during GA biotransformation by resting cells of R. rhodochrous IEGM 1360 was separated using a flash chromatograph (Buchi, Switzerland) on a Sepacore Silica 40g cartridge (26.7 mm × 127 mm) at a substance to sorbent ratio of 1 : 30 (w/w). A mixture of chloroform and isopropanol with a concentration gradient from 100 : 0 to 99 : 1 (v/v) was used as an eluent. Separation yielded 145.5 mg of compound 4. Recrystallization of compound 4 was carried out in the isopropanol–chloroform system (3 : 1, v/v).
3,11-Dioxoolean-12-en-30-oic acid (4). White powder, mp 274.4°C (lit.: 311–313°C (Beseda et al., 2010)), Rf 0.45 (n-hexane–ethyl acetate 4 : 1, v/v), \(\left[ \alpha \right]_{D}^{{20}}\) = +218.2° (с 0.5, CHCl3) (lit.: \(\left[ \alpha \right]_{D}^{{20}}\) = +184.5 (c 0.4, CHCl3) (Beseda et al., 2010)). 1Н NMR (400 MHz, CDCl3, δ, ppm, J/Hz): 5.74 (1H, H-12); 2.96 (1H, m); 2.63 (1H, m); 2.44 (1H, s, H-9); 2.35 (1H, m); 2.22 (1Н, dd, J = 4.0, 16.0 Hz); 1.37; 1.27; 1.22; 1.17; 1.10; 1.06; 0.85 (each 3Н, 7s, 7CH3). 13С NMR (100 MHz, CDCl3, δ, ppm): 217.07 (C-3); 199.56 (C-11); 181.21 (C-30); 169.66 (C-13); 128.48 (C-12); 61.08; 55.52; 48.29; 47.76; 45.30; 43.79; 43.37; 41.01; 39.76; 37.73; 36.75; 34.21; 32.19; 31.89; 30.95; 28.57; 28.39; 26.58; 26.44 (2C); 23.34; 21.43; 18.83; 18.56; 15.61.
In silico analysis of OA, GA and their derivatives. The ecotoxicity and solubility of OA, GA and their derivatives were calculated using the ECOSAR program (Ecological Structure Activity Relationship, EPA, United States). The potential acute toxicity and chronic toxicity to aquatic organisms were predicted based on the available data on the toxic effects of organic compounds of various chemical classes using a computational analysis of structure–function relationship in molecules.
The estimated biological activities of OA, GA and their derivatives were predicted based on their structural formulas using the PASS software (Prediction of Activity Spectra for Substances, http://www.pharmaexpert.ru/passonline/index.php). Exploring the biological potential of substances generated a list of anticipated types of biological activity, including the evaluation of detection (Pa)/non-detection (Pi) pro-babilities of the latter. The highest probability of biological activity was taken as 1.
Phytotoxicity of OA, GA and their derivatives. Phytotoxicity in relation to oats Avena sativa L. was assessed according to the MP 2.1.7.2297-07 Guidelines (2007). The seeds with a 95% germination were used in the experiments. The seeds were germinated for 3 days in sterile Petri dishes with filter paper soaked in distilled water (5 mL). Then the germinated seeds were treated with supernatants obtained after 3 days of OA or GA biotransformations. The level of phytotoxicity was determined after 7 days as the effect of inhibition of the growth of the root system according to Equation (4):
where ЕI is effect of inhibition, %; Lexp is average length of roots in the experiment, cm; Lc is average length of roots in the control, cm.
The phytotoxic effect was considered proven if the El value was 20% or more.
Antimicrobial activity of OA, GA and their derivatives. Minimal inhibitory concentrations (MICs) of OA, GA and their derivatives in relation to bacterial test cultures Bacillus subtilis ATCC 6633, Escherichia coli ATCC 25922, Micrococcus luteus NCIMB 196, and Staphylococcus aureus ATCC 25923 were determined by the method of serial twofold dilutions (Clinical and Laboratory Standards Institute, 2022) using 96-well polystyrene plates. OA, GA and their derivatives dissolved in DMSO (1 mg : 10 μL) were added to the wells containing MPB at an initial concentration of 50 mg/mL, followed by serial twofold dilutions. After that, 10 μL of the bacterial suspension (2 × 106 cells/mL) were inoculated to each well. The plates were incubated for 24 h at optimal temperatures (28 or 37°C) for test cultures. Viability of bacterial cells was assessed by staining with iodonitrotetrazolium chloride. The formation of insoluble formazan and the corresponding purple color indicated the presence of actively respiring cells in the wells. DMSO at the same concentrations was used as a control for the effect of the solvent; antibiotic substances (ampicillin and kanamycin) served as reference drugs.
Statistical analysis. The experiments were carried out in three, five, or ten replications. For statistical analysis of data, the program STATISTICA was used (StatSoft Russia, 2015).
RESULTS AND DISCUSSION
Biotransformation of OA and GA by resting cells of R. rhodochrous IEGM 1360. We have previously shown (Luchnikova et al., 2021) that under growth conditions R. rhodochrous IEGM 1360 catalyzed the directed conversion of 1.0 g/L OA and GA with the formation of 0.9 and 26% 3-oxo derivatives, respectively, for 7 days (Fig. 1). In this research, the influence of the buffer solution acidity and the amount of biomass of resting cells on the process of the biotransformation of triterpenoids was studied. Rhodococci are known to survive at extreme pH values from 1.0 to 11.0, while neutral pH values are optimal (Pátek et al., 2021). According to our data, among the tested acidity conditions, only the buffer solutions with pH 8.0 and 6.0 significantly reduced (up to 3 days) the duration of the biotransformation process of OA and GA with the formation of 14 and 31% of oxidized derivatives, respectively (data not shown).

Scheme of biotransformation of OA (1) and GA (3) by R. rhodochrous IEGM 1360 cells with the formation of 3-oxo-OA (2) and 3-oxo-GA (4), respectively.
One of the advantages of using resting cells is the possibility of strict control of the initial biomass amount throughout the biotransformation process. The absence of active growth of rhodococci during the conversion of OA and GA was confirmed by measuring the optical densities of suspensions of resting cells (OD600), which did not change significantly during the entire experiment (Fig. 2). An increase in the OD600 of the cell suspensions (Fig. 2, day 0) compared to the biotic control is associated with the addition of hydrophobic OA or GA into the transformation medium.

Changes in the optical density (OD600) of suspensions of resting cells of R. rhodochrous IEGM 1360 during biotransformation (1) of OA (a) and GA (b), (2) is biotic control. Arrows indicate the addition of OA and GA.
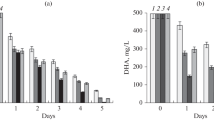
Early studies on the bioconversion of various organic compounds using resting Rhodococcus cells showed the dependence of the catalytic activity of cells on the biomass optical density (Grishko et al., 2013; Nawawi et al., 2016; Ivanova et al., 2022). The data we obtained on the biotransformation of OA and GA confirmed the previously observed dependence (Fig. 3). The OA conversion was found to occur most efficiently (derivative yield, 61%) using a suspension with OD600 2.6 (cell concentration, 19 g/L), while the conversion of GA occurred most efficiently (derivative yield, 100%) using a suspension with OD600 2.2 (cell concentration, 13 g/L). The fact that a more efficient bioconversion of GA compared to OA requires fewer cells per substrate unit seems to be associated with an increased resistance of bacterial cells to this triterpenoid. It should be noted that a subsequent increase in the amount of biomass to OD600 2.8 (cell concentration, 31 g/L, Fig. 3) led to a decrease in the yields of both OA and GA derivatives. This is consistent with the previously obtained results on the biotransformation of betulin by resting cells of R. rhodochrous IEGM 66 (Grishko et al., 2013). The observed phenomenon is apparently due to a decrease in mass transfer with an increase in the cell biomass density and redistribution of the substrate in the buffer system.

Dependence of the formation (1) of 3-oxo-OA (a) and 3-oxo-GA (b) on the amount of biomass of resting cells of R. rhodochrous IEGM 1360 (2) in pH 8.0 (a) and pH 6.0 (b) buffers. The data of the 3rd day of the biotransformation process are shown.
Thus, the experiments showed that the most efficient conversion of OA and GA was provided using suspensions of resting cells of R. rhodochrous IEGM 1360 with OD600 2.6 in pH 8.0 buffer and OD600 2.2 in pH 6.0 buffer, respectively (Fig. S4).
Effects of OA and GA on resting cells of R. rhodochrous IEGM 1360. It should be noted that OA was less soluble in pH 8.0 buffer (4 mg/L) compared to buffers with pH values of 5.0, 6.0, 7.0, or 9.0, while GA was less soluble in pH 6.0 buffer (4 mg/L) compared to buffers with pH values of 5.0, 7.0, 8.0, or 9.0 (Fig. 4). It is possible that the high catalytic activity of bacterial cells to OA and GA in pH 8.0 and pH 6.0 buffers, respectively, was due to the nature of the interaction of rhodococci with the crystalline substrate, namely, the formation of cell aggregates on the surface of OA and GA particles. According to Atrat et al. (1991), the mechanism of interaction between Mycobacterium fortuitum cells and sitosterol particles was characterized by “immobilization of cells on substrate particles,” i.e., the formation of stable multicellular agglomerates on the particle surface. Using electron microscopy, the authors showed that substrate consumption occurred due to direct contact between cells and particles of the substrate, where a multicomponent mobile mesophase was formed. The mesophase consisted of glycolipids, synthetic detergents, sterol, and water and performed the gradual dissolution of the substrate, starting the mechanism of its transformation and transport into the cell (Atrat et al., 1991). Actinobacteria interacting with hydrophobic compounds are known to synthesize glycolipid biosurfactants, for which the function is also to dissolve the substrate and trigger the mechanism of its transport into the cell (Ivshina et al., 1998). It was previously shown that biotransformations of terpenoids betulin and dehydroabietic acid were accompanied by cell adhesion to the surface of substrates and by the formation of an extracellular lipophilic fluid—a biosurfactant (Tarasova et al., 2017; Cheremnykh et al., 2018; Ivanova et al., 2022). Assuming that the transformation of OA and GA by Rhodococcus bacteria proceeds in a similar way, then the high catalytic activity of the cells to crystalline particles of OA and GA may be due to the formation of a multicomponent mobile mesophase or biosurfactants detected using AFM and combined AFM-CLSM scanning (Fig. 5).

Solubility of OA (a) and GA (b) in buffers with various pHs.

AFM and combined AFM-CLSM images of resting cells of R. rhodochrous IEGM 1360 in the presence of OA (a, b) and GA (c, d).
Morphometric studies revealed that OA induced a decrease in the cell surface area-to-volume ratio and an increase in the cell wall roughness of resting cells (Table 1) compared to the growing culture (Luchnikova et al., 2021, 2022). An increase in the degree of roughness of the cell surface may result from the secreted extracellular polymers (see Fig. 5) and changes in the lipid composition of the cell wall that enhance the van der Waals forces facilitating better adhesion of cells to the substrate (Uzoechi and Abu-Lail, 2019). Apparently, the revealed changes in morphometric parameters provided a more efficient contact of cells with the substrates and thereby determined an increased level of bioconversion.
Bioactivity and toxicity of OA and GA derivatives. It is known that 3-oxo-OA has a pronounced in vivo anti-melanoma activity (Huang et al., 2006) and in vitro antileishmanial and antitrypanosomal activities (Funari et al., 2016). Whereas 3-oxo-GA exhibits in vitro inhibitory activity against lipoxygenases; the biosynthesis products of the latter may contribute to the development of inflammatory and autoimmune diseases, bronchial asthma, and cancer (Choudhary et al., 2009). The potential inhibitory activity of 3‑oxo-GA against SARS-CoV-2 protease Mpro has been documented by in silico molecular docking (Florez and Singh, 2020). Using the PASS Online program, the compounds were assessed for their potential bioactivity. It was found that 3-oxo-OA and 3-oxo-GA with a high degree of probability (0.822 and 0.726, respectively) can act as an apoptosis agonist and an antioxidant agent, respectively (Table 2).
Earlier, the inhibitory activity of native OA and GA was shown against pathogenic strains of Staphylococcus aureus, Bacillus subtilis and Pseudomonas aeruginosa, respectively (Duric et al., 2013; Kannan et al., 2019). Even though we did not reveal an antimicrobial potential of 3-oxo derivatives 3 and 4 using the PASS Online program, the experimental in vitro assessment of the antimicrobial activity of the obtained metabolites showed that the C3 oxidation of OA enhanced its inhibitory activity against pathogenic bacteria Micrococcus luteus, Escherichia coli, S. aureus, and B. subtilis. The antimicrobial activity of 3 was higher than that of the widely used antibiotic ampicillin (Table 3).
An in silico analysis of the obtained 3-oxo derivatives using the ECOSAR program showed that compared to the native compounds these triterpenoids may have reduced acute and chronic toxicity to aquatic organisms (Table 4). However, both the native OA and GA and their derivatives are presumably characterized by extremely low solubility in water.
The toxicity of OA, GA and their derivatives in relation to plants was determined using the seeds of oats. After treatment with the studied compounds, the length of roots was measured and proved no significant differences indicative of a phytotoxic effect (Table 5, Fig. 6).

Change in root length of oat Avena sativa L. treated with OA, GA and their transformation products: 1—OA, 2—GA, 3—OA biotransformation products, 4—GA biotransformation products, 5—biotic control, 6—abiotic control (water), 7—medium control.
As a result of the studies, the optimal conditions for C3 oxidative biotransformations of OA and GA (1.0 g/L) were selected using resting cells of R. rhodochrous IEGM 1360. The cell suspensions with OD600 2.6 in pH 8.0 buffer and with OD600 2.2 in pH 6.0 buffer catalyzed the formation of 3-oxo-OA (61%) and 3‑oxo-GA (100%), respectively, for 3 days. The safety of the derivatives to aquatic organisms and plants was shown in silico and in vitro, respectively. The pronounced antimicrobial activity of 3-oxo-OA was revealed in vitro. The data obtained expand our understanding of the catalytic potential of actinobacteria of the genus Rhodococcus and their possible applications in the directed conversion of complex hydrophobic compounds to obtain biologically active derivatives.
REFERENCES
Alho, D.P.S., Salvador, J.A.R., Cascante, M., and Marin, S., Synthesis and antiproliferative activity of novel heterocyclic glycyrrhetinic acid derivatives, Molecules, 2019, vol. 24, p. 766. https://doi.org/10.3390/molecules24040766
Atrat, P., Hosel, P., Richter, W., Meyer, H.W., and Horhold, C., Interactions of Mycobacterium fortuitum with solid sterol substrate particles, J. Basic Microbiol., 1991, vol. 31, pp. 413–422. https://doi.org/10.1002/jobm.3620310605
Beseda, I., Czollner, L., Shah, P.S., Khunt, R., Gaware, R., Kosma, P., Stanetty, C., del Ruiz-Ruiz, M.C., Amer, H., Mereiter, K., Da Cunha, T., Odermatt, A., Claßen-Houben, D., and Jordis, U., Synthesis of glycyrrhetinic acid derivatives for the treatment of metabolic diseases, Bioorg. Med. Chem., 2010, vol. 18, pp. 433–454. https://doi.org/10.1016/j.bmc.2009.10.036
Calixto, J.B., The role of natural products in modern drug discovery, An. Acad. Bras. Cienc., 2019, vol. 91. p. e20190105. https://doi.org/10.1590/0001-3765201920190105
Capel, C.S., De Souza, A.C.D., De Carvalho, T.C., De Sousa, J.P.B., Ambrósio, S.R., Martins, C.H.G., Cunha, W.R., Galán, R.H., and Furtado, N.A.J.C., Biotransformation using Mucor rouxii for the production of oleanolic acid derivatives and their antimicrobial activity against oral pathogens, J. Ind. Microbiol. Biotechnol., 2011, vol. 38, pp. 1493–1498. https://doi.org/10.1007/s10295-010-0935-y
Cheremnykh, K.M., Luchnikova, N.A., Grishko, V.V., and Ivshina, I.B., Bioconversion of ecotoxic dehydroabietic acid using Rhodococcus actinobacteria, J. Hazard. Mater., 2018, vol. 346, pp. 103–112. https://doi.org/10.1016/j.jhazmat.2017.12.025
Choudhary, M.I., Siddiqui, Z.S., and Nawaz, S.A., Microbial transformation of 18β-glycyrrhetinic acid by Cunninghamella elegans and Fusarium lini, and lipoxygenase inhibitory activity of transformed products, Nat. Prod. Res., 2009, vol. 23, pp. 507–513. https://doi.org/10.1080/14786410500463536
Clinical and Laboratory Standards Institute. Performance Standards for Antimicrobial Susceptibility Testing M100, Malvern: Clinical and Laboratory Standards Institute, 2022, 32nd ed.
Dawson, R., Elliott, D.C., Elliott, W.H., and Jones, K.M., Data for Biochemical Research, Oxford: Oxford University Press, 1986, 3rd ed.
Duric, K., Kovac-Besovic, E., Niksic, H., and Sofic, E., Antibacterial activity of methanolic extracts, decoction and isolated triterpene products from different parts of birch, Betula pendula Roth, J. Plant Stud., 2013, vol. 2, no. 2, pp. 61–70. https://doi.org/10.5539/jps.v2n2p61
Funari, C.S., de Almeida, L., Passalaqua, T.G., Martinez, I., Ambrosio, D.L., Cicarelli, R.M.B., Silva, D.H.S., and Graminha, M.A.S., Oleanonic acid from Lippia lupulina (Verbenaceae) shows strong in vitro antileishmanial and antitrypanosomal activity, Acta Amazon., 2016, vol. 46, pp. 411–416. https://doi.org/10.1590/1809-4392201600204
Gong, T., Zheng, L., Zhen, X., He, H.X., Zhu, H.X., and Zhu, P., Microbial transformation of oleanolic acid by Trichothecium roseum, J. Asian Nat. Prod. Res., 2014, vol. 16, pp. 383–386. https://doi.org/10.1080/10286020.2014.884564
Grishko, V.V., Tarasova, E.V., and Ivshina, I.B., Biotransformation of betulin to betulone by growing and resting cells of the actinobacterium Rhodococcus rhodochrous IEGM 66, Process Biochem., 2013, vol. 48, pp. 1640–1644. https://doi.org/10.1016/j.procbio.2013.08.012
Huang, D., Ding, Y., Li, Y., Zhang, W., Fang, W., and Chen, X., Anti-tumor activity of a 3-oxo derivative of oleanolic acid, Cancer Lett., 2006, vol. 233, pp. 289–296. https://doi.org/10.1016/j.canlet.2005.03.019
Ivanova, K.M., Grishko, V.V., and Ivshina, I.B., Highly efficient biodegradation of ecotoxic dehydroabietic acid by resting cells of Rhodococcus rhodochrous IEGM 107, Microbiology (Moscow), 2022, vol. 91, pp. 364–377.
Ivshina, I.B., Kuyukina, M.S., Philp, J.C., and Christofi, N., Oil desorption from mineral and organic materials using biosurfactant complexes produced by Rhodococcus species, World J. Microbiol. Biotechnol., 1998, vol. 14, pp. 711–717. https://doi.org/10.1023/A:1008885309221
Ivshina, I.B., Mukhutdinova, A.N., Tyumina, H.A., Vikhareva, H.V., Suzina, N.E., El’-Registan, G.I., and Mulyukin, A.L., Drotaverine hydrochloride degradation using cyst-like dormant cells of Rhodococcus ruber, Curr. Microbiol., 2015, vol. 70, pp. 307–314. https://doi.org/10.1007/s00284-014-0718-1
Kannan, S., Sathasivam, G., and Marudhamuthu, M., Decrease of growth, biofilm and secreted virulence in opportunistic nosocomial Pseudomonas aeruginosa ATCC 25619 by glycyrrhetinic acid, Microb. Pathog., 2019, vol. 126, pp. 332–342. https://doi.org/10.1016/j.micpath.2018.11.026
Kumar, D. and Dubey, K.K., Hybrid approach for transformation for betulin (an anti-HIV molecule), in New and Future Developments in Microbial Biotechnology and Bioengineering // Eds. Gupta, V., Pandey, A. Amsterdam: Elsevier, 2019. P. 193–203. Luchnikova, N.A., Grishko, V.V., and Ivshina, I.B., Biotransformation of oleanane and ursane triterpenic acids, Molecules, 2020, vol. 25, p. 5526. https://doi.org/10.3390/molecules25235526
Luchnikova, N.A., Grishko, V.V., Kostrikina, N.A., Sorokin, V.V., Mulyukin, A.L., and Ivshina, I.B., Biotransformation of oleanolic acid using Rhodococcus rhodochrous IEGM 757, Catalysts, 2022, vol. 12, p. 1352. https://doi.org/10.3390/catal12111352
Luchnikova, N.A., Ivanova, K.M., Tarasova, E.V., Grishko, V.V., and Ivshina, I.B., Bioconversion of oleanane-type terpenoids by actinobacteria, Proc. 9th Inform. School for Young Scientists, Ekaterinburg, 2021, pp. 15–26. https://doi.org/10.32460/ishmu-2021-9-0002
Ludwig, B., Geib, D., Haas, C., Steingroewer, J., Bley, T., Muffler, K., and Ulber, R., Whole-cell biotransformation of oleanolic acid by free and immobilized cells of Nocardia iowensis: characterization of new metabolites, Eng. Life Sci., 2015, vol. 15, pp. 108–115. https://doi.org/10.1002/elsc.201400121
Ma, C., Nakamura, N., and Hattori, M., Chemical modification of oleanene type triterpenes and their inhibitory activity against HIV-1 protease dimerization, Chem. Pharm. Bull., 2002, vol. 48, pp. 1681–1688.
Maldonado, E., Amador, S., and Juárez-Jaimes, V., Secondary metabolites from Asclepias otarioides, J. Mex. Chem. Soc., 2015, vol. 59, pp. 50–52. https://doi.org/10.29356/jmcs.v59i1.14
Martinez, A., Rivas, F., Perojil, A., Parra, A., Garcia-Granados, A., and Fernandez-Vivas, A., Biotransformation of oleanolic and maslinic acids by Rhizomucor miehei, Phytochemistry, 2013, vol. 94, pp. 229–237. https://doi.org/10.1016/j.phytochem.2013.05.011
Nawawi, N.M., Ahmad, S.A., Maniyam, M.N., and Ibrahim, A.L., Biotransformation of phenol by the resting cells of Rhodococcus sp. NAM 81, Indian J. Fundam. Appl. Life Sci., 2016, vol. 6, pp. 101–107.
Neumann, G., Veeranagouda, Y., Karegoudar, T.B., Sahin, Ö., Mäusezahl, I., Kabelitz, N., Kappelmeyer, U., and Heipieper, H.J., Cells of Pseudomonas putida and Enterobacter sp. adapt to toxic organic compounds by increasing their size, Extremophiles, 2005, vol. 9, pp. 163–168. https://doi.org/10.1007/s00792-005-0431-x
Pátek, M., Grulich, M., and Nešvera, J., Stress response in Rhodococcus strains, Biotechnol. Adv., 2021, p. 107698. https://doi.org/10.1016/j.biotechadv.2021.107698
Singh, S. and Florez, H., Bioinformatic study to discover natural molecules with activity against COVID-19, F1000Research, 2020, vol. 9, art. 1203, pp. 1–15. https://doi.org/10.12688/f1000research.26731.1
Tarasova, E.V., Grishko, V.V., and Ivshina, I.B., Cell adaptations of Rhodococcus rhodochrous IEGM 66 to betulin biotransformation, Process Biochem., 2017, vol. 52, pp. 1–9. https://doi.org/10.1016/j.procbio.2016.10.003
Uzoechi, S.C. and Abu-Lail, N.I., The effects of β-lactam antibiotics on surface modifications of multidrug-resistant Escherichia coli: a multiscale approach, Micros. Microanal., 2019, vol. 25, pp. 135–150. https://doi.org/10.1017/S1431927618015696
Wu, S.Y., Cui, S.C., Wang, L., Zhang, Y.T., Yan, X.X., Lu, H.L., Xing, G.Z., Ren, J., and Gong, L.K., 18β-Glycyrrhetinic acid protects against alpha-naphthylisothiocyanate-induced cholestasis through activation of the Sirt1/FXR signaling pathway, Acta Pharmacol. Sin., 2018, vol. 39, pp. 1865–1873. https://doi.org/10.1038/s41401-018-0110-y
Xu, S.H., Chen, H.L., Fan, Y., Xu, W., and Zhang, J., Application of tandem biotransformation for biosynthesis of new pentacyclic triterpenoid derivatives with neuroprotective effect, Bioorg. Med. Chem. Lett., 2020, vol. 30, p. 126947. https://doi.org/10.1016/j.bmcl.2019.126947
Xu, S.H., Wang, W.W., Zhang, C., Liu, X.F., Yu, B.Y., and Zhang, J., Site-selective oxidation of unactivated C–H sp3 bonds of oleanane triterpenes by Streptomyces griseus ATCC 13273, Tetrahedron, 2017, vol. 73, pp. 3086–3092. https://doi.org/10.1016/j.tet.2017.04.036
Yan, S., Lin, H., Huang, H., Yang, M., Xu, B., and Chen, G., Microbial hydroxylation and glycosidation of oleanolic acid by Circinella muscae and their anti-inflammatory activities, Nat. Prod. Res., 2018, vol. 33, pp. 1849–1855. https://doi.org/10.1080/14786419.2018.1477150
Yu, F., Wang, Q., Zhang, Z., Peng, Y., Qiu, Y., Shi, Y., Zheng, Y., Xiao, S., Wang, H., Huang, X., Zhu, L., Chen, K., Zhao, C., Zhang, C., Yu, M., et al., Development of oleanane-type triterpenes as a new class of HCV entry inhibitors, J. Med. Chem., 2013, vol. 56, pp. 4300–4319. https://doi.org/10.1021/jm301910a
ACKNOWLEDGMENTS
The work was carried out using the equipment of the Core Facilities Centers “Regional Specialised Collection of Alkanotrophic Microorganisms” and “Research of Materials and Matter” at Perm Federal Research Center of the Ural Branch of the Russian Academy of Sciences.
Funding
The research was funded by the Russian Foundation for Basic Research (Grant 20-34-90104) and partially supported by the Ministry of Science and Higher Education of the Russian Federation (State Assignment АААА-А19-119112290008-4, 122010800029-1).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare no conflict of interest. Animals were not used in the experiments.
Supplementary Information
Rights and permissions
About this article

Cite this article
Luchnikova, N.A., Tarasova, E.V., Grishko, V.V. et al. Rhodococcus rhodochrous IEGM 1360, an Effective Biocatalyst of C3 Oxidative Transformation of Oleanane Triterpenoids. Microbiology 92, 204–214 (2023). https://doi.org/10.1134/S0026261722603360
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0026261722603360