
Abstract
Under some pathological conditions, such as pharmacoresistant epilepsy, status epilepticus or certain forms of genetic abnormalities, spiking activity of GABAergic interneurons may enhance excitation processes in neuronal circuits and provoke the generation of ictal discharges. As a result, anticonvulsants acting on the GABAergic system may be ineffective or even increase seizure activity. This paradoxical effect of the inhibitory system is due to ionic imbalances in nervous tissue. This review addresses the mechanisms of ictal discharge initiation in neuronal networks due to the imbalance of chloride and potassium ions, as well as possible ways to regulate ionic concentrations. Both the enhancement (or attenuation) of the activity of certain neuronal ion transporters and ion pumps and their additional expression via gene therapy can be effective in suppressing seizure activity caused by ionic imbalances. The Na+–K+-pump, NKCC1 and KCC2 cotransporters are important for maintaining proper K+ and Cl– concentrations in nervous tissue, having been repeatedly considered as pharmacological targets for antiepileptic exposures. Further progress in this direction is hampered by the lack of sufficiently selective pharmacological tools and methods for providing effective drug delivery to the epileptic focus. The use of the gene therapy techniques, such as overexpressing of the KCC2 transporter in the epileptic focus, seems to be a more promising approach. Another possible direction could be the use of optogenetic tools, namely specially designed light-activated ion pumps or ion channels. In this case, photon energy can be used to create the required gradients of chloride and potassium ions, although these methods also have significant limitations which complicate their rapid introduction into medicine.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
Epilepsy is a common neurological disorder whose principal manifestation is recurrent seizures. Approximately in 30% of patients, epileptic seizures are poorly controlled pharmacologically [1], thus corroborating the need to search for new therapies that would affect alternative targets and pathophysiological mechanisms of seizure generation. Currently, drugs, whose key mechanism of action consists in a modulation of voltage-gated sodium and calcium channels, or a potentiation of inhibitory GABA receptors, or an inhibition of excitatory glutamate receptors, are used to treat epilepsy [2].
Among the numerous abnormalities generated in the epileptic focus, including in pharmacoresistant epilepsy, it is worthwhile to highlight separately the ionic imbalance that leads to impaired functioning of the inhibitory GABAergic system and hyperexcitability of neurons. An intracellular accumulation of chloride ions and increased extracellular concentration of potassium cations exert the most pronounced epileptogenic effect. The ion dynamics in healthy and epileptic tissues is deeply analyzed by Raimondo et al. [3]. The imbalance of chloride ions in epileptic tissue has been discussed in detail in several reviews [4, 5]. The epileptogenic effect of potassium ions was addresses in an article by de Curtis et al. [6]. Model studies also indicate a critical role of ion dynamics in brain tissue in the generation of both interictal and ictal epileptiform discharges [7, 8].
Thus, neuronal hyperexcitability can be reduced and the activity of the GABAergic system in the epileptic focus can be restored by a targeted alteration of the ionic balance in the given brain region. This review is aimed to describe and analyze the most promising ways to affect the ionic balance in the epileptic focus, which may pave avenues for the development of new approaches to treat pharmacoresistant epilepsy. Below, we will consider consecutively what changes in ion dynamics occur during epileptic activity, what consequences they lead to, and how the ion dynamics could be manipulated.
POTASSIUM IMBALANCE DURING SEIZURE ACTIVITY
The resting potential of a neuron is primarily determined by a gradient of potassium ions between outside and inside of the cell. Normally, potassium ion concentration is 3 mM in the extracellular environment of the brain and 140 mM in mammalian neurons. According to the Nernst equation, potassium reversal potential E rev depends on their concentration on the inner [K +]i and outer [K +]o sides of the membrane and can be calculated by the formula:
, (Eq. 1)
where z—ionic charge, F—Faraday constant, R—universal gas constant, T—temperature.
The driving force of the electrochemical gradient of potassium ions is the difference between the membrane potential V(t) and potassium reversal potential E rev:
. (Eq. 2)
According to Eq. 2, a twofold increase in the extracellular concentration of potassium ions will elicit a 19-mV membrane depolarization.
The extracellular potassium concentration is usually recorded by a potassium-sensitive electrode [9]. The method of making such electrodes for neurophysiological experiments is described in detail elsewhere [10, 11].
An increase in the concentration of potassium ions during the generation of epileptiform discharges in vitro was first described back in the 1970s [12–14]. Later on, this phenomenon was independently confirmed in different laboratories. Figure 1 exemplifies a simultaneous record of the extracellular concentration of potassium ions using a monopolar microelectrode, as well as spiking activity or postsynaptic currents using a patch clamp technique, in rat hippocampal-entorhinal cortex slices in the 4-aminopyridine model of epileptiform activity (reproduced from [11]).
The extracellular concentration of potassium ions at any given time is calculated by the formula:
, (Eq. 3)
where [K +]o,baseline—potassium ion concentration in the bathing solution, S—calibration coefficient, as determined experimentally for each electrode by measuring its potential V(t) in solutions with different potassium concentrations [K +]o.
In the absence of spiking activity in the neuronal network, the extracellular concentration of potassium ions does not change, corresponding to the value in the bathing solution (2.5 mM in the above example). During epileptic discharges, the extracellular concentration of potassium ions increases manifold, reaching about 10 mM in the tonic phase of the ictal discharge and depolarizing the membrane, thus making neurons more excitable.

Simultaneous records of the extracellular concentration of potassium ions using a potassium-sensitive electrode (green curve) and postsynaptic currents of a representative neuron (black curve) in the Wistar rat entorhinal cortex in a 4-aminopyridine model of epileptiform activity in vitro (a). The holding potential for the recoding of postsynaptic currents was V hold = –27 mV. The ictal discharge, marked with a blue line, is zoomed in (b) (reproduced from [11]).
EXCESS INTRACELLULAR CONCENTRATION OF CHLORIDE IONS IN EPILEPTIC ACTIVITY
The inhibitory function in the brain is performed by the GABAergic system. As a neurotransmitter, GABA has a rapid and strong hyperpolarizing effect on the postsynaptic neuron upon binding to the GABAA receptor, which represents an ionotropic (chloride) channel. As a rule, the activation of these receptors entails a flow of chloride ions into the cytoplasm along the electrochemical gradient. However, inward chloride current only occurs when the reversal potential, determined predominantly by the chloride equilibrium potential, is lower than the resting membrane potential of the cell [15]. Otherwise, GABA will exert no inhibition and can even depolarize the postsynaptic neuron.
Since the reversal potential is largely determined by changes in the intracellular concentration of chloride ions, it was necessary to develop a convenient method for recording. A non-toxic method for measuring the intracellular concentration of chloride ions was devised in 2000 by Kuner and Augustine [16], based on the Fӧrster (fluorescence) resonance energy transfer (FRET), an energy transfer between two light-sensitive molecules (chromophores). The essence of this physical phenomenon is that a donor chromophore, which is initially in the excited state, may transfer energy to an acceptor chromophore through nonradiative dipole–dipole coupling. A characteristic feature of this process is the quenching of donor’s fluorescence and the appearance of longer-wavelength acceptor’s fluorescence. When studying protein–protein interactions, the authors noticed that the efficiency of FRET between cyan and yellow fluorescent proteins depended on chloride ions. Therefore, the authors proposed a radiometric indicator of chloride ions in the form of a compound of yellow and cyan fluorescent proteins, Clomeleon, and successfully tested it on primary culture of hippocampal neurons [16].
Glykys et al. [17] were the first to measure the concentration of chloride ions during ictal discharge generation after Clomeleon expression in the organotypic hippocampal culture by combining two-photon microscopy and extracellular recording [17]. While in the resting state, the concentration of chloride ions was 25.4 ± 7.4 mM, during ictal discharge generation, it increased to 40.8 ± 13.8 mM. A high intracellular concentration of chloride ions is maintained throughout epileptiform activity generation and recovers to the baseline level only in the prolonged absence of synchronous population activity (Fig. 2).
The alternative ways to assess the concentration of chloride ions using other sensors have been developed. For example, ClopHensorN, which is not only [Cl–]i-, but also pH-sensitive, is used quite widely [18–22]. Burman et al. [22] measured the intracellular chloride concentration dynamics during epileptiform discharge generation in slices of the organotypic hippocampal culture. It was shown that during epileptiform activity, the intracellular concentration of chloride ions increases; accordingly, upon the generation of epileptiform discharges, the reversal potential increases by 30–40 mV and returns to the baseline level after the complete cessation of epileptiform activity.

Schematic representation of the dynamics of the intracellular chloride concentration [Cl–]i during ictal discharges in an in vitro model of epileptiform activity. During the ictal discharge, a representative cortical neuron generates spiking activity, which is presented by the record of the membrane potential Vm. The blue numbers denote: 1—ictal discharge tonic phase, 2—ictal discharge clonic phase, 3—neuronal postictal hyperpolarization, 4—resting state (reproduced from [17]).
EPILEPTOGENIC EFFECTS OF CHLORIDE–POTASSIUM IMBALANCE
Normally, chloride ions in mature neurons are transported in the extracellular space by the K+–Cl– cotransporter (KCC2): the electrochemical gradient of potassium ions facilitates chloride extrusion together with the potassium ion [23]. Thus, KCC2 prevents the accumulation of chloride ions in neurons due to activity of the inhibitory system and an increase in the reversal potential for GABAA receptors.
The functional activity of KCC2 is regulated by a number of mechanisms and usually declines sharply upon high activation of neuronal networks. Recent data suggest that the rate of ion transport, as well as the stability and translocation of KCC2 along the cell surface and plasmalemma, are rapidly and reversibly modulated by (de)phosphorylation of some serine, threonine and tyrosine residues in the C-terminus of this protein [24].
This mechanism for chloride extrusion from cells is quite vulnerable in various damages to the nervous system, interruptions in its work lead to increased excitability of neuronal networks and are epileptogenic for brain tissue. Kaila et al. [25], in their review, suggest the following scenario of such epileptogenesis. During recurrent seizures, there occurs a progressive internalization of postsynaptic GABAA receptors [26], on the one hand, and a rapid decrease in KCC2 expression [27, 28], on the other hand, which leads jointly to a long-term accumulation of chloride ions in neurons and subsequent attenuation of inhibition. The specific molecular mechanism of decreased KCC2 expression is not fully elucidated, but the pathological KCC2 inhibitors, best-known to date, are excess glutamate [29] and high levels of the brain-derived neurotrophic factor (BDNF) [30].
In addition to acquired disorders of chloride balance, congenital disorders may also occur. It is noteworthy that mutations in the SLC12A5 gene that encodes the KCC2 cotransporter have been found in some forms of hereditary epilepsy. For example, heterozygous missense polymorphisms in the SLC12A5 gene were found in an Australian family with hereditary febrile seizures and in a Franco-Canadian cohort with severe genetic generalized epilepsy (GGE), which cause a reduced KCC2-dependent capacity for chloride extrusion [31]. A recessive loss-of-function mutation in SLC12A5 has been found in patients with a severe early-onset Mendelian disease termed epilepsy of infancy with migrating focal seizures (EIMFS) [32]. It has also been shown that KCC2 rs2297201 gene polymorphism may serve as a genetic prognostic marker of febrile seizures [33]; for more details on epileptogenic mutations in the KCC2-encoding gene, see the reviews [31, 34].
The proepileptogenic effect of high extracellular potassium concentrations is supported by ample experimental data. For example, the increasing of the potassium chloride concentration in the bathing solution from 2.5 to 10 mM leads to the generation of epileptiform activity in acute slices of the juvenile mouse hippocampus [35]. During pre- or interictal discharges, mainly provided by the synchronous activity of interneurons, the extracellular concentration of potassium ions increases [11, 36], which, in turn, can provoke ictal discharge generation. The main sources of extracellular potassium are various types of potassium channels and the above-described KCC2 transporter.
For example, during action potential generation, potassium ions flow out of the cell through voltage-gated potassium channels [37], while KCC2 ensures a co-transport of potassium and chloride ions in the extracellular space. As a result, the reversal potential for potassium currents changes in all neurons, including pyramidal cells, and accordingly, the membrane potential becomes more depolarized in all neurons, leading to hyperexcitability of brain tissue.
The effective removal of excess potassium ions from the extracellular space during neuronal activity is of paramount importance. Astrocytes play a key role in this process. The normalization of the extracellular concentration of potassium ions is provided by the Na+–K+ pump and inward-rectifier potassium channels (Kir), as well as fast spatial buffering provided by the gap junctions between astrocytes [38]. Dysfunctions of any of these mechanisms can lead to epileptogenesis. There is evidence that mutations in astrocytic Kir4.1 potassium channels are present in patients with epilepsy [39–41]. Life-compatible mutations in the Na+–K+ pump subunits have also been identified in epileptic patients [42]. In juvenile myoclonic epilepsy, mutations in the connexin 36 gene, which encodes the protein responsible for the formation of gap junctions between astrocytes, have been described [43].
GABAergic MECHANISMS OF EPILEPTIFORM ACTIVITY GENERATION
Ionic imbalance can alter the function of GABAergic interneurons. Normally, the brain GABAergic system performs an inhibitory function and therefore exerts a pronounced antiepileptic effect. However, there is a lot of experimental and clinical evidence of its proepileptic activity in some pathological states [4, 36, 44–49]. The mechanisms of epileptic activity generation due to the work of the GABAergic system are discussed in detail in a number of previously published reviews [4, 5], so below we will only outline the most frequently occurring of them.
The ictal discharge can be triggered by brief synchronous activation followed by deactivation of GABAergic inhibitory interneurons under conditions of high epilepsy readiness. In this case, synchronized generation of action potentials by pyramidal neurons (post-inhibitory rebound synchronization, PIRS) follows immediately after inhibitory neuronal shutdown. Such an effect was documented in in vitro optogenetic experiments with short-term 30-ms photostimulation of GABAergic interneurons in the upper layers of the juvenile mouse somatosensory cortex (VGAT-ChR2 strain) in the 4-aminopyridine model. Photostimulation evokes simultaneous spiking activity of GABAergic interneurons, while light shutdown causes synchronous cessation of this activity. Immediately after photostimulation turn-off, the ictal (or interictal) discharge was generated in the brain slice [47]. Using a perforated patch and multielectrode recordings, the authors demonstrated that during photostimulation of interneurons, there is hyperpolarization of pyramidal neurons, whereas the cessation of photostimulation evokes depolarization of pyramidal neurons and the development of the epileptic event. Thus, simultaneous shutdown of multiple interneurons entails synchronous activation of pyramidal neurons, which can be realized into an ictal or interictal discharge. A similar pattern was observed in human brain tissue obtained from the epileptic focus after local GABA application (100 mM), as well as in vivo, in the anesthetized adult VGAT-ChR2 mouse during photostimulation of interneurons [47]. An analogous result was demonstrated in vitro, when only parvalbumin-positive fast-spiking interneurons were stimulated [50].
Another mechanism can be involved during a longer synchronized spiking activity of interneurons (and optionally, of pyramidal neurons). Such a neuronal network activity increases the extracellular concentration of potassium ions so much that it brings the membrane potential of neurons closer to the threshold of action potential generation. As a result, a sufficient number of pyramidal neurons can be activated to generate the ictal discharge. A reasonable number of experimental data argue in favor of this mechanism: an appreciable jump in the extracellular concentration of potassium ions is recorded during the generation of GABA/glutamate interictal discharges [36, 51]; in the 4-aminopyridine model of epileptiform activity in entorhinal cortex slices, the ictal discharge is preceded by GABA/glutamate preictal discharges, while the extracellular concentration of potassium ions often has no time to return to the baseline level between preictal discharges and provides thereby a greater jump in their concentration [11, 52]. Thus, a concentration threshold value sufficient for the generation of the ictal discharge can be reached.
Simultaneously, chloride ions can accumulate in a pyramidal neuron due to long-term activation of GABA receptors, which increases the GABA reversal potential. Therefore, a hyperpolarizing effect of GABA can change into a depolarizing. We suggest that a local change in the concentration of chloride ions, as well as some specialized inhibitory synapses located nearby the initial axonal segment of the postsynaptic pyramidal neuron, may play an important role in the development of the epileptogenic effect provided by GABAergic interneurons. A number of our and someone else’s experimental data, as well as model-based calculations, support this suggestion. For example, in the 4-aminopyridine model of epileptiform activity in juvenile rat hippocampal-entorhinal cortex slices, during the generation of preictal discharges, we repeatedly recorded low-threshold action potentials in pyramidal neurons [52], which may be due to the fact that they are evoked by interneurons.
One of the subtypes of parvalbumin-expressing interneurons, chandelier cells, forms special axo-axonic synapses, providing GABA release nearby the initial axonal segment of pyramidal neurons [53–55]. It was shown that the application of the GABAA receptor agonist muscimol to this axonal region decreases the probability of action potential generation by a neuron at a low intracellular concentration of chloride ions and increases it at a high concentration [56]. There is experimental evidence that the amount of the KCC2 cotransporter on the axons is significantly less than on the dendrites and soma [57, 58], whereas NKCC1 cotransporter expression in this region is rather high. These features of local cotransporter expression ensure the accumulation of chloride ions in the initial segment of the axon [59]. Since the axonal volume is small, long-term GABAergic activation leads to the local accumulation of chloride ions, thus providing a depolarizing effect of GABA. In this case, the activity of GABAergic chandelier cells may lead to the synchronized excitation of pyramidal neurons and initiation of an ictal discharge. However, this hypothesis requires further experimental proof.
Along with chandelier cells, another subtype of parvalbumin-expressing interneurons, basket cells that form synaptic contacts mainly on the soma and proximal dendrites of pyramidal neurons, may also play an important role in these processes [60, 61]. Optogenetic depolarization of parvalbumin interneurons in the pilocarpine model of focal epilepsy in vivo changes their effect from anti- to pro-epileptic within seconds [4]. The authors also note that the pro-epileptic effect is absent during photostimulation of SOM-interneurons, whose synapses are mainly located on the dendrites of pyramidal neurons, and that proepileptic effect of the photostimulation of parvalbumin interneurons disappears when the KCC2 transporter is overexpressed [4].
PHARMACOLOGICAL REGULATION OF IONIC BALANCE IN THE CENTRAL NERVOUS SYSTEM
While ionic imbalance has an epileptogenic effect in the nervous tissue, pharmacological exposures aimed at restoring ion balance may become one of the promising strategies in the normalization of brain functions.
Pharmacological regulation of chloride balance
The most important role in the natural regulation of intracellular chloride concentration is played by KCC2 and NKCC1 cotransporters whose action is mutually opposed: KCC2 expulses excess chloride ions from a neuron, whereas NKCC1 facilitates their intracellular accumulation. In early ontogenesis, NKCC1 prevails, however, as neurons mature, the contribution of KCC2 increases (Fig. 3). These changes are attributed to functional attenuation of STE20/SPS1-related proline-alanine-rich protein kinase (SPAK) which phosphorylates both KCC2 and NKCC1, although having opposite effects on their functions: it decreases KCC2 activity and increases NKCC1 activity [62]. During the development of epilepsy, NKCC1 activity may become prevailing [63]. Therefore, NKCC1 and KCC2 inhibition can be expected to exert anti- and proepileptic effects, respectively, whereas an unequivocal antiepileptic effect can only be expected upon KCC2 enhancement.
The current set of pharmacological agents affecting cation-chloride cotransporters is very limited, being mainly represented by diuretics (furosemide and bumetanide) and their derivatives. Bumetanide is commonly used to inhibit NKCC1. Bumetanide has a ~500-fold higher affinity for NKCC1 (K i ~0.1 µM) than for KCC2 (K i ~25–50 µM), and therefore low bumetanide concentrations (2–10 µM) can be used to inhibit NKCC1 in vitro without affecting KCC2 appreciably [65]. However, at higher concentrations, bumetanide blocks both transporters. Furosemide is even less specific and affects not only the cation-chloride cotransporters but also the Na+-independent Cl–/HCO3 – exchanger (AE3), some subtypes of GABAA receptors, and carbonic anhydrase [66].
Although some studies have noted the antiepileptic properties of these diuretics (see review [67]), their use as antiepileptic drugs is complicated by a poor permeability across the blood-brain barrier and a large number of side effects, because the NKCC1 transporter is expressed in many cell types.
Specific KCC2 inhibitors have been recently developed: VU0463271 [68] and VU0240551 [69]. We studied the effect of VU0463271 on epileptiform activity in hippocampal-entorhinal cortex slices of rats aged 3 and 8 weeks. Despite the fact that by the age of 3 weeks, rats should already have a maximum level of KCC2 expression corresponding to that in adult animals, KCC2 inhibition exerted differential effects: while at the age of 3 weeks, we found no significant changes in the shape of epileptic activity, in 8-week-old rats, KCC2 inhibition led to a replacement of ictal activity by status epilepticus [52]. Hamidi and Avoli have obtained a similar result in the 4-aminopyridine model in entorhinal-perirhinal cortex slices using VU0240551 (10 µM) [70]. Thus, the experimental data confirm the important role of KCC2 in the regulation of epileptic activity.
A promising strategy for enhancing KCC2 functions, which would potentially have an antiepileptic effect, could be a direct or indirect targeted phosphorylation of KCC2 at some sites via upstream regulatory kinases [24]. For example, there has been identified a PKC-dependent enhancement of KCC2 functions due to its phosphorylation at S940 in the C-terminus [71]. The activation of group I metabotropic glutamate receptors (mGluRs) can lead to the same effect. In experiments with gramicidin-perforated patch-clamp, it has been shown that tonic coactivation of mGluR1 and mGluR5 increases the strength of inhibitory synapses through activation of the PKC-dependent pathway in pyramidal neurons of the hippocampal CA3 region [72]. However, group I mGluR agonists are proconvulsants and lead to epileptogenesis, as they simultaneously modulate neuronal excitability and NMDA receptor activity [73], so that the therapeutic potential of this approach is currently unobvious.
Pharmacological regulation of potassium balance
Potassium removal from the extracellular space is mainly provided by the Na+–K+ pump and Kir channels. If the Na+–K+ pump is responsible for the import of excess K+ from the extracellular space into the cytoplasm, then pump activation should have an antiepileptic effect and, conversely, pump inhibition should have a proepileptic effect.
Since Na+–K+ pump activity increases with increasing intracellular Na+ concentration, the sodium ionophore monensin is sometimes used as a Na+–K+ pump modulator in in vitro studies [74, 75]. However, we have failed to find any references in which monensin was used as an experimental antiepileptic agent. Our unpublished experimental data showed that monensin has a very narrow therapeutic window. In a 4-aminopyridine model of epileptiform activity in mouse hippocampal-entorhinal cortex slices, monensin (2–3 µM) prevented the generation of ictal discharges in some cases, but the increasing of the monensin concentration to 5 µM entailed neuronal death.
In contrast to the activating effect on the Na+–K+ pump, the effect of its inhibition has been studied better and is well consistent with model-based calculations [8]. Intraventricular injection of ouabain has been shown to induce an epileptic seizure in rats [76]. In in vitro models, ouabaine-induced Na+–K+ pump attenuation also has a pronounced proepileptic effect [52].
Astrocytes are also responsible for the removal of potassium ions from the extracellular space. Astrocytes perform spatial potassium buffering; they intake extracellular K+ secreted by neurons and release it in regions with lower K+ levels (e.g., in microcapillaries), combining into a syncytium via gap junctions [77]. K+ entry into astrocytes is mainly mediated by Kir channels containing four Kir4.1 subunits (in the hippocampus) or a combination of Kir4.1/Kir5.1 subunits (in the neocortex) [78–80].
Electrophysiological studies have demonstrated that Kir currents are significantly reduced in hippocampal samples of patients with refractory temporal lobe epilepsy [81], which may be due to reduced expression levels of Kir4.1 channels [82]. Kir4.1 channel dysfunctions has been identified in a number of hereditary forms of neurological disorders accompanied by epileptic seizures, e.g., EAST syndrome consisting of epilepsy, ataxia, sensorineural deafness, and salt-wasting renal tubulopathy, and SeSAME syndrome characterized by seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance [83, 84]. Although studies on epilepsy models in some cases show, on the contrary, an increase in Kir4.1 expression (e.g., in the lithium-pilocarpine model, its expression was found to increase in the brain cortex, striatum, and hypothalamus), this phenomenon may be compensatory in nature [85]. Studies using animals with conditional knockout targeting astrocytic Kir4.1 revealed a clear link between Kir4.1 dysfunction and seizure predisposition [86, 87].
It is therefore reasonable to assume that the amplification of Kir currents via positive modulators or by increasing the expression of Kir4.1 channels in astrocytes will have a pronounced antiepileptic effect. However, no specific modulators of Kir4.1 channels have long been known. Only a few Kir4.1 antagonists have been identified to date, such as VU0134992 (IC50 = 0.97 µM) [88], pentamidine (IC50 = 97 nM) [89] and some others, as reviewed by Ohno et al. [90]. It has also been found that many drugs used in clinics can reduce Kir currents when administered at therapeutic doses. For example, such a side effect has been described for antidepressants [91] and anticancer drugs based on divalent platinum complexes [92, 93].
Unfortunately, we have failed to find any data on positive modulators of Kir4.1 channels. Instead, a number of widely used antiepileptic drugs (valproate, phenytoin, phenobarbital) can increase the expression of astrocytic Kir4.1 in the limbic regions, as shown in experiments on rats [94]; this effect is probably one of the components of their antiepileptic action.
Thus, there are virtually no available pharmacological tools to date that would allow effective manipulation of ionic balance in the nervous tissue. Moreover, the above transporters and channels (or their close analogs) are present not only in the CNS but also in other systems of the organism, ensuring a number of vital functions, so that a wide spectrum of side effects of pharmacological exposures on the Na+–K+ pump, cation-chloride cotransporters and Kir channels can be anticipated.

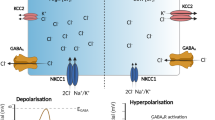
Chloride concentration regulatory mechanisms underlying GABA effects in immature (left) and mature (right) CNS neurons. NKCC1 is the key Na+–K+–2Cl– cotransporter in immature neurons, which regulates Cl– influx. KCC2 is the principal K+–Cl– cotransporter in mature CNS neurons, which mediates Cl– efflux. Under several pathophysiologic conditions, such as epilepsy, neurons undergo “recapitulation” and dedifferentiation to some earlier developmental stages. For other explanations, see the text (reproduced from [63]).
GENE THERAPY TO REGULATE IONIC BALANCE
Experimental approaches to gene therapy of pharmacoresistant epilepsy
There are four types of approaches to gene therapy: (1) gene replacement in monogenic diseases, (2) gene addition in complex pathologies, (3) modification of gene expression levels and RNA targeting with small molecules, and (4) genome editing [95]. As for epilepsy, the first approach has not practically been developed because idiopathic forms of epilepsy, caused by a single-gene mutation and thus a dysfunction of a certain channel or receptor, are relatively rare to occur. In addition, genetic material delivery simultaneously to vast areas of the brain is difficult methodologically. The third and fourth approaches are also not yet implemented for the experimental treatment of epilepsy. The second approach is considered the most promising and can be used to treat pharmacoresistant focal forms of epilepsy. In this option, the required genes are to be delivered to a target cell in order to locally reduce the activity of neuronal networks in the epileptic focus.
Viral constructs are most commonly used for gene delivery. Among the various viruses tested for gene therapy, the most promising candidates for being used in the CNS are constructs derived from adeno-associated viruses (AAV), lentiviruses, and herpes viruses [96]. AAV-derived constructs are currently considered the safest: viral DNA can be almost completely removed during construct assembly, while the viral construct per se is relatively non-toxic to cells and has low immunogenicity. However, their relatively small size imposes a significant limitation on the widespread application of AAV constructs [97]. Since the capacity of AAV constructs does not exceed 4.5–5 thousand base pairs, while the sizes of the coding sequence of many target protein genes are quite comparable or even larger, by far not all genes can be delivered to the CNS in this way. Therefore, the constructs based on lentiviruses or herpes viruses are often used in experimental studies, although having both a higher capacity and a higher risk of side effects [96].
Currently, the main gene therapy techniques for pharmacoresistant epilepsy are aimed at increasing the expression of inhibitory peptides, such as galanin [98] and NPY [99, 100], or suppressing the excitability of neurons by overexpression of potassium channels therein [101–103]. Among the close approaches aimed at reducing neuronal network hyperactivation are chemo- and optogenetic methods surveyed in an article by Walker and Kullmann [104].
Genetic methods for chloride balance regulation in neurons
As described above, chloride accumulation in neurons during epileptic activity leads to a dysfunction of the GABAergic system and self-sustaining of epileptic activity. KCC2 functional enhancement may prevent the impairment of inhibitory functions. There are relatively few experimental works that would aim specifically at modulating the ionic balance of chloride ions. We have managed to find only several studies in which the effect of KCC2 overexpression on epileptic activity was studied [105, 106].
The overexpression of KCC2, delivered by a lentiviral construct under the CaMKII promoter, has been studied in the brain cortex of PV-ChR2 transgenic mice. KCC2 overexpression of the KCC2 transporter delivered by the lentiviral construct under the promoter was performed in the cortex of the PV-ChR2 transgenic mouse. In this murine strain, channelrhodopsin-2 is light-gated and expressed in parvalbumin-positive interneurons [105]. The authors have demonstrated that in PV-ChR2 mice with a pilocarpine-induced status epilepticus and normal KCC2 level, photostimulation of the cortex has a pronounced proictal, instead of anticonvulsant, effect if starting 2 or more seconds following ictal discharge development in the cortex. This observation is in full agreement with the ideas on the chloride accumulation in neurons during an epileptic seizure and the transition from the inhibitory to excitatory action of GABA. KCC2 overexpression in the pyramidal neurons of the cerebral cortex prevented the development of the proictal action of parvalbumin interneurons and retained the inhibitory action of GABA. However, KCC2 overexpression per se did not affect the course of epileptic seizures [105].
A similar conclusion was drawn in another experimental study in which kainate seizures were modeled in a mouse strain with doxycycline-inducible KCC2 overexpression. KCC2 overexpression also had no effect on the severity of epileptic seizures but increased the efficacy of the positive GABA receptor modulator diazepam in stopping the seizures recorded in the EEG [106]. Thus, co-affecting of KCC2 and GABA receptors may be an effective anticonvulsant therapeutic strategy.
Several optogenetic tools are now available to induce a directed (inward/outward) transport of chloride ions to/from a neuron. For example, halorhodopsin (a light-driven chloride pump) imports one chloride ion into the cell when exposed to a single quantum of 560-nm light [107]; the chloride channel opens a pore for chloride currents along an electrochemical gradient when exposed to 490-nm light [108, 109]; the Cl-out complex in which the proton pump provides a driving force chloride extrusion from a neuron through the chloride channel [110].
Similar to the action of the GABAergic system, the chloride pump and chloride channel can exert a proepileptic effect due to chloride accumulation in neurons under conditions of seizure activity. Therefore, optogenetic activation of light-gated chloride channels can evoke both suppression and enhancement of neuronal activity [111, 112].
The Cl-out complex may prove to be instrumental in normalizing the intracellular chloride concentration in epileptic tissue with an impaired gradient of chloride ions. Alfonsa et al. [110] reported that the activation of the Cl-out complex had an antiepileptic effect in the case when epileptic activity was evoked by a high chloride concentration in neurons, as detected in mouse hippocampal slices using KCC2 blockade. Network activity was recorded using extracellular leads in the CA1 pyramidal layer, with electrical stimulation performed to monitor neuronal excitability. Next, a serial photostimulation was performed simultaneously with 488 nm and 561 nm light to activate the chloride channelrhodopsin and proton pump, respectively. Thereby, the authors demonstrated that Cl-out complex activation reduces excitability and, in general, has an antiepileptic effect [110].
Genetic methods to regulate the extracellular potassium concentration
There are quite a lot of effective gene therapy options for epilepsy that enhance the functions of different types of neuronal potassium channels [101–103]. Although additional activation of potassium channels can increase the extracellular concentration of potassium cations, this effect is not predominant. Therefore, in the conclusive section of the review, we will consider only those approaches that serve to normalize the extracellular potassium concentration. The enhancing of Kir currents by increasing the expression of Kir4.1 channels in astrocytes can become an effective strategy for doing this.
A prerequisite for this approach is the reduction of Kir currents in hippocampal samples of patients with refractory temporal lobe epilepsy [81]. We could not find any literature data on experimental validation of this method for epilepsy treatment; however, the increasing of Kir4.1 expression in striatal astrocytes proved effective in treating Huntington’s disease in its R6/2 and Q175 HD transgenic mouse models [113]. The authors showed that the additional expression of Kir4.1 channels through viral delivery to striatal astrocytes restored Kir currents in these cells, resulting in a decreased extracellular potassium concentration and membrane depolarization, as well as an improvement of other functional characteristics of striatal neurons. Such a treatment somewhat attenuated movement disorders and improved the survival rate of R6/2 mice [113].
It should nevertheless be kept in mind that this approach may have limitations. For example, the overamplification of Kir currents can lead to negative consequences. It has been revealed that gain-of-function mutations in the KCNJ10 gene, encoding Kir4.1 channels, are able to promote the formation of neurological syndromes that combine autism spectrum disorders and epilepsy [114]. In this case, the amplification of Kir currents can be assumed to prevent natural depolarization of neurons and inactivation of sodium channels due to accumulation of extracellular potassium, which causes a depolarization block and interrupts spiking activity. However, the existing models of ictal activity provide no direct evidence to support this developmental variant [8]. It is likely that epileptic activity in this case may be due to functional disorders in neuron–glia interrelationships. A decrease in the extracellular potassium concentration leads to hyperpolarization of astrocytes, thus attenuating gliotransmission that normally inhibits glutamate release by pyramidal neurons [115].
CONCLUSION
In epileptic tissue, long-term neuronal hyperactivity leads to a dysfunction of the GABAergic system: it may exert a proepileptic, instead of an inhibitory, effect. These disorders are underlain by the electrolyte imbalance of chloride anions and/or potassium cations in extra- and intracellular environments. Model-based and experimental studies indicate that the rebalancing of these ions may prove to be the most effective approach to treating appropriate forms of epilepsy. In most cases, this requires to restore or enhance the attenuated function of one or another ion channel, transporter, or pump. Unfortunately, the available arsenal of pharmacological tools is extremely scarce and not effective enough.
We believe that the gene therapy methods, allowing the functions of certain proteins to be enhanced by their additional expression in target cells, may well prove to be the most promising. Although the field of gene therapy for epilepsy is intensively developing, the tools aimed at sustaining the ionic balance in epileptic tissue are still relatively understudied. Among the available therapeutic options, KCC2 transporter and Kir4.1 channel overexpression techniques, which allow a restoration of chloride anion and potassium cation concentrations, attract particular attention. However, these therapeutic tools require further experimental verification for the efficacy and safety of these tools in various models of epilepsy and convulsive states.
REFERENCES
Fattorusso A, Matricardi S, Mencaroni E, Dell’Isola GB, Di Cara G, Striano P, Verrotti A (2021) The Pharmacoresistant Epilepsy: An Overview on Existant and New Emerging Therapies. Front Neurol 12: 1030. https://doi.org/10.3389/FNEUR.2021.674483
Löscher W, Klitgaard H, Twyman RE, Schmidt D (2013) New avenues for anti-epileptic drug discovery and development. Nat Rev Drug Discov 12: 757–776. https://doi.org/10.1038/nrd4126
Raimondo JV, Burman RJ, Katz AA, Akerman CJ (2015) Ion dynamics during seizures. Front Cell Neurosci 9: 1–14. https://doi.org/10.3389/fncel.2015.00419
Magloire V, Mercier MS, Kullmann DM, Pavlov I (2019) GABAergic Interneurons in Seizures: Investigating Causality With Optogenetics. Neuroscientist 25: 344–358. https://doi.org/10.1177/1073858418805002
Khazipov R (2016) GABAergic Synchronization in Epilepsy. Cold Spring Harb Perspect Med 6: a022764. https://doi.org/10.1101/CSHPERSPECT.A022764
de Curtis M, Uva L, Gnatkovsky V, Librizzi L (2018) Potassium dynamics and seizures: Why is potassium ictogenic? Epilepsy Res 143: 50–59. https://doi.org/10.1016/J.EPLEPSYRES.2018.04.005
Chizhov AV, Amakhin DV, Zaitsev AV (2017) Computational model of interictal discharges triggered by interneurons. PLoS One 12: e0185752. https://doi.org/10.1371/journal.pone.0185752
Chizhov AV, Zefirov AV, Amakhin DV, Smirnova EY, Zaitsev AV (2018) Minimal model of interictal and ictal discharges “Epileptor-2”. PLOS Comput Biol 14: e1006186. https://doi.org/10.1371/journal.pcbi.1006186
Ammann D, Chao P, Simon W (1987) Valinomycin-based K+ selective microelectrodes with low electrical membrane resistance. Neurosci Lett 74: 221–226. https://doi.org/10.1016/0304-3940(87)90153-4
Codadu NK, Parrish RR, Trevelyan AJ (2019) Region-specific differences and areal interactions underlying transitions in epileptiform activity. J Physiol 597: 2079–2096. https://doi.org/10.1113/JP277267
Chizhov AV, Amakhin DV, Smirnova EY, Zaitsev AV (2022) Ictal wavefront propagation in slices and simulations with conductance-based refractory density model. PLOS Comput Biol 18: e1009782. https://doi.org/10.1371/journal.pcbi.1009782
Moody WJ, Futamachi KJ, Prince DA (1974) Extracellular potassium activity during epileptogenesis. Exp Neurol 42: 248–263. https://doi.org/10.1016/0014-4886(74)90023-5
Prince DA, Lux HD, Neher E (1973) Measurement of extracellular potassium activity in cat cortex. Brain Res 50: 489–495. https://doi.org/10.1016/0006-8993(73)90758-0
Heinemann U, Lux HD, Gutnick MJ (1977) Extracellular free calcium and potassium during paroxysmal activity in the cerebral cortex of the cat. Exp Brain Res 27–27: 237–243. https://doi.org/10.1007/BF00235500
Williams JR, Sharp JW, Kumari VG, Wilson M, Payne JA (1999) The Neuron-specific K-Cl Cotransporter, KCC2. J Biol Chem 274: 12656–12664. https://doi.org/10.1074/jbc.274.18.12656
Kuner T, Augustine GJ (2000) A Genetically Encoded Ratiometric Indicator for Chloride. Neuron 27: 447–459. https://doi.org/10.1016/S0896-6273(00)00056-8
Glykys J, Dzhala V, Egawa K, Balena T, Saponjian Y, Kuchibhotla KV, Bacskai BJ, Kahle KT, Zeuthen T, Staley KJ (2014) Local impermeant anions establish the neuronal chloride concentration. Science 343: 670–675. https://doi.org/10.1126/science.1245423
Arosio D, Ricci F, Marchetti L, Gualdani R, Albertazzi L, Beltram F (2010) Simultaneous intracellular chloride and pH measurements using a GFP-based sensor. Nat Methods 7: 516–518. https://doi.org/10.1038/nmeth.1471
Raimondo J (2013) A genetically-encoded chloride and pH sensor for dissociating ion dynamics in the nervous system. Front Cell Neurosci 7: 202. https://doi.org/10.3389/fncel.2013.00202
Sulis Sato S, Artoni P, Landi S, Cozzolino O, Parra R, Pracucci E, Trovato F, Szczurkowska J, Luin S, Arosio D, Beltram F, Cancedda L, Kaila K, Ratto GM (2017) Simultaneous two-photon imaging of intracellular chloride concentration and pH in mouse pyramidal neurons in vivo. Proc Natl Acad Sci USA 114: E8770–E8779. https://doi.org/10.1073/pnas.1702861114
Ponomareva D, Petukhova E, Bregestovski P (2021) Simultaneous Monitoring of pH and Chloride (Cl–) in Brain Slices of Transgenic Mice. Int J Mol Sci 22: 13601. https://doi.org/10.3390/ijms222413601
Burman RJ, Selfe JS, Lee JH, van den Berg M, Calin A, Codadu NK, Wright R, Newey SE, Parrish RR, Katz AA, Wilmshurst JM, Akerman CJ, Trevelyan AJ, Raimondo JV (2019) Excitatory GABAergic signalling is associated with benzodiazepine resistance in status epilepticus. Brain 142: 3482–3501. https://doi.org/10.1093/brain/awz283
Rahmati N, Hoebeek FE, Peter S, De Zeeuw CI (2018) Chloride Homeostasis in Neurons With Special Emphasis on the Olivocerebellar System: Differential Roles for Transporters and Channels. Front Cell Neurosci 12: 101. https://doi.org/10.3389/fncel.2018.00101
Kahle KT, Deeb TZ, Puskarjov M, Silayeva L, Liang B, Kaila K, Moss SJ (2013) Modulation of neuronal activity by phosphorylation of the K–Cl cotransporter KCC2. Trends Neurosci 36: 726–737. https://doi.org/10.1016/j.tins.2013.08.006
Kaila K, Ruusuvuori E, Seja P, Voipio J, Puskarjov M (2014) GABA actions and ionic plasticity in epilepsy. Curr Opin Neurobiol 26: 34–41. https://doi.org/10.1016/j.conb.2013.11.004
Wasterlain CG, Liu H, Naylor DE, Thompson KW, Suchomelova L, Niquet J, Mazarati AM, Baldwin RA (2009) Molecular basis of self-sustaining seizures and pharmacoresistance during status epilepticus: The receptor trafficking hypothesis revisited. Epilepsia 50: 16–18. https://doi.org/10.1111/j.1528-1167.2009.02375.x
Puskarjov M, Ahmad F, Kaila K, Blaesse P (2012) Activity-Dependent Cleavage of the K-Cl Cotransporter KCC2 Mediated by Calcium-Activated Protease Calpain. J Neurosci 32: 11356–11364. https://doi.org/10.1523/JNEUROSCI.6265-11.2012
Lee HHC, Jurd R, Moss SJ (2010) Tyrosine phosphorylation regulates the membrane trafficking of the potassium chloride co-transporter KCC2. Mol Cell Neurosci 45: 173–179. https://doi.org/10.1016/j.mcn.2010.06.008
Lee HHC, Deeb TZ, Walker JA, Davies PA, Moss SJ (2011) NMDA receptor activity downregulates KCC2 resulting in depolarizing GABAA receptor–mediated currents. Nat Neurosci 14: 736–743. https://doi.org/10.1038/nn.2806
Rivera C, Li H, Thomas-Crusells J, Lahtinen H, Viitanen T, Nanobashvili A, Kokaia Z, Airaksinen MS, Voipio J, Kaila K, Saarma M (2002) BDNF-induced TrkB activation down-regulates the K+–Cl– cotransporter KCC2 and impairs neuronal Cl– extrusion. J Cell Biol 159: 747–752. https://doi.org/10.1083/jcb.200209011
Kahle KT, Khanna AR, Duan J, Staley KJ, Delpire E, Poduri A (2016) The KCC2 Cotransporter and Human Epilepsy. Neuroscience 22: 555–562. https://doi.org/10.1177/1073858416645087
Stödberg T, McTague A, Ruiz AJ, Hirata H, Zhen J, Long P, Farabella I, Meyer E, Kawahara A, Vassallo G, Stivaros SM, Bjursell MK, Stranneheim H, Tigerschiöld S, Persson B, Bangash I, Das K, Hughes D, Lesko N, Lundeberg J, Scott RC, Poduri A, Scheffer IE, Smith H, Gissen P, Schorge S, Reith MEA, Topf M, Kullmann DM, Harvey RJ, Wedell A, Kurian MA (2015) Mutations in SLC12A5 in epilepsy of infancy with migrating focal seizures. Nat Commun 6: 8038. https://doi.org/10.1038/ncomms9038
Dimitrijevic S, Jekic B, Cvjeticanin S, Tucovic A, Filipovic T, Novaković I, Ivić B, Nikolic D (2022) KCC2 rs2297201 Gene Polymorphism Might be a Predictive Genetic Marker of Febrile Seizures. ASN Neuro 14: 175909142210932. https://doi.org/10.1177/17590914221093257
Duy PQ, David WB, Kahle KT (2019) Identification of KCC2 Mutations in Human Epilepsy Suggests Strategies for Therapeutic Transporter Modulation. Front Cell Neurosci 13: 515. https://doi.org/10.3389/FNCEL.2019.00515/BIBTEX
Filatov G, Krishnan GP, Rulkov NF, Bazhenov M (2011) Dynamics of epileptiform activity in mouse hippocampal slices. J Biol Phys 37: 347–360. https://doi.org/10.1007/S10867-011-9216-X/FIGURES/5
Librizzi L, Losi G, Marcon I, Sessolo M, Scalmani P, Carmignoto G, de Curtis M (2017) Interneuronal Network Activity at the Onset of Seizure-Like Events in Entorhinal Cortex Slices. J Neurosci 37: 10398–10407. https://doi.org/10.1523/JNEUROSCI.3906-16.2017
González OC, Krishnan GP, Timofeev I, Bazhenov M (2019) Ionic and synaptic mechanisms of seizure generation and epileptogenesis. Neurobiol Dis 130: 104485. https://doi.org/10.1016/J.NBD.2019.104485
Bellot-Saez A, Stevenson R, Kékesi O, Samokhina E, Ben-Abu Y, Morley JW, Buskila Y (2021) Neuromodulation of Astrocytic K+ Clearance. Int J Mol Sci 22: 2520. https://doi.org/10.3390/ijms22052520
Ferraro TN, Golden GT, Smith GG, Martin JF, Lohoff FW, Gieringer TA, Zamboni D, Schwebel CL, Press DM, Kratzer SO, Zhao H, Berrettini WH, Buono RJ (2004) Fine mapping of a seizure susceptibility locus on mouse Chromosome 1: nomination of Kcnj10 as a causative gene. Mamm Genome 2004 154 15: 239–251. https://doi.org/10.1007/S00335-003-2270-3
Olsen ML, Sontheimer H (2008) Functional implications for Kir4.1 channels in glial biology: from K+ buffering to cell differentiation. J Neurochem 107: 589–601. https://doi.org/10.1111/J.1471-4159.2008.05615.X
Köhling R, Wolfart J (2016) Potassium Channels in Epilepsy. Cold Spring Harb Perspect Med 6: a022871. https://doi.org/10.1101/cshperspect.a022871
Gallanti A, Tonelli A, Cardin V, Bussone G, Bresolin N, Bassi MT (2008) A novel de novo nonsense mutation in ATP1A2 associated with sporadic hemiplegic migraine and epileptic seizures. J Neurol Sci 273: 123–126. https://doi.org/10.1016/J.JNS.2008.06.006
Hempelmann A, Heils A, Sander T (2006) Confirmatory evidence for an association of the connexin-36 gene with juvenile myoclonic epilepsy. Epilepsy Res 71: 223–8. https://doi.org/10.1016/j.eplepsyres.2006.06.021
D’Antuono M, Louvel J, Köhling R, Mattia D, Bernasconi A, Olivier A, Turak B, Devaux A, Pumain R, Avoli M (2004) GABAA receptor‐dependent synchronization leads to ictogenesis in the human dysplastic cortex. Brain 127: 1626–1640. https://doi.org/10.1093/BRAIN/AWH181
Mattia D, Olivier A, Avoli M (1995) Seizure‐like discharges recorded in human dysplastic neocortex maintained in vitro. Neurology 45: 1391–1395. https://doi.org/10.1212/WNL.45.7.1391
Avoli M, De Curtis M, Gnatkovsky V, Gotman J, Köhling R, Lévesque M, Manseau F, Shiri Z, Williams S (2016) Specific imbalance of excitatory/inhibitory signaling establishes seizure onset pattern in temporal lobe epilepsy. J Neurophysiol 115: 3229–3237. https://doi.org/10.1152/JN.01128.2015/ASSET/IMAGES/LARGE/Z9K0071636960005.JPEG
Chang M, Dian JA, Dufour S, Wang L, Moradi Chameh H, Ramani M, Zhang L, Carlen PL, Womelsdorf T, Valiante TA (2018) Brief activation of GABAergic interneurons initiates the transition to ictal events through post-inhibitory rebound excitation. Neurobiol Dis 109: 102–116. https://doi.org/10.1016/J.NBD.2017.10.007
Elahian B, Lado NE, Mankin E, Vangala S, Misra A, Moxon K, Fried I, Sharan A, Yeasin M, Staba R, Bragin A, Avoli M, Sperling MR, Engel J, Weiss SA (2018) Low-voltage fast seizures in humans begin with increased interneuron firing. Ann Neurol 84: 588–600. https://doi.org/10.1002/ANA.25325
Miri ML, Vinck M, Pant R, Cardin JA (2018) Altered hippocampal interneuron activity precedes ictal onset. Elife 7: e40750. https://doi.org/10.7554/eLife.40750
Sessolo M, Marcon I, Bovetti S, Losi G, Cammarota M, Ratto GM, Fellin T, Carmignoto G (2015) Parvalbumin-Positive Inhibitory Interneurons Oppose Propagation But Favor Generation of Focal Epileptiform Activity. J Neurosci 35: 9544–9557. https://doi.org/10.1523/JNEUROSCI.5117-14.2015
Yekhlef L, Breschi GL, Lagostena L, Russo G, Taverna S (2015) Selective activation of parvalbumin- or somatostatin-expressing interneurons triggers epileptic seizurelike activity in mouse medial entorhinal cortex. J Neurophysiol 113: 1616–1630. https://doi.org/10.1152/jn.00841.2014
Smirnova EY, Sinyak DS, Chizhov AV, Zaitsev AV (2021) Age-Dependent Generation of Epileptiform Activity in the 4-Aminopyridine Model with Slices of the Rat Entorhinal Cortex. J Evol Biochem Physiol 57: 230–240. https://doi.org/10.1134/S0022093021020058
Somogyi P, Freund TF, Hodgson AJ, Somogyi J, Beroukas D, Chubb IW (1985) Identified axo-axonic cells are immunoreactive for GABA in the hippocampus visual cortex of the cat. Brain Res 332: 143–149. https://doi.org/10.1016/0006-8993(85)90397-X
Zaitsev AV, Povysheva NV, Gonzalez-Burgos G, Rotaru D, Fish KN, Krimer LS, Lewis DA (2009) Interneuron Diversity in Layers 2–3 of Monkey Prefrontal Cortex. Cereb Cortex 19: 1597–1615. https://doi.org/10.1093/cercor/bhn198
Schneider-Mizell CM, Bodor AL, Collman F, Brittain D, Bleckert A, Dorkenwald S, Turner NL, Macrina T, Lee K, Lu R, Wu J, Zhuang J, Nandi A, Hu B, Buchanan J, Takeno MM, Torres R, Mahalingam G, Bumbarger DJ, Li Y, Chartrand T, Kemnitz N, Silversmith WM, Ih D, Zung J, Zlateski A, Tartavull I, Popovych S, Wong W, Castro M, Jordan CS, Froudarakis E, Becker L, Suckow S, Reimer J, Tolias AS, Anastassiou CA, Seung HS, Reid RC, Costa NM da (2021) Structure and function of axo-axonic inhibition. Elife 10: e73783. https://doi.org/10.7554/eLife.73783
Ruiz A, Fabian-Fine R, Scott R, Walker MC, Rusakov DA, Kullmann DM (2003) GABAA Receptors at Hippocampal Mossy Fibers. Neuron 39: 961–973. https://doi.org/10.1016/S0896-6273(03)00559-2
Báldi R, Varga C, Tamás G (2010) Differential distribution of KCC2 along the axo-somato-dendritic axis of hippocampal principal cells. Eur J Neurosci 32: 1319–1325. https://doi.org/10.1111/j.1460-9568.2010.07361.x
Szabadics J, Varga C, Molnár G, Oláh S, Barzó P, Tamás G (2006) Excitatory effect of GABAergic axo-axonic cells in cortical microcircuits. Science 311: 233–5. https://doi.org/10.1126/science.1121325
Khirug S, Yamada J, Afzalov R, Voipio J, Khiroug L, Kaila K (2008) GABAergic Depolarization of the Axon Initial Segment in Cortical Principal Neurons Is Caused by the Na-K-2Cl Cotransporter NKCC1. J Neurosci 28: 4635–4639. https://doi.org/10.1523/JNEUROSCI.0908-08.2008
Dudok B, Klein PM, Soltesz I (2022) Toward Understanding the Diverse Roles of Perisomatic Interneurons in Epilepsy. Epilepsy Curr 22: 54–60. https://doi.org/10.1177/15357597211053687
Jiang X, Lachance M, Rossignol E (2016) Involvement of cortical fast-spiking parvalbumin-positive basket cells in epilepsy. Prog Brain Res 226: 81–126. https://doi.org/10.1016/bs.pbr.2016.04.012
Moore YE, Kelley MR, Brandon NJ, Deeb TZ, Moss SJ (2017) Seizing Control of KCC2: A New Therapeutic Target for Epilepsy. Trends Neurosci 40: 555–571. https://doi.org/10.1016/j.tins.2017.06.008
Liu R, Wang J, Liang S, Zhang G, Yang X (2019) Role of NKCC1 and KCC2 in Epilepsy: From Expression to Function. Front Neurol 10: 1407. https://doi.org/10.3389/fneur.2019.01407
Buchin A, Chizhov A, Huberfeld G, Miles R, Gutkin BS (2016) Reduced efficacy of the KCC2 cotransporter promotes epileptic oscillations in a subiculum network model. J Neurosci 36: 11619–11633. https://doi.org/10.1523/JNEUROSCI.4228-15.2016
Payne JA, Rivera C, Voipio J, Kaila K (2003) Cation-chloride co-transporters in neuronal communication, development and trauma. Trends Neurosci 26: 199–206. https://doi.org/10.1016/S0166-2236(03)00068-7
Uwera J, Nedergaard S, Andreasen M (2015) A novel mechanism for the anticonvulsant effect of furosemide in rat hippocampus in vitro. Brain Res 1625: 1–8. https://doi.org/10.1016/j.brainres.2015.08.014
Löscher W, Puskarjov M, Kaila K (2013) Cation-chloride cotransporters NKCC1 and KCC2 as potential targets for novel antiepileptic and antiepileptogenic treatments. Neuropharmacology 69: 62–74. https://doi.org/10.1016/j.neuropharm.2012.05.045
Delpire E, Baranczak A, Waterson AG, Kim K, Kett N, Morrison RD, Daniels JS, Weaver CD, Lindsley CW (2012) Further optimization of the K-Cl cotransporter KCC2 antagonist ML077: Development of a highly selective and more potent in vitro probe. Bioorg Med Chem Lett 22: 4532–4535. https://doi.org/10.1016/j.bmcl.2012.05.126
Deisz RA, Wierschke S, Schneider UC, Dehnicke C (2014) Effects of VU0240551, a novel KCC2 antagonist, and DIDS on chloride homeostasis of neocortical neurons from rats and humans. Neuroscience 277: 831–841. https://doi.org/10.1016/j.neuroscience.2014.07.037
Hamidi S, Avoli M (2015) KCC2 function modulates in vitro ictogenesis. Neurobiol Dis 79: 51–58. https://doi.org/10.1016/j.nbd.2015.04.006
Lee HHC, Walker JA, Williams JR, Goodier RJ, Payne JA, Moss SJ (2007) Direct Protein Kinase C-dependent Phosphorylation Regulates the Cell Surface Stability and Activity of the Potassium Chloride Cotransporter KCC2. J Biol Chem 282: 29777–29784. https://doi.org/10.1074/jbc.M705053200
Banke TG, Gegelashvili G (2008) Tonic activation of group I mGluRs modulates inhibitory synaptic strength by regulating KCC2 activity. J Physiol 586: 4925–4934. https://doi.org/10.1113/JPHYSIOL.2008.157024
Ure J, Baudry M, Perassolo M (2006) Metabotropic glutamate receptors and epilepsy. J Neurol Sci 247: 1–9. https://doi.org/10.1016/j.jns.2006.03.018
Kueh D, Barnett WH, Cymbalyuk GS, Calabrese RL (2016) Na(+)/K(+) pump interacts with the h-current to control bursting activity in central pattern generator neurons of leeches. Elife 5: e19322. https://doi.org/10.7554/eLife.19322
Picton LD, Nascimento F, Broadhead MJ, Sillar KT, Miles GB (2017) Sodium Pumps Mediate Activity-Dependent Changes in Mammalian Motor Networks. J Neurosci 37: 906–921. https://doi.org/10.1523/JNEUROSCI.2005-16.2016
Donaldson J, Minnich JL, Barbeau A (1972) Ouabain-induced Seizures in Rats: Regional and Subcellular Localization of 3 H-Ouabain Associated with Na+–K+ -ATPase in Brain. Can J Biochem 50: 888–896. https://doi.org/10.1139/o72-124
Kinboshi M, Ikeda A, Ohno Y (2020) Role of Astrocytic Inwardly Rectifying Potassium (Kir) 4.1 Channels in Epileptogenesis. Front Neurol 11: 1832. https://doi.org/10.3389/fneur.2020.626658
Larsen BR, MacAulay N (2014) Kir4.1-mediated spatial buffering of K+ : Experimental challenges in determination of its temporal and quantitative contribution to K+ clearance in the brain. Channels 8: 544–550. https://doi.org/10.4161/19336950.2014.970448
Moroni RF, Inverardi F, Regondi MC, Pennacchio P, Frassoni C (2015) Developmental expression of Kir4.1 in astrocytes and oligodendrocytes of rat somatosensory cortex and hippocampus. Int J Dev Neurosci 47: 198–205. https://doi.org/10.1016/j.ijdevneu.2015.09.004
Hibino H, Fujita A, Iwai K, Yamada M, Kurachi Y (2004) Differential Assembly of Inwardly Rectifying K+ Channel Subunits, Kir4.1 and Kir5.1, in Brain Astrocytes. J Biol Chem 279: 44065–44073. https://doi.org/10.1074/jbc.M405985200
Steinhäuser C, Seifert G, Bedner P (2012) Astrocyte dysfunction in temporal lobe epilepsy: K+ channels and gap junction coupling. Glia 60: 1192–1202. https://doi.org/10.1002/glia.22313
Heuser K, Eid T, Lauritzen F, Thoren AE, Vindedal GF, Taubøll E, Gjerstad L, Spencer DD, Ottersen OP, Nagelhus EA, Lanerolle NC de (2012) Loss of Perivascular Kir4.1 Potassium Channels in the Sclerotic Hippocampus of Patients With Mesial Temporal Lobe Epilepsy. J Neuropathol Exp Neurol 71: 814–825. https://doi.org/10.1097/NEN.0b013e318267b5af
Reichold M, Zdebik AA, Lieberer E, Rapedius M, Schmidt K, Bandulik S, Sterner C, Tegtmeier I, Penton D, Baukrowitz T, Hulton S-A, Witzgall R, Ben-Zeev B, Howie AJ, Kleta R, Bockenhauer D, Warth R (2010) KCNJ10 gene mutations causing EAST syndrome (epilepsy, ataxia, sensorineural deafness, and tubulopathy) disrupt channel function. Proc Natl Acad Sci U S A 107: 14490–14495. https://doi.org/10.1073/pnas.1003072107
Sala-Rabanal M, Kucheryavykh LY, Skatchkov SN, Eaton MJ, Nichols CG (2010) Molecular Mechanisms of EAST/SeSAME Syndrome Mutations in Kir4.1 (KCNJ10). J Biol Chem 285: 36040–36048. https://doi.org/10.1074/jbc.M110.163170
Nagao Y, Harada Y, Mukai T, Shimizu S, Okuda A, Fujimoto M, Ono A, Sakagami Y, Ohno Y (2013) Expressional analysis of the astrocytic Kir4.1 channel in a pilocarpine–induced temporal lobe epilepsy model. Front Cell Neurosci 7: 104. https://doi.org/10.3389/fncel.2013.00104
Chever O, Djukic B, McCarthy KD, Amzica F (2010) Implication of Kir4.1 Channel in Excess Potassium Clearance: An In Vivo Study on Anesthetized Glial-Conditional Kir4.1 Knock-Out Mice. J Neurosci 30: 15769–15777. https://doi.org/10.1523/JNEUROSCI.2078-10.2010
Kucheryavykh YV, Kucheryavykh LY, Nichols CG, Maldonado HM, Baksi K, Reichenbach A, Skatchkov SN, Eaton MJ (2007) Downregulation of Kir4.1 inward rectifying potassium channel subunits by RNAi impairs potassium transfer and glutamate uptake by cultured cortical astrocytes. Glia 55: 274–281. https://doi.org/10.1002/glia.20455
Kharade SV, Kurata H, Bender AM, Blobaum AL, Figueroa EE, Duran A, Kramer M, Days E, Vinson P, Flores D, Satlin LM, Meiler J, Weaver CD, Lindsley CW, Hopkins CR, Denton JS (2018) Discovery, Characterization, and Effects on Renal Fluid and Electrolyte Excretion of the Kir4.1 Potassium Channel Pore Blocker, VU0134992. Mol Pharmacol 94: 926–937. https://doi.org/10.1124/mol.118.112359
Aréchiga-Figueroa IA, Marmolejo-Murillo LG, Cui M, Delgado-Ramírez M, van der Heyden MAG, Sánchez-Chapula JA, Rodríguez-Menchaca AA (2017) High-potency block of Kir4.1 channels by pentamidine: Molecular basis. Eur J Pharmacol 815: 56–63. https://doi.org/10.1016/J.EJPHAR.2017.10.009
Ohno Y, Kunisawa N, Shimizu S (2021) Emerging Roles of Astrocyte Kir4.1 Channels in the Pathogenesis and Treatment of Brain Diseases. Int J Mol Sci 22: 10236. https://doi.org/10.3390/ijms221910236
Ohno Y, Hibino H, Lossin C, Inanobe A, Kurachi Y (2007) Inhibition of astroglial Kir4.1 channels by selective serotonin reuptake inhibitors. Brain Res 1178: 44–51. https://doi.org/10.1016/j.brainres.2007.08.018
Leo M, Schmitt L-I, Kutritz A, Kleinschnitz C, Hagenacker T (2021) Cisplatin-induced activation and functional modulation of satellite glial cells lead to cytokine-mediated modulation of sensory neuron excitability. Exp Neurol 341: 113695. https://doi.org/10.1016/j.expneurol.2021.113695
Leo M, Schmitt L-I, Steffen R, Kutritz A, Kleinschnitz C, Hagenacker T (2021) Modulation of Glutamate Transporter EAAT1 and Inward-Rectifier Potassium Channel Kir4.1 Expression in Cultured Spinal Cord Astrocytes by Platinum-Based Chemotherapeutics. Int J Mol Sci 22: 6300. https://doi.org/10.3390/ijms22126300
Mukai T, Kinboshi M, Nagao Y, Shimizu S, Ono A, Sakagami Y, Okuda A, Fujimoto M, Ito H, Ikeda A, Ohno Y (2018) Antiepileptic Drugs Elevate Astrocytic Kir4.1 Expression in the Rat Limbic Region. Front Pharmacol 9: 845. https://doi.org/10.3389/fphar.2018.00845
Wang D, Gao G (2014) State-of-the-art human gene therapy: part II. Gene therapy strategies and clinical applications. Discov Med 18: 151–161.
Ingusci S, Cattaneo S, Verlengia G, Zucchini S, Simonato M (2019) A Matter of Genes: The Hurdles of Gene Therapy for Epilepsy. Epilepsy Curr 19: 38–43. https://doi.org/10.1177/1535759718822846
Wang D, Tai PWL, Gao G (2019) Adeno-associated virus vector as a platform for gene therapy delivery. Nat Rev Drug Discov 18: 358–378. https://doi.org/10.1038/s41573-019-0012-9
McCown TJ (2006) Adeno-associated Virus-Mediated Expression and Constitutive Secretion of Galanin Suppresses Limbic Seizure Activity in Vivo. Mol Ther 14: 63–68. https://doi.org/10.1016/J.YMTHE.2006.04.004
Noè F, Pool AH, Nissinen J, Gobbi M, Bland R, Rizzi M, Balducci C, Ferraguti F, Sperk G, During MJ, Pitkänen A, Vezzani A (2008) Neuropeptide Y gene therapy decreases chronic spontaneous seizures in a rat model of temporal lobe epilepsy. Brain 131: 1506–1515. https://doi.org/10.1093/BRAIN/AWN079
Cattaneo S, Verlengia G, Marino P, Simonato M, Bettegazzi B (2021) NPY and Gene Therapy for Epilepsy: How, When,... and Y. Front Mol Neurosci 13: 608001. https://doi.org/10.3389/fnmol.2020.608001
Bernard C (2012) Treating epilepsy with a light potassium diet. Sci Transl Med 4: 161fs40. https://doi.org/10.1126/scitranslmed.3005297
Wykes RC, Heeroma JH, Mantoan L, Zheng K, MacDonald DC, Deisseroth K, Hashemi KS, Walker MC, Schorge S, Kullmann DM (2012) Optogenetic and potassium channel gene therapy in a rodent model of focal neocortical epilepsy. Sci Transl Med 4: 161ra152. https://doi.org/10.1126/scitranslmed.3004190
Snowball A, Chabrol E, Wykes RC, Shekh-Ahmad T, Cornford JH, Lieb A, Hughes MP, Massaro G, Rahim AA, Hashemi KS, Kullmann DM, Walker MC, Schorge S (2019) Epilepsy Gene Therapy Using an Engineered Potassium Channel. J Neurosci 39: 3159–3169. https://doi.org/10.1523/JNEUROSCI.1143-18.2019
Walker MC, Kullmann DM (2020) Optogenetic and chemogenetic therapies for epilepsy. Neuropharmacology 168: 107751. https://doi.org/10.1016/j.neuropharm.2019.107751
Magloire V, Cornford J, Lieb A, Kullmann DM, Pavlov I (2019) KCC2 overexpression prevents the paradoxical seizure-promoting action of somatic inhibition. Nat Commun 10: 1225. https://doi.org/10.1038/s41467-019-08933-4
Cheung DL, Cooke MJ, Goulton CS, Chaichim C, Cheung LF, Khoshaba A, Nabekura J, Moorhouse AJ (2022) Global transgenic upregulation of KCC2 confers enhanced diazepam efficacy in treating sustained seizures. Epilepsia 63: e15–e22. https://doi.org/10.1111/epi.17097
Han X, Boyden ES (2007) Multiple-Color Optical Activation, Silencing, and Desynchronization of Neural Activity, with Single-Spike Temporal Resolution. PLoS One 2: e299. https://doi.org/10.1371/journal.pone.0000299
Berndt A, Lee SY, Ramakrishnan C, Deisseroth K (2014) Structure-guided transformation of channelrhodopsin into a light-activated chloride channel. Science 344: 420–424. https://doi.org/10.1126/science.1252367
Berndt A, Lee SY, Wietek J, Ramakrishnan C, Steinberg EE, Rashid AJ, Kim H, Park S, Santoro A, Frankland PW, Iyer SM, Pak S, Ährlund-Richter S, Delp SL, Malenka RC, Josselyn SA, Carlén M, Hegemann P, Deisseroth K (2016) Structural foundations of optogenetics: Determinants of channelrhodopsin ion selectivity. Proc Natl Acad Sci USA 113: 822–829. https://doi.org/10.1073/pnas.1523341113
Alfonsa H, Lakey JH, Lightowlers RN, Trevelyan AJ (2016) Cl-out is a novel cooperative optogenetic tool for extruding chloride from neurons. Nat Commun 7: 13495. https://doi.org/10.1038/ncomms13495
Malyshev AY, Roshchin MV, Smirnova GR, Dolgikh DA, Balaban PM, Ostrovsky MA (2017) Chloride conducting light activated channel GtACR2 can produce both cessation of firing and generation of action potentials in cortical neurons in response to light. Neurosci Lett 640: 76–80. https://doi.org/10.1016/j.neulet.2017.01.026
Messier JE, Chen H, Cai Z-L, Xue M (2018) Targeting light-gated chloride channels to neuronal somatodendritic domain reduces their excitatory effect in the axon. Elife 7: e38506. https://doi.org/10.7554/eLife.38506
Tong X, Ao Y, Faas GC, Nwaobi SE, Xu J, Haustein MD, Anderson MA, Mody I, Olsen ML, Sofroniew MV, Khakh BS (2014) Astrocyte Kir4.1 ion channel deficits contribute to neuronal dysfunction in Huntington’s disease model mice. Nat Neurosci 17: 694–703. https://doi.org/10.1038/nn.3691
Sicca F, Ambrosini E, Marchese M, Sforna L, Servettini I, Valvo G, Brignone MS, Lanciotti A, Moro F, Grottesi A, Catacuzzeno L, Baldini S, Hasan S, D’Adamo MC, Franciolini F, Molinari P, Santorelli FM, Pessia M (2016) Gain-of-function defects of astrocytic Kir4.1 channels in children with autism spectrum disorders and epilepsy. Sci Rep 6: 34325. https://doi.org/10.1038/srep34325
Niday Z, Tzingounis AV (2018) Potassium Channel Gain of Function in Epilepsy: An Unresolved Paradox. Neurosci 24: 368–380. https://doi.org/10.1177/1073858418763752
Funding
The reported study was funded by the Russian Foundation for Basic Research (RFBR), project no. 19-315-60016.
Author information
Authors and Affiliations
Contributions
The authors equally contributed to the writing of this review.
Corresponding author
Ethics declarations
CONFLICT OF INTEREST
The authors declare that they have neither evident nor potential conflict of interest related to the publication of this article.
Additional information
Translated by A. Polyanovsky
Russian Text © The Author(s), 2022, published in Zhurnal Evolyutsionnoi Biokhimii i Fiziologii, 2022, Vol. 58, No. 5, pp. 349–366https://doi.org/10.31857/S0044452922050096.
Rights and permissions
Open Access. This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

{kind=link}
{kind=link}
{kind=link}
Cite this article
Proskurina, E.Y., Zaitsev, A.V. Regulation of Potassium and Chloride Concentrations in Nervous Tissue as a Method of Anticonvulsant Therapy. J Evol Biochem Phys 58, 1275–1292 (2022). https://doi.org/10.1134/S0022093022050015
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0022093022050015