
The interlayer thermal conductivity of two asymmetric bilayer carbon structures has been studied within the nonorthogonal tight binding model. One layer of the first structure proposed in this work for the first time is graphene and the second layer is Stone–Wales graphene, which is recently proposed carbon allotrope. The second asymmetric structure is bilayer graphene, where one layer consists of 12C isotope and the second layer consists of rarer 13C isotope. It has been shown that the interlayer thermal conductivity of asymmetric structures is more than an order of magnitude lower than that for their symmetric analogs, bilayer graphene and Stone–Wales bilayer graphene, with the same isotope composition. A high interlayer thermal conductivity of symmetric structures compared to asymmetric ones is due to the resonant interaction of phonon subsystems of individual layers (phonon spectra of individual layers in symmetric structures coincide, whereas these spectra in asymmetric structures are different). It has been shown that the graphene layer in the unstrained graphene–Stone–Wales graphene structure is flat, whereas the Stone–Wales graphene layer is corrugated. Both layers of this structure biaxially stretched by 5% become flat. The interlayer attraction energy, interlayer distance, activation energy of parallel shear of the layers, and the elastic modulus under vertical compression have been determined for unstrained and deformed structures.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
The fabrication of graphite monolayer called graphene [1] having a high strength [2] and a high conductivity [3] stimulated active search for other planar carbon structures. Graphane [4], graphyne [5], graphdiyne [6], diamane on the SiC (0001) substrate [7], fluoridated diamond (F-diamane) on the CuNi (111) substrate [8], and quasi-two-dimensional composite of polyaniline and graphene oxide [9] were synthesized. A number of stable two-dimensional carbon allotropes were determined within various models of the interatomic interaction. Metallic R-10 graphene [10], carbon monolayer specified by the authors of [11] as 123-E8Y24-1, two \(s{{p}^{2}} + s{{p}^{3}}\)-hybridized structures with unique electronic properties [12], PHH-graphene [13], etc., were proposed in 2021. Azugraphene [14] and Stone–Wales graphene (SWG) [15], which were predicted in 2019, have the highest thermodynamic stability among all known single-layer carbon structures. Stone–Wales graphene can be obtained by forming numerous periodically arranged Stone–Wales (SW) defects in conventional graphene [16]. An SW defect is formed in graphene by rotating a C–C bond by 90°; in this case, a pair of pentagonal rings and a pair of heptagonal rings are formed from four hexagonal rings (see Fig. 1). Аzugraphene is less energetically favorable compared to SWG. According to the density functional theory calculations, the binding energy of azugraphene is 16 meV/atom above that of SWG. According to the ab initio calculations [15], the binding energy of SWG is 149 meV/atom above that of graphene. This excess determined in [17] within the nonorthogonal tight binding model [18] is 161 meV/atom.

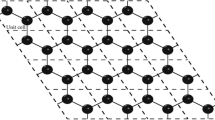
Unit cell of asymmetric bilayer graphene: (gray circles) lower graphene sheet and (black circles) Stone–Wales graphene.
Attention is paid not only to single-layer but also to bilayer planar carbon structures. Bilayer graphene (BG) with a controlled band gap width was synthesized in 2006 [19]. The bilayer carbon structure, which is formed by two graphene sheets with an angle of 1.1° between them and was classified in [20] as a “strange metal,” has an extraordinary temperature dependence of the conductivity at low temperatures and is a superconductor with the critical temperature Tc = 1.7 K [21]. The authors of [22, 23] mentioned that a BG can be used to create a new type of field-effect transistors. Bilayer graphene can also be used as a humidity sensor [24]. The properties of perforated BG with interlayer bridges were studied in [25], the authors of [26] examined the conductivity of BG ribbons, and the interlayer thermal conductivity of BG was determined in [27].
A bilayer structure can be formed not only from graphene but also from its allotropes and derivatives. A strong covalent bond between hydrogenated graphene sheets was predicted in the structure called by the authors of [28] diamane. Аzugraphene and SWG can also form a bilayer material [14, 29]. In contrast to diamane, the interlayer bond in bilayer azugraphene and bilayer Stone–Wales graphene (BSWG) is weak (similar to BG).
The aim of this work is to study an asymmetric bilayer carbon structure called asymmetric bilayer graphene (ABG), one layer of which is graphene and the second layer is Stone–Wales graphene. As mentioned in [30], it is promising to study graphene with numerous Stone–Wales defects densely located on it. However, the authors of [30] indicate that the formation of such defects is more difficult than, e.g., the formation of graphene divacancies. To the best of my knowledge, any practically implementable method of the creation of the structure has not yet been proposed. An anomalously high binding energy of this structure, whose thermodynamic stability is only slightly lower than that of graphene, allows one to expect that SWG could be synthesized. In addition, the creation of ABG can also be promoted by the combination of the following factors. The first factor is the possibility of forming corrugations in the desired direction, which is controlled by the uniaxial compression of BG [27]. The second factor is the preferential adsorption of hydrogen atoms on the convex surface of a graphene corrugation. This tendency follows from the scanning tunneling microscopy data obtained in [31], where local adsorbed structures of unilaterally hydrated graphene on the SiC substrate were revealed. Hydrogen was collected in stable clusters predominantly on convex (owing to the substrate profile) areas of the graphene surface. Third, hydrogen adsorbed in desired areas can serve as a catalyst, which noticeably reduces the activation energy of the formation of SW defects. In particular, the activation energy of the formation of Stone–Wales defects in pure graphene determined within the nonorthogonal tight binding model is 8.03 eV. The adsorption of a hydrogen atom on a Stone–Wales defect reduces this energy to 6.51 eV [32], which corresponds to a value of 6.8 eV obtained within the density functional theory [33]. The mutual attraction of SW defects at distances exceeding the sizes of the unit cell of graphene and their repulsion at small distances can also lead to the aggregation of these defects into ordered dense clusters [34].
CALCULATION METHODS
The interatomic interaction was described by a potential based on the nonorthogonal tight binding model [18], which is less accurate than ab initio methods but requires less computer resources. The FO-RTRAN implementation of the interatomic potential [18] was reported in [35]. The results of this approach are in good agreement with density functional theory results for structures containing carbon atoms in states with various hybridization types [36]. This model was successfully applied to study many systems consisting of carbon, hydrogen, nitrogen, and oxygen atoms (see, e.g., [17, 27, 29, 37–39] and references therein). The adequacy of this approach was also confirmed in [40]. An insignificant modification of this potential performed in [27] makes it possible to take into account the van der Waals interlayer interaction in graphite and bilayer graphene. This modification is also used in this work.
Figure 1 shows the front view of the unit cell of ABG, where large gray and small black circles indicate atoms in graphene and SWG, respectively. The black thick straight segment indicates the C–C bond; the rotation of this bond by an angle of 90° transforms the upper layer of Stone–Wales graphene to conventional graphene.
All details of the method to numerically study the structure of ABG are similar to those used in [27, 29] for BG and BSWG, respectively. This allows one to correctly compare the characteristics of all three bilayer structures. The calculation ABG cluster consists of 2 × 2 = 4 unit cells and contains 128 atoms. The boundary conditions are periodic in the \((X,Y)\) plane and are free in the direction of the vertical Z axis. The periodicity cell is formed by two vectors \({\mathbf{V}_{1}} = ({{X}_{1}},{{Y}_{1}},0)\) and \({\mathbf{V}_{2}} = ({{X}_{2}},{{Y}_{2}},0)\). To determine the stable configuration of ABG, the gradient minimization of the binding energy of the structure in the coordinates of the atoms and in the sizes of the periodic calculation cell was performed. The interplanar spacing and elastic modulus \({{C}_{{ZZ}}} = {{\partial }^{2}}{{E}_{b}}{\text{/}}\partial \varepsilon _{{ZZ}}^{2}\), where Eb is the binding energy of ABG (in eV/atom) and \({{\varepsilon }_{{ZZ}}}\) is the normal diagonal component of the strain tensor, were determined for the resulting equilibrium structure. Similar to [27, 29], the graphene sheet initially had zero temperature and SWG was heated to 77, 300, 1000, 3000, 5000, and 7000 K. the melting temperature of single-layer graphene and SWG determined within the nonorthogonal tight binding model are 5100 and 3800 K, respectively [17, 39]. Since the melting temperature of SWG is lower than that of graphene, several behaviors of ABG are possible for temperatures of 5000 and 7000 K. If the thermal decay time of the SWG layer is shorter than the heat transfer time between the layers of ABG, this decay should be expected. Otherwise, because of the redistribution of the thermal energy between the layers, the temperature of the “hot” layer is approximately halved to 2500 and 3500 K and melting becomes impossible. In this case, one can expect the stratification of the structure or parallel shift of layers. The dynamic simulation performed in this work allows one to determine which of the listed scenarios is implemented. Inhomogeneous heating can be implemented, e.g., by passing a pulsed current through the upper layer if the lower “cold” layer has a low conductivity because of functionalization (existence of regions doped with fluorine [41, 42], chlorine [43], or hydrogen [5] that can block the current flow through this layer).
Heating was simulated by the corresponding stochastic displacement of SWG atoms and assigning them random velocities satisfying a Maxwell distribution. The dynamic simulation of ABG was performed within a microcanonical ensemble [44]. The time evolution of motion of atoms was calculated by the Verlet method with a step of 0.3 fs.
The characteristic time \(\tau \) of establishing the equilibrium temperature common for both layers of ABG through interlayer heat transfer was determined by the algorithm of the exponential interpolation of the time dependence of the kinetic energies of both layers, which was described in detail in [27]. Four variants of motion of the system differing in the initial stochastic distribution of velocities and displacements of atoms were calculated for each temperature. As a result, the dispersion of the physical quantities under study was determined.
RESULTS AND DISCUSSION. STATIC CHARACTERISTICS
The gradient relaxation of the potential energy Eb in the coordinates of all atoms and the coordinates of the vectors \({\mathbf{V}_{1}} = ({{X}_{1}},{{Y}_{1}},0)\) and \({\mathbf{V}_{2}} = ({{X}_{2}},{{Y}_{2}},0)\), which form the periodicity cell, provided the geometrical and energy characteristics of the structure of ABG (see Tables 1 and 2). The phonon spectrum of this structure, where imaginary frequencies are absent, confirms that the resulting configuration is a local minimum of the energy surface of the cluster in the coordinate space of atoms. Furthermore, this structure has the minimum potential energy among all configurations obtained by the parallel displacement of SWG in the \((X,Y)\) plane above the graphene sheet (see Fig. 2).

Potential energy contours (eV/atom) under the displacement of the upper layer (Stone–Wales graphene) with respect to the lower one (graphene) by the translation vector with the coordinates \((X,Y)\) with respect to the equilibrium asymmetric bilayer graphene structure. The interval between contours is 0.05 meV/atom.
Several local minima (black areas) whose energies coincide with the initial energy are observed in Fig. 2 because of the periodicity of the structure under study. The light dashed line in Fig. 2 is one of the optimal grazing trajectories of SWG over the lower graphene sheet. The difference of the potential energy from the initial value at the motion in this trajectory is no more than \({{E}_{{{\text{shift}}}}} = 0.7{\kern 1pt} \) meV/atom. The arrows mark the points of maximum energy barriers in this trajectory preventing grazing.
The interlayer distance d in ABG presented in Table 1 is determined as
where \(Z{{g}_{i}}\) and \(ZSW{{g}_{i}}\) are the vertical coordinates of atoms in graphene and SWG, respectively. The interlayer interaction energy \({{E}_{{{\text{lay}}}}}\) in Table 2 is determined by the formula
where EABG is the potential energy of the ABG structure consisting of 128 atoms and EG and ESWG are the potential energies of single cells of graphene and SWG, respectively, each consisting of 64 atoms.
The characteristics of the structure biaxially stretched by 5% (diagonal components of the strain tensor are \({{\varepsilon }_{{XX}}} = {{\varepsilon }_{{YY}}} = 0.05\)) were also studied. The results of the calculations of the deformed structure are also summarized in Tables 1 and 2. According to these tables, the interlayer distance d in the unstrained ABG structure is much larger than that in the unstrained BG and BSWG structures, whereas the vertical stiffness CZZ, interlayer attraction energy Elay, and shift activation energy Eshift in the unstrained ABG structure are lower than the respective parameters in the unstrained BG and BSWG structures. This difference is explained by the corrugated shape of the upper SWG sheet in the ABG structure (see Figs. 3a and 3b). The different densities of the unstrained graphene and SWG sheets indicate the presence of corrugation on SWG and its absence on graphene. Indeed, the p-eriodicity cells in BG and BSWG are smaller and larger than that in ABG (see the X components of the vectors V1 and V2 in Table 1). For this reason, the graphene and SWG layers in ABG are stretched and compressed, respectively. Compression results in corrugation not only in single-layer but also in bilayer graphene and SWG structures [27, 29]. Under biaxial stretching of ABG by 5%, both parts of this structure are stretched (see the X components of the vectors V1 and V2 in Table 1 at \({{\varepsilon }_{{XX}}} = {{\varepsilon }_{{YY}}} = 0.05\)). In this case, the upper part of the structure becomes flat (see Fig. 3c), and the differences of the quantities d, CZZ, Elay, and Eshift from the respective parameters in BG and BSWG are much smaller. The study of dynamic processes of heat redistribution in nonuniformly heated structures provides a different picture.

(a) Front and (b) side views of the equilibrium structure and (с) side view of the structure of asymmetric bilayer graphene biaxially stretched by 5%.
RESULTS AND DISCUSSION. DYNAMIC CHARACTERISTICS
The molecular dynamics results for the characteristic thermalization time τ in the BG, BSWG, and ABG structures are summarized in Table 3, as well as in Figs. 4a and 4b. The thermalization time τ in the ABG structure at a cryogenic temperature is much longer than those in the BG and BSWG structures. For this reason, data for the ABG structure at T = 77 K are not shown in Figs. 4a and 4b. The thermalization time τ in the ABG structure at temperatures below the melting temperature of the heated layer is also much longer than those in the BG and BSWG structures.

(Color online) Temperature dependences of the interlayer heat transfer time in (black solid lines and squares) asymmetric bilayer graphene, (red dashed lines and triangles) bilayer graphene, and (blue solid lines and circles) bilayer Stone–Wales graphene for (a) the unstrained crystal and (b) the crystal biaxially stretched by 5%.
An anomalously long thermalization time τ in the unstrained ABG structure cannot be due to the corrugated surface of SWG because corrugations are absent in the deformed structure (see Fig. 3c), and the thermalization time τ at low temperatures remains long. The most probable reason for this effect can be the resonant interaction of the phonon subsystems of the weakly coupled hot and cold parts of bilayer structures. The phonon spectrum of an individual upper layer of symmetric BG and BSWG structures is identical to the spectrum of an individual lower layer. The interlayer van der Waals interaction is much weaker than covalent bonds inside the layer. Consequently, at the initial stage when the energy of the upper layer exceeds the energy of the lower layer, the hot and cold layers can be considered as the transmitter and receiver of phonons, respectively. In this case, the energy transfer intensity is maximal under the resonance condition of the coincidence of frequencies of the receiver and transmitter. The phonon spectrum of an individual upper layer of ABG is significantly different from the spectrum of an individual lower layer (see Fig. 5). Resonance between phonon subsystems is absent; as a result, the thermal energy transfer intensity decreases (the heat transfer time τ increases).

(Color online) Normalized phonon densities of states of (black solid line) graphene and (red dashed line) Stone–Wales graphene. The phonon density of states obtained in this work within the nonorthogonal tight binding model for a cluster consisting of 64 12C atoms qualitatively corresponds to that obtained in [45] within the density functional theory.
To confirm the resonant character of the interlayer thermal conductivity in symmetric bilayer structures, the thermalization time τ in unstrained bilayer graphene with an inhomogeneous isotopic composition was determined for four variants of thermal relaxation in unstrained bilayer graphene with the initial heating of the hot plane to a temperature of 77 K. The initial thermal displacements and velocities of atoms in these variants coincide with those used in [27]. However, unlike [27], where both graphene sheets consisted of the 12C isotope, in the case under consideration, only the hot plane consists of this isotope, whereas the cold plane is formed by the rarer 13C isotope. An increase in the mass proportionally reduced the phonon frequencies of a single lower plane by ~4%, which violated the resonance condition between phonon subsystems of two layers. The performed simulation showed that the interlayer heat transfer time in this case is (564 ± 93) ps, which is about 22 times longer than that in the BG structure with a homogeneous isotopic composition (see Table 3). The resonant character of the interaction of phonon subsystems in symmetric structures can also explain a decrease in the difference between τ values in ABG and symmetric structures with increasing temperature (see Table 3 and Fig. 4). Indeed, nonresonant heat transfer between modes with different frequencies, which is due to anharmonic processes, coexist in symmetric structures with resonant heat transfer between modes with the same frequency. The contribution to heat transfer from anharmonic processes that do not require the coincidence of phonon frequencies increases with the temperature. This mechanism diminishes the difference between the symmetric resonant and asymmetric systems at temperatures up to 3000 K. At a temperature of 7000 K in strained and unstrained structures, as well as at a temperature of 5000 K, the melting of the hot layer plays a decisive role in interlayer thermal conductivity in the strained ABG structure. The character of melting in the SWG structure is similar to that presented in [17, 27, 29, 34, 39] and involves the formation of multiatomic rings, which are further transformed to long carbine chains of sp-hybridized atoms. The bilayer structure can hardly exist after melting, although the attachment of atoms of the hot plane to the cold plane in the ABG structure was not observed, as at the melting of the BG and BSWG structures [27, 29].
The behavior of the unstrained ABG structure at a temperature of 5000 K was surprising. Melting is absent in all four variants, but there are (i) the parallel shift of the planes with respect to each other and (ii) the intense annealing of Stone–Wales defects down to their disappearance. This annealing transforms the ABG structure to the BG structure and, thereby, the interlayer heat transfer times in the initially different structures are the same (see Table 3). The shift of the planes is observed in the temperature interval of 300–3000 K, but the annealing of defects in the thermalization time τ is not detected. The shift is absent at a temperature of 77 K. The strained ABG structure demonstrates the shift observed in four variants at T = 3000 K and only in one variant at T = 1000 K, whereas the shift is absent at lower temperatures. The temperature intervals of appearance of shift in the strained and unstrained ABG crystals are different because the shift activation energies in these structures are different, 1.2 and 0.7 meV/atom, respectively.
CONCLUSIONS
The study of the static characteristics of ABG has shown that the interlayer distance, the vertical stiffness, and the activation energy of the parallel shift of the layers in the unstrained structure are much different from the respective parameters of the BG and BSWG structures, whereas this difference in stretched systems is smaller. This is explained by the different surface densities of graphene and SWG; as a result, SWG is compressed and corrugations are formed in it. Under stretching, corrugations disappear, and the structure becomes planar, similar to BG and BSWG.
The molecular dynamics study showed that resonance between phonon systems of individual layers plays a significant role in symmetric bilayer structures. The interlayer heat transfer time in an asymmetric bilayer structure (ABG or bilayer graphene with a nonuniform isotopic composition), where the phonon spectra of the layers are different is noticeably longer than this time in symmetric BG and BSWG structures. Since the difference between the phonon spectra decisively affects the interlayer thermal conductivity, it would be of interest to study the effect of the asymmetric functionalization of bilayer graphene with fluorine, chlorine, or hydrogen on the interlayer heat transfer time. Such a study can be of practical significance for the estimate of cooling regimes of fast electronics elements. If the single-sided deposition of these atoms on the BG structure (for the creation of dielectric regions) significantly reduces the thermal conductivity, this will complicate the creation of fast logical elements.
It has been shown that heating results in the following structural changes in ABG: grazing of layers with respect to each other at temperatures up to room temperature and the melting (fragmentation) of the heated layer at high temperatures. The complete annealing of Stone–Wales defects has been detected in the unstrained ABG crystal at T = 5000 K. It is noteworthy that this effect has not been observed at the melting of single- and bilayer SWG [17, 29]. For this reason, it would be interesting to study in detail the possibility of the annealing of an individual Stone–Wales defect in the BG crystal uniaxially deformed in various directions.
Change history
06 December 2022
An Erratum to this paper has been published: https://doi.org/10.1134/S0021364022340069
REFERENCES
K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva, and A. A. Firsov, Science (Washington, DC, U. S.) 306, 666 (2004).
A. E. Galashev and O. R. Rakhmanova, Phys. Usp. 57, 970 (2014).
K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, M. I. Katsnelson, I. V. Grigorieva, S. V. Dubonos, and A. A. Firsov, Nature (London, U.K.) 438, 197 (2005).
J. O. Sofo, A. S. Chaudhari, and G. D. Barber, Phys. Rev. B 75, 153401 (2007).
Q. Li, Y. Li, Y. Chen, L. Wu, C. Yang, and X. Cui, Carbon 136, 248 (2018).
Y. Li, L. Xu, H. Liu, and Y. Li, Chem. Soc. Rev. 43, 2572 (2014).
Y. Gao, T. Cao, F. Cellini, C. Berger, W. A. de Heer, E. Tosatti, E. Riedo, and A. Bongiorno, Nat. Nanotechnol. 13, 133 (2018).
P. V. Bakharev, M. Huang, M. Saxena, S. W. Lee, S. H. Joo, S. O. Park, J. Dong, D. Camacho-Mojica, S. Ji, Y. Kwon, M. Biswal, F. Ding, S. K. Kwak, Z. Lee, and R. S. Ruoff, arXiv: 1901.02131 (2019).
G. M. de Araujo, L. Codognoto, and F. R. Simões, J. Solid State Electrochem. (2020). https://doi.org/10.1007/s10008-020-04517-1
Zh.-L. Lv, Q. Lu, D.-H. Huang, and F.-T. Liu, Diamond Relat. Mater. 114, 108315 (2021).
P. L. Yan, T. Ouyang, and J. X. Zhong, Nanoscale 13, 3564 (2021).
W. Zhang, C. C. Chai, and Y. T. Yang, Appl. Surf. Sci. 537, 147885 (2021).
J. Li, Sh. Li, T. Ouyang, Ch. Zhang, Ch. Tang, Ch. He, and J. Zhong, J. Phys. Chem. Lett. 12, 732 (2021).
J. Liu and H. Lu, RSC Adv. 9, 34481 (2019).
H. Yin, X. Shi, C. He, M. Martinez-Canales, J. Li, C. J. Pickard, C. Tang, T. Ouyang, C. Zhang, and J. Zhong, Phys. Rev. B 99, 041405 (2019).
A. J. Stone and D. J. Wales, Chem. Phys. Lett. 128, 501 (1986).
L. A. Openov and A. I. Podlivaev, JETP Lett. 109, 710 (2019).
M. M. Maslov, A. I. Podlivaev, and K. P. Katin, Mol. Simul. 42, 305 (2016).
T. Ohta, A. Bostwick, T. Seyller, K. Horn, and E. Rotenberg, Science (Washington, DC, U. S.) 313, 951 (2006).
Y. Cao, D. Chowdhury, D. Rodan-Legrain, O. Rubies-Bigorda, K. Watanabe, T. Taniguchi, T. Senthi, and P. Jarillo-Herrero, Phys. Rev. Lett. 124, 076801 (2020).
Y. Cao, V. Fatemi, S. Fang, K. Watanabe, T. Taniguchi, E. Kaxiras, and P. Jarillo-Herrero, Nature (London, U.K.) 556, 43 (2018).
Y. Zhang, T. Tang, C. Girit, Z. Hao, M. C. Martin, A. Zettl, M. F. Crommie, Y. R. Shen, and F. Wang, Nature (London, U.K.) 459, 820 (2009).
G. Fiori and G. Iannaccone, IEEE Electron Dev. Lett. 30, 261 (2009).
M.-C. Chen, C.-L. Hsu, and T.-J. Hsueh, IEEE Electron Dev. Lett. 35, 590 (2014). https://doi.org/10.1038/nmat2082
A. A. Artyukh and L. A. Chernozatonskii, JETP Lett. 109, 472 (2019).
V. A. Demin, D. G. Kvashnin, P. Vancho, G. Mark, and L. A. Chernozatonskii, JETP Lett. 112, 305 (2020).
A. I. Podlivaev, K. S. Grishakov, K. P. Katin, and M. M. Maslov, JETP Lett. 113, 169 (2021).
L. A. Chernozatonskii, P. B. Sorokin, A. G. Kvashnin, and D. G. Kvashnin, JETP Lett. 90, 134 (2009).
A. I. Podlivaev, K. S. Grishakov, K. P. Katin, and M. M. Maslov, JETP Lett. 114, 143 (2021).
F. L. Thiemann, P. Rowe, A. Zen, E. A. Müller, and A. Michaelides, Nano Lett. 21, 8143 (2021).
R. Balog, B. Jørgensen, J. Wells, E. Lægsgaard, P. Hofmann, F. Besenbacher, and L. Hornekær, J. Am. Chem. Soc. 131, 8744 (2009).
A. I. Podlivaev and L. A. Openov, Phys. Solid State 57, 2562 (2015).
A. J. M. Nascimento and R. W. Nunes, Nanotechnology 24, 435707 (2013).
A. I. Podlivaev and L. A. Openov, JETP Lett. 101, 173 (2015).
K. P. Katin, K. S. Grishakov, A. I. Podlivaev, and M. M. Maslov, J. Chem. Theory Comput. 16, 2065 (2020).
K. P. Katin and M. M. Maslov, J. Phys. Chem. Solids 108, 82 (2017).
A. I. Podlivaev and K. P. Katin, JETP Lett. 92, 52 (2010).
M. M. Maslov, K. P. Katin, A. I. Avkhadieva, and A. I. Podlivaev, Russ. J. Phys. Chem. B 33, 152 (2014).
L. A. Openov and A. I. Podlivaev, Phys. Solid State 58, 847 (2016).
Yu. S. Nechaev, E. A. Denisov, N. A. Shurygina, A. O. Cheretaeva, E. K. Kostikova, and S. Yu. Davydov, JETP Lett. 114, 337 (2021).
J. T. Robinson, J. S. Burgess, C. E. Junkermeier, S. C. Badescu, T. L. Reinecke, F. K. Perkins, M. K. Zalalutdniov, J. W. Baldwin, J. C. Culbertson, P. E. Sheehan, and E. S. Snow, Nano Lett. 10, 3001 (2010).
K. S. Grishakov, K. P. Katin, V. S. Prudkovskiy, and M. M. Maslov, Appl. Surf. Sci. 463, 1051 (2019).
B. Li, L. Zhou, D. Wu, H. Peng, K. Yan, Y. Zhou, and Z. Liu, ACS Nano 5, 5957 (2011).
E. M. Pearson, T. Halicioglu, and W. A. Tiller, Phys. Rev. A 32, 3030 (1985).
F. Liu, P. Ming, and J. Li, Phys. Rev. B 76, 064120 (2007).
ACKNOWLEDGMENTS
I am grateful to K.P. Katin for discussion of the results.
Funding
This work was supported by the National Research Nuclear University MEPhI (program Prioritet 2030).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The author declares that he has no conflicts of interest.
Additional information
Translated by R. Tyapaev
The original online version of this article was revised: Due to a retrospective Open Access order.
Rights and permissions
Open Access. This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Podlivaev, A.I. Bilayer Graphene–Stone–Wales Graphene: Structure, Stability, and Interlayer Thermal Conductivity. Jetp Lett. 115, 348–355 (2022). https://doi.org/10.1134/S0021364022200024
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0021364022200024