
Abstract
Demyelinating diseases of the central nervous system are caused by an autoimmune attack on the myelin sheath surrounding axons. Myelin structural proteins become antigenic, leading to the development of myelin lesions. The use of highly specialized laboratory diagnostic techniques for identification of specific antibodies directed against myelin components can significantly improve diagnostic approaches. Myelin oligodendrocyte glycoprotein (MOG) antibody-associated disease (MOGAD) currently includes demyelinating syndromes with known antigens. Based on the demonstrated pathogenic role of human IgG against MOG, MOGAD was classified as a distinct nosological entity. However, generation of multiple MOG isoforms by alternative splicing hinders antigen detection even with the most advanced immunofluorescence techniques. On the other hand, MOG conformational changes ensure the structural integrity of other myelin proteins and maintain human-specific mechanisms of immune autotolerance.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
Demyelinating disorders of the central nervous system (CNS) have always been in the spotlight due to their clinical diversity and relatively high incidence. Demyelinating diseases are caused by neuroinflammation induced by the autoimmune attack of peripheral immune cells on the CNS antigens. They are associated with the formation in the gray and white matter of the brain and/or spinal cord of foci characterized by myelin damage, inflammatory perivascular infiltration by T and B lymphocytes and macrophages, activation of microglia in the disease acute phase, and subsequent alleviation of inflammatory processes with the onset of myelin sheath repair. Depending on the extent of myelin sheath damage and functional and quantitative viability of myelin-forming cells, remyelination can be complete, partial, or ineffective [1]. Different inflammatory demyelination mechanisms exist. The attack of antibodies on oligodendrocytes and myelin components can result in the development of primary demyelination. Secondary demyelination occurs due to the damage to other CNS cells and, as a result of developing inflammation, secondary destruction of myelin takes place. The underlying causes of autoimmune diseases of the CNS can be identified using highly specific cell-based assays (CBAs). Understanding the role of autoantibodies in neurological diseases has significantly altered clinical practices over the past decade, while identification of autoantibodies has made it possible to reclassify some of these diseases. For instance, the discovery of antibodies targeting aquaporin-4 (AQP4), a water channel protein of astrocytes, in patients with neuromyelitis optica or antibodies against N-methyl-D-aspartate glutamate receptors (NMDARs) associated with the most prevalent autoimmune encephalitis allowed to classify these diseases as distinct nosological entities. Most important, the discovery of antibodies targeting neurons, myelin components, or glial cells has significantly expanded existing diagnostic approaches and therapeutic options [2, 3].
CNS DISEASES ASSOCIATED WITH ANTI-MOG ANTIBODIES – NEW NOSOLOGICAL ENTITIES WITH IDENTIFIED ANTIGEN
To date, the most common and well-studied chronic demyelinating disease of the CNS is multiple sclerosis (MS), which is characterized by the appearance of multiple lesions mainly in the white matter of the brain and spinal cord due to the development of autoimmune inflammation. In the majority of cases, progression of MS is marked by relapses accompanied by the emergence of new neurological symptoms or worsening of the existing ones. Over time, progression of neurological deficits in MS acquires a steady character and leads to serious disabilities, mainly in young and middle-aged people. However, the precise mechanisms initiating the production of specific antibodies and activation of effector lymphocytes in MS have yet to be determined [4]. Most researchers believe that MS has a primary autoimmune etiology. Identification of its immunological causes and molecular targets has been in the research focus over the past few decades, resulting in the discovery of strong genetic associations with the immune regulatory mechanisms and elucidation of mechanisms of immune attack on the elements of the CNS. Observations of the natural progression of MS, however, have revealed inconsistencies that cast doubt on the assumption that MS is a solely autoimmune disorder. According to the alternative theory, MS is a neurodegenerative disorder and the initial failure occurs in the CNS cells and structures. This model states that the event initiating the disease is primary cytodegeneration resulting in the release of highly antigenic components which then trigger the immunopathological cascade [5]. Currently, the diagnosis of MS is based on clinical manifestations, neuroimaging data, and detection of intrathecal synthesis of oligoclonal immunoglobulins, emphasizing the role of B lymphocytes in the disease pathogenesis [6]. CNS demyelinating syndromes that do not meet the criteria for MS have been considered as its atypical versions. Improving the methods of laboratory diagnostics, in particular, immunofluorescent CBAs, makes it possible to detect antibodies against antigenic components of myelin with a high accuracy.
A great success in the search for specific pathogenic antibodies in the atypical MS variants was achieved in 2004, when such autoantibodies were identified in patients with neuromyelitis optica, a disease manifested by unilateral or bilateral optic neuritis and longitudinal widespread transverse myelitis involving more than three vertebral segments [7]. The detected antibodies were directed against AQP4, a water channel protein expressed in the perivascular zone on the astrocytic pedicles around blood vessels. The discovery of antibody to AQP4 (AQP4-IgG) as a target antigen unequivocally confirmed neuromyelitis optica as a separate nosological entity distinct from MS [8]. Over time, AQP4-IgG was identified by CBA not only in patients with the optic-spinal phenotype, but also in patients with demyelinating lesions in the brain, diencephalon, area postrema, and brainstem. Later, the term NMOSD (neuromyelitis optica spectrum disorder) was introduced, which combined both AQP4-IgG seropositive and seronegative forms of the disease with a typical clinical and radiological picture [9]. It should be noted that demyelination caused by AQP4-IgG is secondary, since the main pathogenic events are associated with the development of astrocytopathy and damage to the blood–brain barrier (BBB). An increase in the BBB permeability initiates penetration of effector lymphocytes, antibodies, and granulocytes into the CNS. AQP4-IgG is synthesized by plasmablasts on the periphery, which may be due to the wide distribution of AQP4 outside the CNS (collecting tubules of the kidneys, stomach parietal cells, epithelial cells of the bronchial mucosa, skeletal muscles). The pro-inflammatory cytokine profile, antibody- and complement-dependent cellular cytotoxicity, and impaired water and electrolyte homeostasis lead to the secondary myelin damage [10]. Therefore, no antibodies against the myelin sheath components are secreted in NMOSD. Myelin damage occurs at the sites of high expression of the AQP4 protein (optic nerves, spinal canal, circumventricular organs, i.e., CNS zones closely adjacent to the ependymal lining). However, the uncertainty of diagnostics in the case of negative serological results has forced researchers to continue the search for other immunological markers of CNS demyelinating syndromes.
Later, the antibodies against myelin oligodendrocyte glycoprotein (MOG) have been identified in the serum. Anti-MOG antibodies (MOG-IgG) are found in a large percent of children with acute disseminated encephalomyelitis (ADEM). In adult population, recurrent optic neuritis is a condition in which elevated MOG-IgG titers are most prevalent, followed by myelitis of varying degree of severity, encephalomyelitis, stem encephalitis, ADEM-like phenotypes, and cortical encephalitis. More than 40% NMOSD patients with the AQP4-IgG seronegative status have high MOG-IgG titers [11]. Because of this, myelin oligodendrocyte glycoprotein antibody-associated disease (MOGAD) was initially considered as one of the NMOSD phenotypes. First of all, this was due to the coincidence of their clinical manifestations, especially in adult patients. However, histological studies demonstrated different immunopathological patterns in these diseases. MOGAD is characterized by perivenous inflammatory demyelination with the infiltration by macrophages, B lymphocytes, CD4+ T cells, and less prominent deposition of activated complement and immunoglobulin in comparison to the inflammation in NMOSD [12]. In the majority of MOGAD cases, the histopathological picture was consistent with type II demyelination, which is typical of MS or ADEM (“sleeves of demyelination”). Moreover, no astrocyte destruction or loss of AQP4 expression was observed in MOGAD. Reactive hypertrophied astrocytes, apoptotic oligodendrocytes with compacted nuclei, and intact pre-oligodendrocytes that did not express MOG were detected in the sections of biopsied material. Demyelinating lesions in MOGAD demonstrated a more pronounced loss of MOG staining than of other myelin proteins, and MOG-laden macrophages were identified, suggesting MOG-dominant myelin loss. Moreover, axons with edema were detected very infrequently; in the majority of cases, the axons were intact. In rare severe forms of MOGAD, axonal spheroids were detected [13, 14].
International diagnostic criteria for MOGAD were first published in 2018 and then updated in 2023. Currently, MOGAD is recognized as a distinct nosological entity, distinct from other known neuroinflammatory diseases, including MS and NMOSD [15, 16]. In fact, MOGAD is the first autoimmune demyelinating disease of the CNS in which primary demyelination develops and an antigen/antibody is clearly identified. Distinguishing MOGAD from the spectrum of other autoimmune demyelinating syndromes, with known or unknown antigens, is very important in terms of differential diagnosis, prognosis, and treatment. However, for a long time, it has been discussed whether IgG-MOG detected in the serum of patients is pathogenic per se or represent an immunological epiphenomenon of prior demyelination and a clinically significant biomarker of the disease.
MOG STRUCTURE AND FUNCTIONS
Myelin is a complex multilayer structure synthesized by oligodendrocytes that is composed of multiple cytoplasmic and transmembrane proteins interacting with highly structured lipids and glycolipids. These connections maintain the integrity, structure, and function of the myelin sheath surrounding axons. The majority of myelin proteins belong to protein families represented by several isoforms resulting from alternative splicing of a single gene [17]. The majority of CNS myelin proteins belong to the two families: myelin basic protein (MBP) and proteolipid protein (PLP). Minor components include glycoproteins, such as myelin-associated glycoprotein (MAG) and MOG, which, together with other myelin components, play an essential role in the formation and maintenance of the myelin sheath [18].
MOG was first identified in the late 1970s, when it was shown that a CNS myelin component named M2 (distinct from MBP and PLP) induced immune response in a form of demyelination in guinea pigs [19]. Using the mouse monoclonal antibody (mAb) 8-18C5, the M2 antigen was then identified in the rat cerebellum tissue. Based on the tissue and cellular location, molecular weight, and immunological cross-reactivity, it was demonstrated that mAb 8-18C5 recognized a unique myelin antigen similar to MOG. Using ultrastructural immunocytochemistry with 8-18C5, it was shown that MOG is expressed on the bodies, processes, and uncompacted abaxonal (attached to the basement membrane) and adaxonal (separated from the axonal membrane by the periaxonal space) myelin membranes of oligodendrocytes [20]. The MOG protein has a molecular weight of 26-28 kDa and consists of 218 amino acids (with additional 29 residues of the signal peptide). The MOG epitope interacting with mAb 8-18C5 was found only in mammals, in contrast to the evolutionarily older MBP and PLP [21, 22].
MOG is a type I integral membrane protein that belongs to the immunoglobulin (Ig) superfamily. It consists of the extracellular variable (IgV) domain, the transmembrane hydrophobic domain, a short cytoplasmic loop, and the second hydrophobic region within the membrane bilayer (transmembrane region), followed by the cytoplasmic end. This structure is unique, since other members of this superfamily either have one transmembrane domain or are attached to the membrane surface by a glycosylphosphatidylinositol anchor [23]. Using recombinant MOG, it was found that the 8-18C5 epitope was in the IgV-like domain, but at that time, all attempts to refine the binding site for the 8-18C5 mAb were unsuccessful. In this connection, it was suggested that MOG undergoes dimerization and its dimeric form partially masks the 8-18C5 epitope, which is conformational [24].
Demyelination in MOGAD develops due to the formation of a ternary complex consisting of the T-cell receptor (TCR), MOG, and human leukocyte antigen (HLA) [25]. T cells identify three epitopes in the MOG extracellular domain (residues 1-22, 35-55, and 92-106) as autoantigens and initiate the process of antibody generation. MOG1-22, MOG35-55, and MOG92-106 have been shown to be encephalitogenic epitopes. It was also found that the FG loop (residues 92-106) acts as an epitope for the interaction with mAb 8-18C5, the residues His103 and Ser104 being essential for the antibody binding [26]. MOG 35-55 has been used as an immunodominant epitope in a large number of animal models. At the same time, human MOG 35-55 showed mild encephalitogenic properties compared to the same fragment of rat MOG. The immunodominant peptide MOG 35-55 is a part of a highly conserved region of the MOG protein (residues 20-50) with a single amino acid polymorphism at position 42 [the only difference between the human and animal (mouse, rat) MOG proteins]. MOG proteins of humans and higher primates have proline in position 42, while rodent proteins have serine, which explains why human MOG epitopes cannot be identified by rodent antibodies [27]. The pathogenic activity of IgG-MOG was analyzed in vivo by transferring affinity-purified antibodies from MOGAD patients to experimental animals. Moreover, selected antibodies recognized different MOG epitopes (CC′ and FG loop) with the cross-reactivity to rodent MOGs. In this case, other antibody conformations were excluded. After intrathecal injection of IgG-MOG from patients into Lewis rats, the animals developed a demyelinating disease. The antibodies, together with related MOG-specific T cells, increased T cell infiltration and induced demyelination associated with the deposition of C9neo antigen (marker of lytic complement complex that is deposited exclusively in areas of active myelin destruction), resembling type II demyelination pattern characteristic of MS [28]. Human MOG is a heterogeneous protein, and IgG-MOGs derived from MOGAD patients recognized different epitopes in animal MOG proteins. The study using single-point MOG mutants identified seven different binding patterns for IgG-MOG in patients with MOGAD. Mutations were most often found in the CC′ loop (residues 41-46) and FG loop [29].
Glycosylation introduces a significant structural diversity and regulates biological activity of proteins. Post-translational modifications of proteins at N-glycosylation sites affect protein folding, localization, and function. The binding of antibodies to an antigen often depends on the position of glycosylated residue [30]. MOG has one N-linked glycosylation site (Asn31). Glycosylation reduces the flexibility of MOG in both unbound and Fab-bound states. The presence of glycan enhances stability of the MOG protein and helps to maintain its almost native folded conformation [31]. In some patients, attachment of glycan chain to MOG created a steric hindrance to antigen recognition [32].
The functions of MOG remain poorly understood. MOG is located on the myelin sheath outer surface and has an extracellular domain, which makes it accessible to potential antibodies. Other components of myelin are difficult to interact with antibodies and T cells because of their location. PLP is also a transmembrane protein, but it is extremely hydrophobic and hidden within dense, multilayered myelin. MBP is attached to the inner surface of the cell membrane and resides predominantly in the cytoplasm. MAG is found in the innermost layer of myelin sheets, which remains in close contact with the axonal membrane [33].
MOG expression begins with the onset of myelination and thus can serve as a differentiation marker of oligodendrocyte maturation [34]. During myelination, there is an increase in the content of MBP, which is responsible for stabilization of microtubules in oligodendrocytes. In turn, excessive stabilization of microtubules leads to the retraction of normal myelin sheath. Addition of the anti-MOG antibody 8-18C5 causes local MBP degradation and results in microtubule depolymerization. It is known that continuous turnover of microtubules is necessary for myelination, and it is likely that the interaction of signals from these two proteins maintains the dynamic balance of microtubules in oligodendrocytes [35].
It was suggested that MOG does not play a direct role in myelination, but rather mediates interactions between myelin and immune system. One of the unique functions of CNS myelin is its ability to directly activate the classical complement pathway. Therefore, CNS myelin should contain a protein capable of binding the C1q complement component and triggering the following cascade of events. Highly purified native MOG and recombinant extracellular IgV-like MOG domain have been shown to bind C1q in a dose-dependent manner [36].
The composition and function of myelin also depend on the adhesion mechanisms mediated by the epitopes of human natural killer-1 (HNK-1), a unique trisaccharide containing sulfated glucuronic acid and predominantly expressed on cells of the nervous system. These molecules, originally identified as adhesion markers, mediate interaction of neurons with glial cells and growth of astrocytic processes. It was shown that some MOG (and MAG) molecules are glycosylated with HNK-1. Therefore, MOG can provide contact between adjacent myelinated fibers [37].
MOG ALTERNATIVE SPLICING
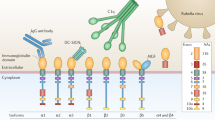
The human MOG gene is located in the major histocompatibility complex (MHC) locus on chromosome 6p21.3-p22 and is a member of the immunoglobulin superfamily. Detailed mapping of the MOG gene has shown that it lies 60 kilobases telomeric to HLA-F [38]. It is hypothesized that this close proximity to the MHC locus mediates MOG involvement in the initiation of autoimmune response. In 2006, the first complete analysis of alternative splicing of the MOG gene in five representative species (from rodents to humans) was performed. It was found that, unlike other myelin proteins, the most complex MOG splicing patterns were unique to higher mammals (humans and primates). The structural and topographic features of MOG splice variants can be important for the phenotypic expression of autoimmunity [39]. The human MOG gene contains 10 exons and demonstrates a complex pattern of alternative splicing resulting in 17 MOG isoforms. At present, there are six most studied MOG isoforms that were classified as alpha (α1, α2, and α3) and beta (β1, β2, β3) based on the exon composition and molecular weight and that differ in the composition of the intracellular (cytoplasmic) C-terminal fragment. All mRNA splicing variants contain exon 1 (signal peptide), exon 2 (Ig-like domain), exon 4 (first transmembrane domain), and small exons 5 and 6 (cytoplasmic region). Exons 5, 6, 7, 9, and 10a/b encode the hydrophilic domains found on the cytoplasmic surface of the lipid bilayer. Exon 8 encodes the membrane-associated intracellular portion of the α1 and β1 transcripts. Exons 10a and 10b encode the C-terminal amino acids that define the α and β isoforms.
Incorporation of exon 3 into two MOG splicing variants causes premature termination of protein translation due to the presence of multiple in-frame stop codons, resulting in two shortest MOG isoforms that have identical amino acid sequences and represent soluble Ig domains (since their transmembrane and cytoplasmic regions are not translated) [40, 41].
It should be noted that compared to the full-length MOG isoforms (α1 and β1), other isoforms are not translated or only poorly translated and therefore, cannot be detected by enzyme-linked immunosorbent assay (ELISA) and immunoblotting. Soluble MOG isoforms can most likely be detected only in the cerebrospinal fluid. Indeed, the results of a recent study suggest that a number of patients with MOGAD synthesize MOG-IgG intrathecally. Interestingly, it is adjusted cerebrospinal fluid/serum MOG-IgG index, but not serum MOG-IgG titer, that indicates a less favorable course of the disease [42].
The studies of alternative splicing of the MOG mRNA during human brain development demonstrated that the main α1 isoform of the MOG mRNA is expressed already at the 21st week of prenatal development. Expression of MOG α1 mRNA was even more pronounced in the brain biopsy of a 2-year-old child, when detection of other isoforms has only started to be possible. These data show that expression of additional MOG isoforms was initiated as the organism developed and positively correlated with the stages of myelination and acquisition of coordinated CNS activity at this age (e.g., speech development) [43]. These changes in the MOG expression are similar to the shift in the expression of the two PLP gene isoforms in the process of human brain development, since the PLP isoform DM20 dominates in the prenatal period, while PLP is more strongly expressed starting from the second year of life, when the amount of PLP is 50%. In this context, the discovery of MOG splicing variants during late development suggests that their role can be specific and indicates myelin maturity. It is also interesting that some isoforms can be re-expressed during remyelination [44].
ANALYSIS OF IgG RESPONSE TO MOG ISOFORMS
Due to the conformational variability of MOG, there are different patterns of its binding with MOG-Ig. Most often, the epitopes are located in the loops between the β-sheets of the extracellular Ig domain of human MOG. In the retrospective study [45], serum samples from 202 patients with positive MOG-IgG serostatus were analyzed for their reactivity with MOG isoforms using live MOG α1 CBA and HEK293 cells transfected with other MOG isoforms. Cell surface expression of the MOG isoforms was demonstrated by performing live CBA with serial dilutions of the humanized mouse mAb 8-18C5. Despite small differences in the transfection efficiency, MOG isoforms showed comparable specific binding to 8-18-C5. Therefore, all extracellular domains of different MOG isoforms were equally available for antibody binding. According to the results of this study, the strongest reactivity of IgG was directed against the longest MOG isoforms α1 (currently used as a standard in the CBA for MOG-IgG) and β1, while the shorter isoforms α2, α3, β2, and β3 were recognized less frequently; however, this does not exclude their pathogenicity in the development of MOGAD. Three different antibody binding patterns have been identified in MOGAD: (i) binding to MOG α1 and β1 isoforms only (α1, β1), (ii) preferential binding of MOG α1 and β1 isoforms with recognition of at least one other MOG α isoform, but not β isoforms (α1-3, β1) and (iii) binding to all MOG isoforms (α1-3, β1-3). MOG-IgG with α, β1 and α1-3, β1 patterns were more common in children, while MOG-IgG with α1-3, β1-3 pattern were found mostly in adults. This observation was consistent with the earlier data showing dominant expression of MOG α1 and β1 isoforms in early human development [46]. No differences or correlations between the identified antibody binding patterns and clinical manifestations of MOGAD were found. Existence of differential MOG isoform-binding patterns can explain some of the conflicting results obtained in CBAs with fixed cells, which might have failed to detect IgG against some alternative MOG isoforms [45].
Both nucleotide and amino acid sequences of MOG are highly conserved among various species, which is also the case for other myelin genes. However, a number of studies have shown that the alternative splicing patterns of the MOG gene are more complex in humans than in mice. It appears that exon 2, which codes for the Ig-like domain, have undergone adaptive shuffling in the process of evolution, since it was also found in some other genes. In particular, MOG gene exon 2 is homologous (46% identity) to the Ig-like domain of the gene for butyrophilin, the main glycoprotein of milk fat globules. Butyrophilin is absent in myelin and its expression is associated exclusively with lactation. Hence, it is likely that the observed homology resulted from exon shuffling, a process by which a common Ig-like domain has become associated with other unrelated functional exons [47]. These evolutionary events provided an opportunity for differential expression of MOG isoforms to serve as an additional source of phenotypic diversity among species. Similar to substitutions in gene nucleotide sequences leading to the synthesis of functionally different proteins, expression of different MOG isoforms might ensure different biological roles of this protein in different species or in the same species in normal and pathological conditions.
MOGAD ENDOPHENOTYPES
The clinical phenotypes MOGAD differ in different age groups. Children under 12 years of age mainly suffer from ADEM with large asymmetric foci of brain damage, which in 10-20% of cases can be accompanied or followed by the acute attack of optic neuritis. In adult patients (older than 12 years old), ADEM-like phenotype is much less common; both the onset of MOGAD and subsequent relapses are dominated by the unilateral or bilateral forms of optic neuritis with or without damage to the spinal cord, brain stem, or combined with atypical demyelination at other sites [48]. The mechanisms underlying the pathogenic effects of human IgG-MOG and their correlation with the age-related disease characteristics remain poorly understood [49]. It is known that the effector functions of antibodies are largely mediated by the IgG fragment crystallizable region (Fc) that binds to the Fcγ receptors (FcγRs) on the surface of natural killer (NK) cells and macrophages. The Fc fragment also binds the C1q component of complement and plays an important role in the initiation of enzymatic cascade resulting in the formation of the membrane attack complex (MAC) and generation of pores in the pathogen’s cell membrane, a terminal event in the complement-dependent cell cytotoxicity [50].
Depending on the effector function of IgG-MOG, two MOGAD endophenotypes can be distinguished. Endophenotype 1 (pro-inflammatory) is characterized by strongly positive titers of MOG-IgG with a high ability to bind FcγR and mediate antibody-dependent activation of NK cells in both children and adults, while endophenotype 2 exhibits lower MOG-IgG titers and lower FcγR-binding capacity. The endophenotypes also differ in the mechanism of IgG-MOG Fc fragment glycosylation. The glycan linked to the heavy chain of the Fc fragment is crucial for maintaining both the pro-inflammatory and anti-inflammatory functions of the IgG [51]. Agalactosylation of the Fc fragment dominates in the MOGAD endophenotype 1, while sialylation of Fc fragment is more common in the endophenotype 2. It is noteworthy that the correlation between the titer, binding to FcγR, and glycosylation profile of the Fc fragment is more pronounced in the acute stage than during the remission of MOGAD and does not depend on age. Therefore, the active phase of MOGAD is characterized by active humoral response with a higher affinity of FcγR binding due to the antibody-mediated activation of NK cells, as well as agalactosylation and asialylation of the IgG-MOG Fc fragment, which enhances the pro-inflammatory activity of antibodies [52].
In children with MOGAD, the ability of IgG-MOG to mediate the NK cell-related cytotoxicity is less pronounced in comparison with adults, due to the prevalence of fucosylation of the IgG Fc fragment, because Fc-fucose prevents interaction between IgG and FcγR, thus reducing the ability of antibodies to activate NK cells [53]. Indeed, these results are consistent with the data on the reduction of the Fc fragment fucosylation over the lifetime [54].
On the other hand, the presence of N-acetylglucosamine at the N-glycosylation site (Asn31) in MOG promotes antibody-dependent cell cytotoxicity after antigen interaction with the antibody [55]. Moreover, glycosylation of the Fab fragment in IgG-MOG positively correlates with age. Therefore, IgG-MOG glycoforms can significantly affect the pro-inflammatory activity of IgG-MOG. Changes in the antibody glycosylation can lead to a decrease in the antibody titer during remission, while persistent seropositive results can be a predictor of the recurrent course of MOGAD [56]. The difference in the IgG-MOG glycosylation in children and adults can, among other things, determine the clinical diversity of MOGAD. This assumption is consistent with the experimental data obtained in mouse models which demonstrated that galactosylation or sialylation changed the IgG structure and increase its affinity for activating FcγR [57].
Other well-known autoimmune diseases, such as rheumatoid arthritis, systemic lupus erythematosus, and nonspecific ulcerative colitis (Crohn’s disease), are also characterized by increase in the content of agalactosylated glycoforms of both serum and antigen-specific IgG, and the level of these glycoforms correlates with the disease clinical manifestation. Although the Fc-linked carbohydrates contain only a small fraction of sialylated structures, it was reported that the content of asialylated glycovariants of antibodies increases in patients with systemic diseases of connective tissue and precedes the relapse of the disease [58]. Therefore, elevated levels of agalactosylated and asialylated IgG can be associated with the clinical activity of MOGAD, which is corroborated by observations made in other autoimmune diseases.
DETECTION OF SERUM MOG-IgG
The first attempts to detect MOG-IgG in the serum of adult patients were performed using ELISA, yielding inconsistent data that showed no association with any form of demyelination. An attempt to search for MOG-IgG in atypical demyelination in adults was caused by the fact that in pediatric patients, MOG-IgG were detected in more than 50% ADEM cases, and the use of ELISA provided sufficiently high specificity and sensitivity of their detection. However, in adult patients, this technique was less promising. Over time, numerous attempts have been made to identify MOG-IgG in adult patients with atypical demyelinating syndromes other than MS. In summary, since 2018, testing for MOG-IgG has been recommended only by CBA in live or fixed cells followed by immunofluorescence microscopy (CBA-IF) or flow cytometry (CBA-FACS). For the purpose of international standardization, Reindl et al. [59] published the study on the reproducibility of various methods for MOG-IgG determination that was conducted at five national testing centers in 2020. Comparative analysis revealed an excellent agreement (96%) between the live CBAs for MOG-IgG for samples previously identified as clearly positive or negative from four different testing centers. The agreement was lower (90%) with fixed CBA-IF due to the loss of some conformational epitopes during fixation (Euroimmun center). Importantly, the majority of negative results in fixed assays were from patients with a typical MOGAD clinical phenotype that showed high titers of MOG-IgG when reassessed with live CBAs. Therefore, it should be taken into account that the use of a commercial (fixed) CBAs can lead to diagnostic errors in 10% cases. ELISA showed no concordance with CBAs for detection of human MOG-IgG in both positive and negative serum. Later, it was found that MOG is a glycoprotein with a complex conformational structure that can form various splice variants, and given that ELISA denatures native tertiary structure of the protein, detection errors can occur in a large percentage of cases. This emphasizes again that ELISA should not be used for MOG-IgG detection in humans [59].
BIOCHEMICAL FEATURES OF MOGAD: CYTOKINES AND NEUROFILAMENTS
Any type of neuroinflammation is accompanied by changes in the cytokine profile. In demyelinating syndromes, changes in the levels of cytokines, chemokines, myelin decay products, glial cells, and axons in the cerebrospinal fluid (CSF) are of particular interest. A comparative study of the cytokine panel, MBP (marker of myelin damage), and glial fibrillary acidic protein (GFAP, marker of astrocyte damage) in MS, NMOSD, and MOGAD revealed a number of differences between these diseases. Thus, elevated concentrations of cytokines secreted by T helper 17 (Th17) cells, such as IL-6, IL-8, and GM-CSF (granulocyte-macrophage colony-stimulating factor), were found in the CSF of patients with MOGAD and active NMOSD. In addition, there was an increased production of IFN-γ (interferon-gamma), TNF-α (tumor necrosis factor-alpha), and IL-10 compared with MS patients. Also, patients with NMOSD and MOGAD exhibited higher concentration of MBP in the CSF. Compared to MOG and MS, the content of GFAP was significantly higher in NMOSD and correlated with the IL-6 and MBP levels. Therefore, cytokines produced by Th17 cells may play an important role in the pathogenesis of both diseases associated with the production of antibodies against MOG and AQP-4. Although the exact mechanisms remain unknown, it can be assumed that naive CD4+ T cells, which play a leading role in the initiation of autoimmune responses, go through a dichotomy path under the influence of IL-6 and differentiate more towards Th17 cells than regulatory T cells providing immunological tolerance. This results in the overproduction of GM-CSF and IL-17A, leading to the recruitment of inflammatory cells, including polymorphonuclear cells (neutrophils), in the CNS. Indeed, routine CSF analysis in MOGAD and NMOSD often reveals moderate pleocytosis, which is rarely seen in MS. IL-6 also stimulates plasmablasts and B cells, this promoting production of autoantibodies. IL-6 increases permeability of the BBB, thus facilitating penetration of pro-inflammatory factors into the CNS [60]. Previously, the role of IL-6 as a pleiotropic cytokine in the pathogenesis of IgG-AQP4-associated syndromes was proven in [61]. Clinical studies have shown that a humanized monoclonal antibody inhibiting the IL-6 signaling pathway significantly reduced the risk of recurrence in AQP4-IgG positive NMOSD patients [62]. Given that the cytokine profiles in MOGAD and NMOSD are very similar, it can be expected that the therapeutic targets may be the same.
Assays for quantification of neurofilament light chains (NfLs), components of the neuronal cytoskeleton that play an important role in axonal growth and stability, are now available. NfLs are biomarkers of axonal damage in various neurological diseases [63]. In particular, NfL serum levels are elevated in patients with MS and correlate with the clinical and radiological activity, severity, and rate of disability progression [64]. In NMSOD, the level of NfLs increases during the periods of disease relapses and correlates with the EDSS (Expanded Disability Status Scale) score [65]. Measuring NfLs in the serum of MOGAD patients revealed that the NfL content reaches its maximum at the disease onset and then decreases with time; even during relapses, the concentration of NfLs increases only slightly. Hence, the axonal damage in MOGAD is mainly due to the first attack of the disease, which once again emphasizes the difference between MOGAD and other diseases, such as MS and NMSOD, and presumably reflects a more favorable prognosis for the patients. At the same time, it indicates an importance of timely and appropriate treatment starting from the very onset of the disease [66].
MECHANISMS OF MYELIN RECOVERY IN DEMYELINATING DISEASES
Regeneration of myelin in the CNS after an episode of demyelination is mainly mediated by oligodendrocyte progenitor cells (OPCs), multipotent parenchymal stem cells. After the myelin damage, OPCs rapidly proliferate in the areas of myelin damage, where they give the rise to new myelinating oligodendrocytes [67]. OPCs are highly proliferative, motile bipolar cells that express large amounts of gangliosides. Specification and differentiation of OPCs are regulated by oligodendrocyte transcription factors 1 and 2 [68]. OPCs make up 5-8% of all CNS cells and are diffusely distributed in the adult brain, with the largest concentration in the subventricular zone, optic nerve, motor cortex, corpus callosum, and cerebellum that serve as reservoirs of new oligodendrocytes [69]. In healthy brain, oligodendrocytes proliferate constantly to maintain their cell density, but much more slowly than during the childhood or upon damage [70]. It is believed that formation of new myelinating oligodendrocytes is an important mechanism of neuroplasticity due to their participation in the regulation of CNS homeostasis by affecting glutamatergic neurotransmission. OPCs are the only glial cells that form synaptic contacts with neurons. Synaptic activity influences OPC proliferation and differentiation into mature oligodendrocytes, which form new myelin. OPCs are activated by the glutamate released by neurons due to the expression of AMPA glutamate receptors. However, the surface density of AMPA receptors significantly reduces after OPC differentiation into mature oligodendrocytes. Therefore, the short-term signaling mediated by the AMPA receptors is important for the induction of OPC differentiation and initiation of active axonal myelination [71]. Newly formed oligodendrocytes also play an important role in the formation and transformation of glial scars [72]. After myelin damage, mature OPCs differentiate into oligodendrocytes capable of axonal remyelination and restoration of nearly normal nerve conduction [73]. Like other regenerative processes, remyelination occurs most efficiently at the early active stages of formation of demyelination lesion characterized by the abundance of T and B lymphocytes and macrophages containing the products of myelin degradation and often containing OPCs that form new myelin sheaths. The density of OPCs in these zones is comparable to their density in the surrounding white matter. In contrast, in chronic inflammation foci, mature myelinating oligodendrocytes are extremely rare, supposedly because of the block of differentiation and arrest of OPC development at the preoligodendrocyte stage [74]. The reasons why OPCs fail to complete differentiation into mature oligodendrocytes might be related to intrinsic changes in the OPC population or specific changes in the lesions during chronic inflammation. For example, in progressive forms of MS, experimental depletion of immune cells in demyelination foci negatively affected remyelination, because the signals from these cells stimulate the activation and recruitment of OPCs and regulate their differentiation [75]. The state of incomplete remyelination and partial gliosis of the demyelination focus can be observed in MS due to the fact that in MS, demyelinating lesions undergo a number of changes over time. The first changes occur when the BBB permeability increases, immune cells rush into the CNS, and the inflammatory infiltrate is distributed perivascularly. Next, myelinating oligodendrocytes begin to enter the site of damage and initiate the restoration of myelin. Due to the decrease in the activation of glial cells and in production of inflammatory mediators, expression of tight junction proteins is restored, whereas decrease in the expression of adhesion molecules on endotheliocytes prevents the penetration of activated lymphocytes into the CNS. As the inflammation subsides and the integrity of the BBB is restored, the remaining immune cells move to the lesion periphery, where they contribute to the slow expansion of the peripheral zone of the focus and gradual increase in its size. During this period, gliosis starts to dominate, and hypertrophied astrocytes, single myelinating oligodendrocytes, areas of myelin decay, and axonopathy can be observed. Accordingly, axons located in such foci almost never remyelinate and a glial scar is formed, which can be visualized by MRI. Such lesions are called black holes, hypointense areas on T1-weighted MRI images, that are observed mainly at the late progressive stages of MS. There is a correlation between the number of black holes, increase in the degree of global brain atrophy, and the EDSS score [76]. However, black holes are rarely formed in NMSOD and MOGAD, due to the absence of chronic smoldering neuroinflammation in these diseases. Clinical manifestations and the following complete or incomplete recovery of neurological functions are associated with the severity of the autoimmune demyelinating process only during the relapse period. In syndromes associated with IgG-APQ4, secondary severe demyelination most often develops due to the BBB structural failure and production of pathogenic IgG-APQ4. In NMSOD, cystic transformation of demyelination foci at the sites of former acute lesions in the spinal cord often takes place, leading to severe neurological symptoms, which, even at the time of the first attack of the disease, can result in significant disability [77]. In MOGAD, formation of black holes and cystic changes in the demyelination areas are extremely rare and only accompany severe ADEM-like phenotypes with very large myelin lesions. In this case, cystic transformation or appearance of a T1-hypointense area can be observed in the center of the focus, where inflammation persists for a long time [78]. In most MOGAD cases, the foci almost completely disappear or significantly decrease in size, which indicates, first of all, consistent remyelination, and apparently, a milder degree of myelin damage in general. Most often, patients experience a complete regression of symptoms after the treatment with glucocorticosteroids. However, despite the recovery and a fairly favorable disease prognosis, MOGAD relapses in more than 50-70% cases (especially in adults). This requires therapy to prevent these relapses, otherwise repeated relapses can lead to extremely negative consequences, such as blindness or movement impairments [79].
CONCLUSION
The discovery of new antibodies has significantly improved our understanding of the pathogenesis of inflammatory demyelinating diseases of the CNS, some of which were identified as new nosological entities. MOGAD is the first demyelinating disease of the CNS in which the antigen has been identified and antibodies produced against MOG have been proven to be pathogenic. The difficulties in identifying MOG-Ig have prompted researchers to gain new knowledge about molecular and biochemical aspects of this disease, which made it possible to investigate the structure of MOG in more detail, partially elucidate the mechanisms of interaction between MOG and antibodies, and show the complexity of alternative splicing of the MOG gene. The data obtained will help in improving the diagnostics of the disease. Although MOGAD has a better prognosis than other demyelinating diseases, it is still relatively common for it to relapse. These are also severe cases of MOGAD, including in children, which require emergency treatment. Therefore, future studies should investigate various aspects of this disease in order to clarify the therapeutic targets and to conduct multicenter randomized trials of drugs that can affect the frequency of MOGAD relapses.
Abbreviations
- ADEM:
-
acute disseminated encephalomyelitis
- AQP4:
-
aquaporin-4
- AQP4-IgG:
-
anti-aquaporin-4 antibody
- BBB:
-
blood–brain barrier
- CBA:
-
cell-based assay
- CSF:
-
cerebrospinal fluid
- ELISA:
-
enzyme-linked immunosorbent assay
- Fc:
-
fragment crystallizable region
- FcγR:
-
natural killer Fc-gamma receptor
- IgG:
-
immunoglobulin G
- IL:
-
interleukin
- mAb:
-
monoclonal antibody
- MBP:
-
myelin basic protein
- MOG:
-
myelin oligodendrocyte glycoprotein
- MOGAD:
-
myelin oligodendrocyte glycoprotein antibody-associated disease
- MOG-IgG:
-
anti-MOG antibody
- MS:
-
multiple sclerosis
- NfL:
-
neurofilament light chain
- NK cell:
-
natural killer cell
- NMOSD:
-
neuromyelitis optica spectrum disorder
- OPC:
-
oligodendrocyte progenitor cell
- PLP:
-
proteolipid protein
References
Emery, B. (2010) Regulation of oligodendrocyte differentiation and myelination, Science, 330, 779-782, https://doi.org/10.1126/science.1190927.
Saikali, P., Cayrol, R., and Vincent, T. (2009) Anti-aquaporin-4 auto-antibodies orchestrate the pathogenesis in neuromyelitis optica, Autoimmun. Rev., 9, 132-135, https://doi.org/10.1016/j.autrev.2009.04.004.
Graus, F., Titulaer, M. J., Balu, R., Benseler, S., Bien, C. G., Cellucci, T., Cortese, I., Dale, R. C., Gelfand, J. M., Geschwind, M., Glaser, C. A., Honnorat, J., Höftberger, R., Iizuka, T., Irani, S. R., Lancaster, E., Leypoldt, F., Prüss, H., Rae-Grant, A., Reindl, M., Rosenfeld, M. R., Rostásy, K., Saiz, A., Venkatesan, A., Vincent, A., Wandinger, K. P., Waters, P., and Dalmau, J. (2016) A clinical approach to diagnosis of autoimmune encephalitis, Lancet Neurol., 15, 391-404, https://doi.org/10.1016/S1474-4422(15)00401-9.
Yamout, B. I., and Alroughani, R. (2018) Multiple sclerosis, Semin. Neurol., 38, 212-225, https://doi.org/10.1055/s-0038-1649502.
Giovannoni, G., Popescu, V., Wuerfel, J., Hellwig, K., Iacobaeus, E., Jensen, M. B., García-Domínguez, J. M., Sousa, L., De Rossi, N., Hupperts, R., Fenu, G., Bodini, B., Kuusisto, H. M., Stankoff, B., Lycke, J., Airas, L., Granziera, C., and Scalfari, A. (2022) Smouldering multiple sclerosis: the “real MS”, Ther. Adv. Neurol. Disord., 25, 17562864211066751, https://doi.org/10.1177/17562864211066751.
McGinley, M. P., Goldschmidt, C. H., and Rae-Grant, A. D. (2021) Diagnosis and treatment of multiple sclerosis: a review, JAMA, 23, 765-779, https://doi.org/10.1001/jama.2020.26858.
Lennon, V. A., Wingerchuk, D. M., Kryzer, T. J., Pittock, S. J., Lucchinetti, C. F., Fujihara, K., Nakashima, I., and Weinshenker, B. G. (2004) A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis, Lancet, 364, 2106-2112, https://doi.org/10.1016/S0140-6736(04)17551-X.
Lennon, V. A., Kryzer, T. J., Pittock, S. J., Verkman, A. S., and Hinson, S. R. (2005) IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel, J. Exp. Med., 202, 473-477, https://doi.org/10.1084/jem.20050304.
Wingerchuk, D. M., Banwell, B., Bennett, J. L., Cabre, P., Carroll, W., Chitnis, T., de Seze, J., Fujihara, K., Greenberg, B., Jacob, A., et al. (2015) International consensus diagnostic criteria for neuromyelitis optica spectrum disorders, Neurology, 85, 177-189, https://doi.org/10.1212/WNL.0000000000001729.
Jarius, S., and Wildemann, B. (2010) AQP4 antibodies in neuromyelitis optica: diagnostic and pathogenetic relevance, Nat. Rev. Neurol., 6, 383-392, https://doi.org/10.1038/nrneurol.2010.72.
Jarius, S., Metz, I., Konig, F. B., Ruprecht, K., Reindl, M., Paul, F., Bruck, W., and Wildemann, B. (2016) Screening for MOG-IgG and 27 other anti-glial and anti-neuronal autoantibodies in “pattern II multiple sclerosis” and brain biopsy findings in a MOG-IgG-positive case, Multiple Sclerosis, 22, 1541-1549, https://doi.org/10.1177/1352458515622986.
Misu, T., Höftberger, R., Fujihara, K., Wimmer, I., Takai, Y., Nishiyama, S., Nakashima, I., Konno, H., Bradl, M., Garzuly, F., Itoyama, Y., Aoki, M., and Lassmann, H. (2013) Presence of six different lesion types suggests diverse mechanisms of tissue injury in neuromyelitis optica, Acta Neuropathol., 125, 815-827, https://doi.org/10.1007/s00401-013-1116-7.
Kaneko, K., Sato, D. K., Nakashima, I., Nishiyama, S., Tanaka, S., Marignier, R., Hyun, J. W., Oliveira, L. M., Reindl, M., Seifert-Held, T., Sepulveda, M., Siritho, S., Waters, P. J., Kurosawa, K., Akaishi, T., Kuroda, H., Misu, T., Prayoonwiwat, N., Berger, T., Saiz, A., and Aoki, M. (2016) Myelin injury without astrocytopathy in neuroinflammatory disorders with MOG antibodies, J. Neurol. Neurosurg. Psychiatry, 87, 1257-1259, https://doi.org/10.1136/jnnp-2015-312676.
Takai, Y., Misu, T., Kaneko, K., Chihara, N., Narikawa, K., Tsuchida, S., Nishida, H., Komori, T., Seki, M., Komatsu, T., Nakamagoe, K., Ikeda, T., Yoshida, M., Takahashi, T., Ono, H., Nishiyama, S., Kuroda, H., Nakashima, I., Suzuki, H., Bradl, M., et al. (2020) Myelin oligodendrocyte glycoprotein antibody-associated disease: an immunopathological study, Brain, 143, 1431-1446, https://doi.org/10.1093/brain/awaa102.
Jarius, S., Paul, F., Aktas, O., Asgari, N., Dale, R. C., de Seze, J., Franciotta, D., Fujihara, K., Jacob, A., Kim, H. J., Kleiter, I., Kümpfel, T., Levy, M., Palace, J., Ruprecht, K., Saiz, A., Trebst, C., Weinshenker, B. G., and Wildemann, B. (2018) MOG encephalomyelitis: international recommendations on diagnosis and antibody testing, J. Neuroinflamm., 15, 134, https://doi.org/10.1186/s12974-018-1144-2.
Banwell, B., Bennett, J. L., Marignier, R., Kim, H. J., Brilot, F., Flanagan, E. P., Ramanathan, S., Waters, P., Tenembaum, S., Graves, J. S., Chitnis, T., Brandt, A. U., Hemingway, C., Neuteboom, R., Pandit, L., Reindl, M., Saiz, A., Sato, D. K., Rostasy, K., Paul, F., and Palace, J. (2023) Diagnosis of myelin oligodendrocyte glycoprotein antibody-associated disease: International MOGAD Panel proposed criteria, Lancet Neurol., 22, 268-282, https://doi.org/10.1016/S1474-4422(22)00431-8.
Baumann, N., and Pham-Dinh, D. (2001) Biology of oligodendrocyte and myelin in the mammalian central nervous system, Physiol. Rev., 81, 871-927, https://doi.org/10.1152/physrev.2001.81.2.871.
Quarles, R. H. (2002) Myelin sheaths: glycoproteins involved in their formation, maintenance and degeneration, Cell. Mol. Life Sci., 59, 1851-1871, https://doi.org/10.1007/pl00012510.
Lebar, R., Boutry, J. M., Vincent, C., Robineaux, R., and Voisin, G. A. (1976) Studies on autoimmune encephalomyelitis in the guinea pig. II. An in vitro investigation on the nature, properties, and specificity of the serum-demyelinating factor, J. Immunol., 116, 1439-1446.
Von Büdingen, H. C., Hauser, S. L., Fuhrmann, A., Nabavi, C. B., Lee, J. I., and Genain, C. P. (2002) Molecular characterization of antibody specificities against myelin/oligodendrocyte glycoprotein in autoimmune demyelination, Proc. Natl. Acad. Sci. USA, 99, 8207-8212, https://doi.org/10.1073/pnas.122092499.
Campagnoni, A. T., and Skoff, R. P. (2001) The pathobiology of myelin mutants reveal novel biological functions of the MBP and PLP genes, Brain Pathol., 11, 74-91, https://doi.org/10.1111/j.1750-3639.2001.tb00383.x.
Slavin, A. J., Johns, T. G., Orian, J. M., and Bernard, C. C. (1997) Regulation of myelin oligodendrocyte glycoprotein in different species throughout development, Dev. Neurosci., 19, 69-78, https://doi.org/10.1159/000111187.
Kroepfl, J. F., Viise, L. R., Charron, A. J., Linington, C., and Gardinier, M. V. (1996) Investigation of myelin/oligodendrocyte glycoprotein membrane topology, J. Neurochem., 67, 2219-2222, https://doi.org/10.1046/j.1471-4159.1996.67052219.x.
Bettadapura, J., Menon, K. K., Moritz, S., Liu, J., and Bernard, C. C. (1998) Expression, purification, and encephalitogenicity of recombinant human myelin oligodendrocyte, glycoprotein, J. Neurochem., 70, 1593-1599, https://doi.org/10.1046/j.1471-4159.1998.70041593.x.
Koukoulitsa, C., Chontzopoulou, E., Kiriakidi, S., Tzakos, A. G., and Mavromoustakos, T. (2020) A journey to the conformational analysis of T-cell epitope peptides involved in multiple sclerosis, Brain Sci., 10, 356, https://doi.org/10.3390/brainsci10060356.
Breithaupt, C., Schäfer, B., Pellkofer, H., Huber, R., Linington, C., and Jacob, U. (2008) Demyelinating myelin oligodendrocyte glycoprotein-specific autoantibody response is focused on one dominant conformational epitope region in rodents, J. Immunol., 181, 1255-1263, https://doi.org/10.4049/jimmunol.181.2.1255.
Bittner, S., Afzali, A. M., Wiendl, H., and Meuth, S. G. (2014) Myelin oligodendrocyte glycoprotein (MOG35-55) induced experimental autoimmune encephalomyelitis (EAE) in C57BL/6 mice, J. Vis. Exp., 86, 51275, https://doi.org/10.3791/51275.
Peschl, P., Schanda, K., Zeka, B., Given, K., Böhm, D., Ruprecht, K., Saiz, A., Lutterotti, A., Rostásy, K., Höftberger, R., Berger, T., Macklin, W., Lassmann, H., Bradl, M., Bennett, J. L., and Reindl, M. (2017) Human antibodies against the myelin oligodendrocyte glycoprotein can cause complement-dependent demyelination, J. Neuroinflamm., 14, 208, https://doi.org/10.1186/s12974-017-0984-5.
Mayer, M. C., Breithaupt, C., Reindl, M., Schanda, K., Rostásy, K., Berger, T., Dale, R. C., Brilot, F., Olsson, T., Jenne, D., Pröbstel, A. K., Dornmair, K., Wekerle, H., Hohlfeld, R., Banwell, B., Bar-Or, A., and Meinl, E. (2013) Distinction and temporal stability of conformational epitopes on myelin oligodendrocyte glycoprotein recognized by patients with different inflammatory central nervous system diseases, J. Immunol., 191, 3594-3604, https://doi.org/10.4049/jimmunol.1301296.
Mayer, M. C., and Meinl, E. (2012) Glycoproteins as targets of autoantibodies in CNS inflammation: MOG and more, Ther. Adv. Neurol. Disord., 5, 147-159, https://doi.org/10.1177/1756285611433772.
Ramya, L. (2020) Role of N-glycan in the structural changes of myelin oligodendrocyte glycoprotein and its complex with an antibody, J. Biomol. Struct. Dynamics, 38, 1649-1658, https://doi.org/10.1080/07391102.2019.1614999.
Marti Fernandez, I., Macrini, C., Krumbholz, M., Hensbergen, P. J., Hipgrave Ederveen, A. L., Winklmeier, S., Vural, A., Kurne, A., Jenne, D., Kamp, F., Gerdes, L. A., Hohlfeld, R., Wuhrer, M., Kümpfel, T., and Meinl, E. (2019) The glycosylation site of myelin oligodendrocyte glycoprotein affects autoantibody recognition in a large proportion of patients, Front. Immunol., 10, 1189, https://doi.org/10.3389/fimmu.2019.01189.
Ambrosius, W., Michalak, S., Kozubski, W., and Kalinowska, A. (2020) Myelin oligodendrocyte glycoprotein antibody-associated disease: current insights into the disease pathophysiology, diagnosis and management, Int. J. Mol. Sci., 22, 100, https://doi.org/10.3390/ijms22010100.
Barkovich, A. J. (2000) Concepts of myelin and myelination in neuroradiology, Am. J. Neuroradiol., 21, 1099-1109.
Dale, R. C., Tantsis, E. M., Merheb, V., Kumaran, R. Y., Sinmaz, N., Pathmanandavel, K., Ramanathan, S., Booth, D. R., Wienholt, L. A., Prelog, K., Clark, D. R., Guillemin, G. J., Lim, C. K., Mathey, E. K., and Brilot, F. (2014) Antibodies to MOG have a demyelination phenotype and affect oligodendrocyte cytoskeleton, Neurol. Neuroimmunol. Neuroinflamm., 1, e12, https://doi.org/10.1212/NXI.0000000000000012.
Johns, T. G., and Bernard, C. C. (1999) The structure and function of myelin oligodendrocyte glycoprotein, J. Neurochem., 72, 1-9, https://doi.org/10.1046/j.1471-4159.1999.0720001.x.
Morise, J., Takematsu, H., and Oka, S. (2017) The role of human natural killer-1 (HNK-1) carbohydrate in neuronal plasticity and disease, Biochim. Biophys. Acta, 1861, 2455-2461, https://doi.org/10.1016/j.bbagen.2017.06.025.
Pham-Dinh, D., Mattei, M. G., Nussbaum, J. L., Roussel, G., Pontarotti, P., Roeckel, N., Mather, I. H., Artzt, K., Lindahl, K. F., and Dautigny, A. (1993) Myelin/oligodendrocyte glycoprotein is a member of a subset of the immunoglobulin superfamily encoded within the major histocompatibility complex, Proc. Natl. Acad. Sci. USA, 90, 7990-7994, https://doi.org/10.1073/pnas.90.17.7990.
Delarasse, C., Della Gaspera, B., Lu, C. W., Lachapelle, F., Gelot, A., Rodriguez, D., Dautigny, A., Genain, C., and Pham-Dinh, D. (2006) Complex alternative splicing of the myelin oligodendrocyte glycoprotein gene is unique to human and non-human primates, J. Neurochem., 98, 1707-1717, https://doi.org/10.1111/j.1471-4159.2006.04053.
Spadaro, M., Winklmeier, S., Beltrán, E., Macrini, C., Höftberger, R., Schuh, E., Thaler, F. S., Gerdes, L. A., Laurent, S., Gerhards, R., Brändle, S., Dornmair, K., Breithaupt, C., Krumbholz, M., Moser, M., Krishnamoorthy, G., Kamp, F., Jenne, D., Hohlfeld, R., Kümpfel, T., and Meinl, E. (2018) Pathogenicity of human antibodies against myelin oligodendrocyte glycoprotein, Ann. Neurol., 84, 315-328, https://doi.org/10.1002/ana.25291.
Reindl, M., and Waters, P. (2019) Myelin oligodendrocyte glycoprotein antibodies in neurological disease, Nat. Rev. Neurol., 15, 89-102, https://doi.org/10.1038/s41582-018-0112-x.
Kwon, Y. N., Kim, B., Kim, J. S., Mo, H., Choi, K., Oh, S. I., Kim, J. E., Nam, T. S., Sohn, E. H., Heo, S. H., Kim, S. B., Park, K. C., Yoon, S. S., Oh, J., Baek, S. H., Kim, B. J., Park, K. S., Sung, J. J., Jung, J. H., Kim, S. J., and Kim, S. M. (2021) Myelin oligodendrocyte glycoprotein-immunoglobulin G in the CSF: clinical implication of testing and association with disability, Neurol. Neuroimmunol. Neuroinflamm., 9, e1095, https://doi.org/10.1212/NXI.0000000000001095.
Pujol, J., Soriano-Mas, C., Ortiz, H., Sebastián-Gallés, N., Losilla, J. M., and Deus, J. (2006) Myelination of language-related areas in the developing brain, Neurology, 66, 339-343, https://doi.org/10.1212/01.wnl.0000201049.66073.8d.
Mathisen, P. M., Kawczak, J. A., Yu, M., Johnson, J. M., and Tuohy, V. K. (2001) Differential DM20 mRNA expression distinguishes two distinct patterns of spontaneous recovery from murine autoimmune encephalomyelitis, J. Neurosci. Res., 64, 542-551, https://doi.org/10.1002/jnr.1106.
Schanda, K., Peschl, P., Lerch, M., Seebacher, B., Mindorf, S., Ritter, N., Probst, M., Hegen, H., Di Pauli, F., Wendel, E. M., Lechner, C., Baumann, M., Mariotto, S., Ferrari, S., Saiz, A., Farrell, M., Leite, M., Irani, S. R., Palace, J., Lutterotti, A., and Reindl, M. (2021) Differential binding of autoantibodies to MOG isoforms in inflammatory demyelinating diseases, Neurol. Neuroimmunol. Neuroinflamm., 8, e1027, https://doi.org/10.1212/NXI.0000000000001027.
Allamargot, C., and Gardinier, M. V. (2007) Alternative isoforms of myelin/oligodendrocyte glycoprotein with variable cytoplasmic domains are expressed in human brain, J. Neurochem., 101, 298-312, https://doi.org/10.1111/j.1471-4159.2006.04296.x.
Eichinger, A., Neumaier, I., and Skerra, A. (2021) The extracellular region of bovine milk butyrophilin exhibits closer structural similarity to human myelin oligodendrocyte glycoprotein than to immunological BTN family receptors, Biol. Chem., 402, 1187-1202, https://doi.org/10.1515/hsz-2021-0122.
Jurynczyk, M., Messina, S., Woodhall, M. R., Raza, N., Everett, R., Roca-Fernandez, A., Tackley, G., Hamid, S., Sheard, A., Reynolds, G., Chandratre, S., Hemingway, C., Jacob, A., Vincent, A., Leite, M. I., Waters, P., and Palace, J. (2017) Clinical presentation and prognosis in MOG-antibody disease: a UK study, Brain, 140, 3128-3138, https://doi.org/10.1093/brain/awx276.
Marignier, R., Hacohen, Y., Cobo-Calvo, A., Pröbstel, A. K., Aktas, O., Alexopoulos, H., Amato, M. P., Asgari, N., Banwell, B., Bennett, J., Brilot, F., Capobianco, M., Chitnis, T., Ciccarelli, O., Deiva, K., De Sèze, J., Fujihara, K., Jacob, A., Kim, H. J., Kleiter, I., et al. (2021) Myelin-oligodendrocyte glycoprotein antibody-associated disease, Lancet Neurol., 20, 762-772, https://doi.org/10.1016/S1474-4422(21)00218-0.
Bournazos, S., Wang, T. T., Dahan, R., Maamary, J., and Ravetch, J. V. (2017) Signaling by antibodies: recent progress, Ann. Rev. Immunol., 35, 285-311, https://doi.org/10.1146/annurev-immunol-051116-052433.
Wang, T. T., and Ravetch, J. V. (2019) Functional diversification of IgGs through Fc glycosylation, J. Clin. Invest., 129, 3492-3498, https://doi.org/10.1172/JCI130029.
Spatola, M., Chuquisana, O., Jung, W., Lopez, J. A., Wendel, E. M., Ramanathan, S., Keller, C. W., Hahn, T., Meinl, E., Reindl, M., Dale, R. C., Wiendl, H., Lauffenburger, D. A., Rostásy, K., Brilot, F., Alter, G., and Lünemann, J. D. (2023) Humoral signatures of MOG-antibody-associated disease track with age and disease activity, Cell Rep. Med., 4, 100913, https://doi.org/10.1016/j.xcrm.2022.100913.
Shields, R. L., Lai, J., Keck, R., O’Connell, L. Y., Hong, K., Meng, Y. G., Weikert, S. H., and Presta, L. G. (2002) Lack of fucose on human IgG1 N-linked oligosaccharide improves binding to human Fcgamma RIII and antibody-dependent cellular toxicity, J. Biol. Chem., 277, 26733-26740, https://doi.org/10.1074/jbc.M202069200.
Gudelj, I., Lauc, G., and Pezer, M. (2018) Immunoglobulin G glycosylation in aging and diseases, Cell. Immunol., 333, 65-79, https://doi.org/10.1016/j.cellimm.2018.07.009.
Mony, J. T., Khorooshi, R., and Owens, T. (2014) MOG extracellular domain (p1-125) triggers elevated frequency of CXCR3+ CD4+ Th1 cells in the CNS of mice and induces greater incidence of severe EAE, Multiple Sclerosis, 20, 1312-1321, https://doi.org/10.1177/1352458514524086.
Oliveira, L. M., Apóstolos-Pereira, S. L., Pitombeira, M. S., Bruel Torretta, P. H., Callegaro, D., and Sato, D. K. (2019) Persistent MOG-IgG positivity is a predictor of recurrence in MOG-IgG-associated optic neuritis, encephalitis and myelitis, Multiple Sclerosis, 25, 1907-1914, https://doi.org/10.1177/1352458518811597.
Quast, I., Keller, C. W., Maurer, M. A., Giddens, J. P., Tackenberg, B., Wang, L. X., Münz, C., Nimmerjahn, F., Dalakas, M. C., and Lünemann, J. D. (2015) Sialylation of IgG Fc domain impairs complement-dependent cytotoxicity, J. Clin. Invest., 125, 4160-4170, https://doi.org/10.1172/JCI82695.
Seeling, M., Brückner, C., and Nimmerjahn, F. (2017) Differential antibody glycosylation in autoimmunity: sweet biomarker or modulator of disease activity?, Nat. Rev. Rheumatol., 13, 621-630, https://doi.org/10.1038/nrrheum.2017.146.
Reindl, M., Schanda, K., Woodhall, M., Tea, F., Ramanathan, S., Sagen, J., Fryer, J. P., Mills, J., Teegen, B., Mindorf, S., Ritter, N., Krummrei, U., Stöcker, W., Eggert, J., Flanagan, E. P., Ramberger, M., Hegen, H., Rostasy, K., Berger, T., Leite, M. I., and Waters, P. (2020) International multicenter examination of MOG antibody assays, Neurol. Neuroimmunol. Neuroinflamm., 7, e674, https://doi.org/10.1212/NXI.0000000000000674.
Kaneko, K., Sato, D. K., Nakashima, I., Ogawa, R., Akaishi, T., Takai, Y., Nishiyama, S., Takahashi, T., Misu, T., Kuroda, H., Tanaka, S., Nomura, K., Hashimoto, Y., Callegaro, D., Steinman, L., Fujihara, K., and Aoki, M. (2018) CSF cytokine profile in MOG-IgG+ neurological disease is similar to AQP4-IgG+ NMOSD but distinct from MS: a cross-sectional study and potential therapeutic implications, J. Neurol. Neurosurg. Psychiatry, 89, 927-936, https://doi.org/10.1136/jnnp-2018-317969.
Fujihara, K., Bennett, J. L., de Seze, J., Haramura, M., Kleiter, I., Weinshenker, B. G., Kang, D., Mughal, T., and Yamamura, T. (2020) Interleukin-6 in neuromyelitis optica spectrum disorder pathophysiology, Neurol. Neuroimmunol. Neuroinflamm., 7, e841, https://doi.org/10.1212/NXI.0000000000000841.
Traboulsee, A., Greenberg, B. M., Bennett, J. L., Szczechowski, L., Fox, E., Shkrobot, S., Yamamura, T., Terada, Y., Kawata, Y., Wright, P., Gianella-Borradori, A., Garren, H., and Weinshenker, B. G. (2020) Safety and efficacy of satralizumab monotherapy in neuromyelitis optica spectrum disorder: a randomised, double-blind, multicentre, placebo-controlled phase 3 trial, Lancet Neurol., 19, 402-412, https://doi.org/10.1016/S1474-4422(20)30078-8.
Khalil, M., Teunissen, C. E., Otto, M., Piehl, F., Sormani, M. P., Gattringer, T., Barro, C., Kappos, L., Comabella, M., Fazekas, F., Petzold, A., Blennow, K., Zetterberg, H., and Kuhle, J. (2018) Neurofilaments as biomarkers in neurological disorders, Nat. Rev. Neurol., 14, 577-589, https://doi.org/10.1038/s41582-018-0058-z.
Bozzetti, S., Ferrari, S., Gajofatto, A., and Mariotto, S. (2021) Neurofilament light chain in demyelinating conditions of the central nervous system: a promising biomarker, Neuroimmunol. Neuroinflamm., 8, 1-13, https://doi.org/10.20517/2347-8659.2020.26.
Watanabe, M., Nakamura, Y., Michalak, Z., Isobe, N., Barro, C., Leppert, D., Matsushita, T., Hayashi, F., Yamasaki, R., Kuhle, J., and Kira, J. I. (2019) Serum GFAP and neurofilament light as biomarkers of disease activity and disability in NMOSD, Neurology, 93, e1299-e1311, https://doi.org/10.1212/WNL.0000000000008160.
Mariotto, S., Gastaldi, M., Grazian, L., Mancinelli, C., Capra, R., Marignier, R., Alberti, D., Zanzoni, S., Schanda, K., Franciotta, D., Calabria, F., Monaco, S., Reindl, M., Ferrari, S., and Gajofatto, A. (2021) NfL levels predominantly increase at disease onset in MOG-Abs-associated disorders, Multiple Scleros. Related Disord., 50, 102833, https://doi.org/10.1016/j.msard.2021.102833.
Franklin, R. J., and Goldman, S. A. (2015) Glia disease and repair-remyelination, Cold Spring Harb. Perspect. Biol., 7, a020594, https://doi.org/10.1101/cshperspect.a020594.
Mitew, S., Hay, C. M., Peckham, H., Xiao, J., Koenning, M., Emery, B. (2014) Mechanisms regulating the development of oligodendrocytes and central nervous system myelin, Neuroscience, 276, 29-47, https://doi.org/10.1016/j.neuroscience.2013.11.029.
Clarke, L. E., Young, K. M., Hamilton, N. B., Li, H., Richardson, W. D., and Attwell, D. (2012) Properties and fate of oligodendrocyte progenitor cells in the corpus callosum, motor cortex, and piriform cortex of the mouse, J. Neurosci., 32, 8173-8185, https://doi.org/10.1523/jneurosci.0928-12.2012.
McTigue, D. M., Wei, P., and Stokes, B. T. (2001) Proliferation of NG2-positive cells and altered oligodendrocyte numbers in the contused rat spinal cord, J. Neurosci., 21, 3392-3400, https://doi.org/10.1523/JNEUROSCI.21-10-03392.2001.
Birey, F., Kloc, M., Chavali, M., Hussein, I., Wilson, M., Christoffel, D. J., Chen, T., Frohman, M. A., Robinson, J. K., Russo, S. J., Maffei, A., and Aguirre, A. (2015) Genetic and stress-induced loss of NG2 glia triggers emergence of depressive-like behaviors through reduced secretion of FGF2, Neuron, 88, 941-956, https://doi.org/10.1016/j.neuron.2015.10.046.
Hughes, E. G., Kang, S. H., Fukaya, M., and Bergles, D. E. (2013) Oligodendrocyte progenitors balance growth with self-repulsion to achieve homeostasis in the adult brain, Nat. Neurosci., 16, 668-676, https://doi.org/10.1038/nn.3390.
Zawadzka, M., Rivers, L. E., Fancy, S. P., Zhao, C., Tripathi, R., Jamen, F., Young, K., Goncharevich, A., Pohl, H., Rizzi, M., Rowitch, D. H., Kessaris, N., Suter, U., Richardson, W. D., and Franklin, R. J. (2010) CNS-resident glial progenitor/stem cells produce Schwann cells as well as oligodendrocytes during repair of CNS demyelination, Cell Stem Cell, 6, 578-590, https://doi.org/10.1016/j.stem.2010.04.002.
Kuhlmann, T., Miron, V., Cui, Q., Wegner, C., Antel, J., and Brück, W. (2008) Differentiation block of oligodendroglial progenitor cells as a cause for remyelination failure in chronic multiple sclerosis, Brain, 131, 1749-1758, https://doi.org/10.1093/brain/awn096.
Foote, A. K., and Blakemore, W. F. (2005) Inflammation stimulates remyelination in areas of chronic demyelination, Brain, 128, 528-539, https://doi.org/10.1093/brain/awh417.
Kuhlmann, T., Ludwin, S., Prat, A., Antel, J., Brück, W., and Lassmann, H. (2017) An updated histological classification system for multiple sclerosis lesions, Acta Neuropathol., 133, 13-24, https://doi.org/10.1007/s00401-016-1653-y.
Kawachi, I., and Lassmann, H. (2017) Neurodegeneration in multiple sclerosis and neuromyelitis optica, J. Neurol. Neurosurg. Psychiatry, 88, 137-145, https://doi.org/10.1136/jnnp-2016-313300.
Denève, M., Biotti, D., Patsoura, S., Ferrier, M., Meluchova, Z., Mahieu, L., Heran, F., Vignal, C., Deschamps, R., Gout, O., Champfleur, N. M., Ayrignac, X., Dallière, C. C., Labauge, P., Dulau, C., Tourdias, T., Dumas, H., Cognard, C., Brassat, D., and Bonneville, F. (2019) MRI features of demyelinating disease associated with anti-MOG antibodies in adults, J. Neuroradiol., 46, 312-318, https://doi.org/10.1016/j.neurad.2019.06.001.
Ramanathan, S., Mohammad, S., Tantsis, E., Nguyen, T. K., Merheb, V., Fung, V. S. C., White, O. B., Broadley, S., Lechner-Scott, J., Vucic, S., Henderson, A. P. D., Barnett, M. H., Reddel, S. W., Brilot, F., Dale, R. C., and Australasian and New Zealand MOG Study Group (2018) Clinical course, therapeutic responses and outcomes in relapsing MOG antibody-associated demyelination, J. Neurol. Neurosurg. Psychiatry, 89, 127-137, https://doi.org/10.1136/jnnp-2017-316880.
Author information
Authors and Affiliations
Contributions
Eliseeva Daria – conceptualization, writing – original draft; Zakharova Maria – conceptualization, writing – review and editing.
Corresponding author
Ethics declarations
The authors declare no conflict of interest in financial or any other sphere. This article does not contain any studies with human participants or animals performed by any of the authors.
Rights and permissions
Open access. This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit https://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Eliseeva, D.D., Zakharova, M.N. Myelin Oligodendrocyte Glycoprotein as an Autoantigen in Inflammatory Demyelinating Diseases of the Central Nervous System. Biochemistry Moscow 88, 551–563 (2023). https://doi.org/10.1134/S0006297923040107
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0006297923040107