
Abstract
Year 2022 marks 25 years since the first mutation in familial autosomal dominant Parkinson’s disease was identified. Over the years, our understanding of the role of genetic factors in the pathogenesis of familial and idiopathic forms of Parkinson’s disease has expanded significantly – a number of genes for the familial form of the disease have been identified, and DNA markers for an increased risk of developing its sporadic form have been found. But, despite all the success achieved, we are far from an accurate assessment of the contribution of genetic and, even more so, epigenetic factors to the disease development. The review summarizes the information accumulated to date on the genetic architecture of Parkinson’s disease and formulates issues that need to be addressed, which are primarily related to the assessment of epigenetic factors in the disease pathogenesis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION. PARKINSON’S DISEASE: CLINICAL PICTURE AND PATHOGENESIS
Parkinson’s disease (PD) is a steadily progressive neurodegenerative disease characterized by a tetrad of motor symptoms, such as resting tremors (primarily of hands), muscle rigidity, bradykinesia, and postural instability. In terms of frequency of occurrence among neurodegenerative diseases, PD is second only to Alzheimer’s disease, and at the age over 80 years, the symptoms of parkinsonism are observed in at least 2% of the population worldwide [1]. It is customary to distinguish between familial and sporadic (idiopathic) forms of PD, even that these forms are virtually indistinguishable phenotypically. Familial forms account for 5 to 10% of all PD cases, although this number might be underestimated. Taking into account some features of inheritance; the proportion of familial PD forms can reach 20%.
The development of motor disorders in PD is due to the selective death of dopaminergic neurons in the substantia nigra pars compacta and a significant decrease in the level of dopamine in the striatum, leading to the disruption of normal functioning of brain basal ganglia and impaired control of motor behavior. In most cases, neuronal death is preceded by the appearance of the so-called Lewy neurites and Lewy bodies – cytoplasmic inclusions consisting mostly of fibrillar alpha-synuclein, ubiquitin, myelin-associated tau protein, and some other proteins. It should be noted that Lewy bodies and Lewy neurites are also found in other brain structures (dorsal motor nucleus, thalamus, amygdala, raphe nucleus, olfactory bulbs, cerebral cortex), and in this regard, PD should not be considered as a disease limited to only one type of neurons and several brain structures [2, 3]. Lewy body-type inclusions have been also found in the peripheral nervous system, e.g., in the neurons of the intestinal submucosa. It is likely that these inclusions can form at the early stage of the pathological process in various parts of the nervous system and gradually spread to other regions of the brain, thus underlying the steady progression of the disease. Notably, at its initial stages, this process does not affect the dopaminergic system of the substantia nigra and striatum or impair motor behavior. The prodromal stage of the disease is associated with the sleep disturbances, olfactory impairments, and dysfunction of the intestine and genitourinary system; however, these changes are not specific and cannot serve as criteria for diagnosing PD. Motor disorders are observed when a significant part (at least 50%) of dopaminergic neurons in the substantia nigra die and dopamine levels in the striatum decrease by 70-80% [4]. The main approaches in the PD therapy, especially those using the dopamine precursor levodopa, are aimed to compensate for the decrease in the dopamine level in the striatum. At the early stages of treatment, dopamine agonists and inhibitors of monoamine oxidase B and catechol-O-methyltransferase can be used. At the later stages, the side effects of levodopa are blocked by certain drugs, such as amantadine (dyskinesia) or apomorphine (development of obsessive-compulsive disorders). In any case, the therapy is symptomatic and does not address the causes of neurodegeneration [5].
MONOGENIC FORMS OF PARKINSON’S DISEASE
The exact genetic nature of the familial form of PD was first demonstrated in 1997, when the p.Ala30Thr missense mutation in the alpha-synuclein gene (SNCA) was detected in a large German family with the autosomal dominant inheritance of PD in four generations, as well as in three unrelated Greek families, in which the disease was observed in two and three generations [6]. Later, several other pathogenetically significant missense mutations in SNCA have been identified (p.Ala30Pro, p.Glu46Lys, p.Gly51Asp, p.Ala53Glu, p.Ala53Thr). It was shown that mutations resulting in the gene dosage change (duplications, triplications, quadruplications), but causing no disturbances in the protein structure can also lead to the PD development [7]. The frequency of mutations in SNCA is low: approximately 0.2% and 1-2% in patients with sporadic and familial forms of the disease, respectively. These mutations serve as a link between the disease forms and indicate a fundamental importance of structural and functional disorders of alpha-synuclein in the pathogenesis of PD [8].
The number of identified genes involved in the familial form of PD has steadily increased, and to date, more than 10 genes have been found, mutations in which unequivocally result in Mendelian PD. Table 1 provides a brief description of genes whose pathogenetic role has been proven and repeatedly confirmed in various studies. However, the discussion about the role of these and other genes in the etiopathogenesis of PD continues.
One of the most striking examples of such discussion is the reassessment of the role of the ubiquitin carboxy-terminal hydrolase UCHL1 (PARK5) gene (not included in Table 1) in the PD pathogenesis. Mutations in this gene were first identified in 1998, when the cosegregating missense mutation p.Ile193Met was detected in a German family with the late-onset PD [9]. However, since then, no familial PD cases associated with a mutation in the UCHL1 gene have been identified [10, 11].
At the same time, a number of polymorphic variants of the UCHL1 gene have been identified, one of which (missense mutation p.Ser18Tyr) has been extensively studied in patients with sporadic PD. Although the results of these association studies were contradictory, a meta-analysis of data from more than 6500 patients using the dominant, recessive, and additive models revealed no association of this polymorphism with the PD development [12]. It should be emphasized that this meta-analysis was first carried out in Caucasian patients and then in a Japanese population [13]. Meta-analysis performed in [14] also excluded the influence of the p.Ser18Tyr polymorphism on the risk of PD development in Asian subjects [14] both in the entire analyzed sample and in ethnically selected subsets.
Analysis of transgenic mice with mutations in the ubiquitin carboxy-terminal hydrolase protein gene yielded conflicting results. Thus, mice with an intragenic deletion in the UCHL1 gene homologue demonstrated a decrease in the level of monoubiquitin and formation of protein inclusions in vivo, but no signs of neurodegeneration in the substantia nigra [15]. At the same time, transgenic mice with the mutant UCHL1 p.193Met gene displayed neurodegeneration in the substantia nigra and impaired spontaneous motor activity at the age of 20 months [16], which was accompanied by the formation of protein inclusions and impaired alpha-synuclein metabolism [17]. In the case of p.Ser18Tyr polymorphism, the 18Tyr variant (unlike the wild-type protein) displayed the antioxidant activity both in vitro in neuronal cell cultures and in vivo when the transgene was introduced into the substantia nigra [18, 19], which reduced the risk of neurodegeneration in carriers of the 18Tyr variant.
Summarizing the above, genetic and molecular biology data fail to provide a clear answer to the question about the role of the UCHL1 gene in the pathogenesis of PD, but so far, its involvement in the disease development seems unlikely.
The second gene not included in Table 1 is the GBA gene coding for glucocerebrosidase (lysosomal hydrolase). Mutations in this gene were first described in patients with Gaucher disease, a systemic disease with impaired hematopoiesis, increased risk of fractures, and pronounced neurological disturbances. Currently, more than 300 pathogenetically significant mutations in GBA have been described. There is no doubt that heterozygous, homozygous, or compound heterozygous mutations in this gene play an important role in the PD development [20-23].
However, even in the patients homozygous for mutations in GBA, the symptoms of parkinsonism develop only in some of mutation carriers regardless of the presence of the clinical phenotype of Gaucher disease. Thus, only about 9% individuals homozygotes for the frequent and relatively mild Asn370Ser mutation (leading to type 1 Gaucher disease without pronounced neurological disturbances) developed parkinsonism [20]. About 40% of Gaucher disease patients displaying parkinsonism had this mutation in the compound heterozygous state; therefore, it was detected in 49% of patients with this phenotype.
Heterozygous carriers of mutations in the GBA gene also demonstrated an increased risk of developing PD – approximately 10% of them developed the disease, and the penetrance of these mutations was estimated as 30% at the age of 80 years [23, 24]. It is most likely that all Gaucher disease-causing mutations increase the risk of developing PD, and the likelihood of developing parkinsonism correlates with the severity of the Gaucher disease clinical picture observed in the carriers of these mutations. To date, more than 130 pathogenetically significant mutations in the GBA gene have been identified in patients with PD, with different mutations dominating in different ethnic groups. Thus, in Ashkenazi Jews, PD is mainly associated with the p.Arg496His, p.Asn370Ser, and 84insGC mutations, while in Caucasians it is associated with the p.Asn370Ser, p.Leu444Pro, p.Arg120Trp, IVS2 + 1G>A, p.His255Gln, p.Asp409His, p.Glu326Lys, and p.Thr369Met mutations. The frequency of GBA mutant variants also differs in different populations, which may affect the assessment of the cumulative risk of developing GBA-associated PD in different ethnic groups. In general, mutations in the GBA gene increase the risk of developing PD, although their penetrance is significantly below 100%.
Mutations in the gene for leucine-rich repeat kinase 2 (LRRK2, or dardarin) also have a reduced penetrance. The LRRK2 gene has been historically regarded as a gene for the autosomal dominant PD form with a penetrance close to 100%. However, out of more than 100 mutations described in this gene, only six (p.Gly2019Ser, p.Arg1441Cys/Gly/His, Tyr1699Cys, Ile2020Thr) have a clear familial cosegregation with the disease. However, even in this case, the mutation penetrance does not reach 100% [25]. Thus, the penetrance of the p.Gly2019Ser mutation is 85% at the age of 80 years [26], and this value differs in different ethnic groups. The penetrance for mutations in codon 1441 is even lower [27] and differs for different missense variants of this mutation. Although it was impossible to accurately estimate the penetrance of the remaining four mutations due to their low frequency, but it is obviously incomplete.
Therefore, the presence of mutations with pathogenetically proven significance in the LRRK2 gene indicates an increased risk of developing PD; however, an accurate assessment of this risk based only on the fact of mutation presence is impossible. This makes it difficult to assess the contribution of mutations in LRRK2 to the risk of developing familial forms of PD, and the estimates available in the literature (according to which mutations in the LRRK2 gene are found in 5% of patients with familial forms of PD and in 1% of patients with idiopathic PD) should be considered as approximate [28].
The situation with the parkin gene (PRKN) is extremely interesting. Homozygous or compound heterozygous diallelic loss-of-function mutations in this gene lead to the development of the autosomal recessive juvenile form of PD, which is characterized by a median age of clinical onset of 31 years. In 16% of patients, the first signs of the disease are observed before the age of 20 years [29]. It is believed that from 10 to 20% of patients with an onset age below 40 years carry the diallelic mutations in the PRKN gene, although the data for various populations are quite different [30-32]. The vast majority of these mutations are large deletions/duplications resulting from unequal homologous crossing over. They include several exons of the PRKN gene and lead to a reading frame shift with the formation of a non-functional variant of the parkin protein. However, such mutations in the heterozygous state were found in patients with sporadic PD [33-35]. Deletions/duplications the PRKN gene were also detected in about 5% of healthy individuals examined in the population studies [36], i.e., their frequency exceeds the population frequency of mutations in the dardarin gene. In this regard, they can be considered as autosomal dominant, leading to the form of PD with the classic onset age of over 55-60 years, or as risk factors for the disease development (by analogy with mutations in the GBA gene). A potential pathogenetic role of these mutations in the heterozygous state was confirmed by the functional activity analysis of the brain nigrostriatal system using fluorodopa [37]. Heterozygous mutation carriers demonstrated a decrease in the dopamine level in the caudate nucleus and putamen, which, however, was less pronounced than in patients with idiopathic PD. This decrease in the dopamine level caused a compensatory activation of the right dorsal premotor cortex area and rostral supplementary motor area [37, 38], which slowed down the development of motor disturbances in heterozygous carriers of parkin gene mutations on the background of impaired dopamine metabolism. A similar picture was observed in heterozygous carriers of mutations in the PINK1 gene, but in both cases, no prospective studies to assess the development of PD in the carriers of heterozygous mutations have been conducted yet [39]. On the other hand, no statistically significant differences were found in the frequency of heterozygous deletions of the parkin gene in the CNV (DNA copy number variation) analysis of large (more than 2000 people) cohorts of PD patients and healthy individuals [40].
In general, it is believed that monogenic forms of PD are observed in 5-10% of patients [41]. The main contribution to the development of autosomal dominant and autosomal recessive monogenic PD forms is made by the LRRK2 and PRKN genes, respectively. It should be emphasized that very few studies have been published on a large-scale analysis of multiple genes. Thus, a meta-analysis covering all major PD genes in Brazilian patients was conducted in [42] revealing that the most frequent point mutations were in the LRRK2 gene (2.5% of patients, with the p.Gly2019Ser mutation being found in 2.2% of cases). Mutations in the parkin gene were found in 8.3% of patients [42]. In Ireland, the analysis was limited to mutations in the PRKN, DJ1, and PINK1 genes and patients with the early onset PD, and only mutations in the PRKN gene were detected in 6.9% of the examined patients [31]. The same three genes were analyzed in patients with the early onset PD from the Central Europe (mainly, Poland); mutations in the PRKN gene were found in 3.1% of the patients [30]. Often, mutational screening is limited only to the p.Gly2019Ser mutation, which has been studied in a large number of world populations. In most of them, its frequency in both familial and sporadic PD did not exceed 3-5% [43].
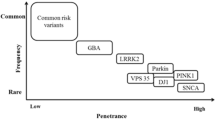
These data indicate a low frequency of pathogenetically significant mutations in major genes for the familial PD forms, pronounced interpopulation heterogeneity, and the need to continue the analysis of mutation spectrum in PD with inclusion of a wider panel of genes, as well to search for new genes for the monogenic PD forms [44, 45]. Some authors offered their own variants of a gene panel for familial PD, but these panels are quite different (figure). As a result, currently available information is insufficient for the implementation of algorithms for genetic testing for the risk of developing PD. A number of commercial panels have been developed, which differ in the number and repertoire of genes; however, according to experts. none of them is sufficiently informative without including additional data describing the family history and phenotypic characteristics of the tested individual. Meanwhile, the minimum panel of five genes common to all panels (PRKN, LRRK2, SNCA, PINK1, PARK7) does not differ in its efficiency from the maximum panel of 43 genes [46].

Lack of full consensus on the genes included in the panels for testing for the monogenic forms of PD. Blue boxes, genes for the monogenic PD forms proposed by different authors [7, 41, 47, 48]; yellow box, genes included in each panel are shown in red; genes included in at least two panels are shown in green
GENOME-WIDE ASSOCIATION STUDIES IN IDIOPATHIC PARKINSON’S DISEASE
An obvious importance of genetic factors in the PD pathogenesis, as well as existence of familial and idiopathic forms of PD with a clear predominance of the latter, have become the basis for genome-wide association studies (GWASs) of the idiopathic forms of the disease. The first studies that were carried out in 2005-2006 on relatively small cohorts of patients using a limited set of DNA markers, have failed to produce reliable results [49, 50]. However, they became the first steps toward developing the GWAS technologies for studying neurodegenerative diseases. The first meta-analysis of the GWAS data became a trial run that used the results of two previously mentioned works [51]. It revealed three single-nucleotide DNA markers associated with the PD development, regardless of the meta-analysis strategy used, but none of these markers reached the level of genome-wide significance (p < 5×10–8). At the same time, it became obvious that the informative value of the analysis would increase with a significant increase in the size of the analyzed populations (from hundreds subjects in the GWASs in 2005-2006 to tens of thousands in later studies) and increase in the density of analyzed DNA markers.
The overall technological development of the GWAS methodology (increase in the information content of microarrays, improvement of statistical analysis methods) has made possible the genotyping of tens of thousands of PD patients and allowed to conduct the associative analysis for both the disease itself and its endophenotypes (age of clinical onset, nature of motor disturbances, presence of concomitant psychoemotional disorders, rate of disease progression). According to the GWAS Catalog (https://www.ebi.ac.uk/), more than 70 association studies have been carried out to date, that reveled the association of PD with more than 500 single-nucleotide DNA markers with the significance level exceeding 10–6 (https://www.ebi.ac.uk/gwas/efotraits/MONDO_0005180). However, only a few genes had several DNA markers each. Taking into account the number of markers associated with the disease and the significance of this association, there are about six protein-coding genes that determine the multifactorial risk of developing PD and/or affect the clinical course of the disease (Table 2).
Two of these genes, SNCA and LRRK2, determine the development of monogenic forms of PD; they also were found to be most strongly associated with the idiopathic form of the disease in terms of the number of associated DNA markers and the relative risk they pose. Mutations in the gene for the microtubule-associated tau protein (MAPT) are also linked to the development of monogenic neurodegenerative diseases, but the clinical manifestations of these mutations are associated primarily with the frontotemporal dementia and essential tremor. Muscle disorders characteristic of PD are secondary to the symptoms of dementia/tremor, and the associations revealed by GWASs reflect the genetic proximity of a wide range of neurodegenerative diseases [52-54].
The role of the other three genes associated with the PD development is studied insufficiently, but the data on their functional activity suggest their association them with PD etiopathogenesis.
The TMEM175 gene encodes a transmembrane lysosomal protein, an ion channel for potassium ions and protons [55]. Modeling of this protein deficiency in a neuronal cell culture in vitro showed that a decrease in the activity of TMEM175 led to the impaired control of lysosomal pH, which, in turn, reduced the activity of lysosomal enzymes (including GBA). When monomeric alpha-synuclein is present, TMEM175 deficiency promoted α-synuclein phosphorylation and accompanied by formation of insoluble inclusions containing hyperphosphorylated alpha-synuclein in the presence of exogenously provided preformed α-synuclein fibrils [56]. Two coding variants, TMEM175 p.M393T and p.Q65P, were associated with PD [57]. Deficiency of TMEM175 in mice in TMEM175 caused the loss of dopaminergic neurons and impairment of motor function [58].The LAMP3 gene associated with the development of PD, encodes a lysosomal membrane-associated protein and affects the processes of lysosomal autophagy and apoptosis. It may also be connected to the lysosomal dysfunction, but its role in the functioning of neuronal cells has been studied very poorly [59-61]. The role in the PD pathogenesis of the BST1/CD157 protein, a glycoprotein from the ADP-ribosyl cyclase superfamily associated primarily with autoimmune, hematological diseases, and tumors, has been poorly studied as well. However, recent data indicate an important role of this protein in the regulation of oxytocin metabolism and development of behavioral disorders [62-64].
Most of DNA markers associated with the PD development have little effect on the risk of developing the disease – for the majority of markers, the odds ratio (OR) is increased or decreased by no more than 1.5 times. The meta-analysis conducted in 2019 [65] of all GWAS data published to date [7.8 million polymorphic DNA markers (including imputing), 1.4 million control DNA samples, 37,700 patients with PD, 18,600 first-degree relatives of patients with PD] identified a total of 90 independent DNA markers belonging to 78 genomic regions associated with the disease development. Thirty-eight of these markers have not been previously described, and were identified due to large sample size, which made it possible for weakly associated markers to achieve the level of genome-wide significance (with a reliability of about 10–8). For these new DNA markers, the polygenic risk score (PRS) for developing PD associated with genome polymorphism was found to be less than 36% (if the estimated incidence in a population of people of 80 years and older is 2%). At the same time, it must be remembered that at present, there is no generally accepted methodology for assessing multilocus risk scores; perhaps, this assessment will change significantly when standards for calculating PRS are introduced into practice [66].
Hence, at present, the PRS for developing PD is low for both monogenic forms and multifactorial variants of the disease. One of the possible reasons is underestimation of the role of rare mutations or polymorphisms in the disease development. Further continuation of research in the field of large-scale sequencing of genomes and exomes of PD patients will allow identification of new loci and genes associated with the PD development. At the same time, the key role in these studies will belong to the integration of the obtained primary data into large international databases, combined with the functional annotation programs for such rare variants. The first example of such database is Gene4PD [47], which integrates the results of all published studies on the pathogenetically significant genetic variants of PD and allows to annotate the identified variants on the same platform using the data on the nature and incidence of genetic variant, functional annotation of variant-associated proteins, and results of transcriptome and methylome studies. Eventually, based on the analysis of variants associated with the PD development, all genes containing these variants will be classified as candidate genes of a high, medium, or low certainty.
As mentioned above, in a number of cases (mutations in the parkin, PINK1, and alpha-synuclein genes), the cause of the disease can be mutations or polymorphisms such as CNV, associated with deletion or duplication of genome fragments larger than 50 bp. Mutations of this type can make an important contribution to the overall genome variability [67], and including them in the analysis of genetic risks of multifactorial diseases can significantly change our estimates of PRS. One of the CNV marker affecting the risk of developing PD is associated with the X-linked dystonia/parkinsonism syndrome. This disease is caused by the insertion of a SINE-VNTR-Alu (SVA)-type complex retrotransposon into intron 32 of the TAF1 gene, which encodes the TATA box-binding protein-associated factor 1. This insertion leads to the disruption of the TAF1 gene expression, which is mostly pronounced for the neuronal mRNA variant. The disease phenotype and the mRNA level are influenced by the number of monomers (CCCCST)n in the VNTR repeat. This number varies from 34 to 52 and affects both the comparative severity of dystonia/parkinsonism and the age of clinical onset of the disease [68-70].
It must be emphasized that in this case, the mutation does not involve the protein-coding gene region, and its identification and confirmation of its pathogenetic role required a complex of genome and transcriptome studies using genome-editing techniques. On one hand, this highlights the difficulty of searching for pathogenetically significant mutations of this type and at least partly explains a relatively low genetic risk estimate obtained based on the analysis of single-nucleotide DNA markers only. On the other hand, this indicates an importance of searching for epigenetic factors associated with the disease development – identification of certain changes in the epigenome will allow a better understanding of genetic factors that cause them.
EPIGENETIC FACTORS IN THE DEVELOPMENT OF PARKINSON’S DISEASE
In recent years, an interest in the role of epigenetic mechanisms and factors in the pathogenesis of neurodegenerative diseases has significantly increased [71, 72]. DNA methylation, histone modification, and expression of various noncoding RNAs (microRNAs, small interfering RNAs, long non-coding RNAs) have been actively analyzed. The epigenome is considered not only as the cause of the disease, but also as a possible therapeutic target.
However, when considering epigenetic mechanisms of PD, it is necessary to take into account the influence of factors limiting the studies.
The first factor is that the vast majority of epigenetic studies are not focused (for obvious reasons) on the substantia nigra and striatum epigenome and are aimed at the analysis of peripheral tissues, primarily blood. Although some studies were conducted using autopsy material, the studied cohorts of patients, who were at the late PD stages (stages four and five according to the Hoehn and Yahr scale), were small and the patients presented various concomitant diseases, which were, in fact, the cause of death [73, 74]. The epigenome is actively studied in model objects (cell lines, including induced pluripotent cells and their derivatives, genetic and drug-induced animal models of PD), but the transfer of these data to humans requires caution and accuracy.
The second factor is a long asymptomatic course of the disease, while all epigenetic studies are carried out in patients with already severe motor impairments. At best, the studied cohorts are formed of patients at the first or early second stage (according to Hoehn and Yahr scale) and include only the patients that have been diagnosed with PD before the start of treatment. This may be important, since epigenetic markers (for example, miRNA expression levels) can respond to therapeutic interventions [75]. If epigenetic changes are detected, the question arises about their causal relationship with the disease, as changes in the epigenome can also be caused by a damage to dopaminergic neurons. But such changes are extremely important, since they can be considered as markers of the course of the pathological process. Identification of causal relationships in this case will also require analysis of various disease models, although it should be mentioned again that the transfer of data from a model to humans requires caution.
The first epigenetic factor studied in PD is DNA methylation. It has been shown that PD is associated with a global decrease in the level of DNA methylation both in the brain (including striatum and substantia nigra) and peripheral tissues [76]. This decrease is due to the interaction between DNA methyltransferase 1 (DNMT1) and alpha-synuclein in the nervous tissue, resulting in DNMT1 accumulation in the cytoplasm [77]. It was also shown that DNMT1 binds to the alpha-synuclein gene. SNCA intron 1 contains binding sites that determine differential methylation of the this gene. At the same time, no association was found between polymorphic variants of the DNMT1 gene and the risk of developing PD [78]. A number of genes have been identified whose methylation levels change in PD. For example, methylation of genes for the dopamine transporter (DAT), catechol-O-methyltransferase (COMT), prion protein (PRNP), and mitochondrial proteins (LARS2, MIR1977, and DDAH2) is reduced [79]. However, results obtained in the analysis of methylation are poorly reproducible. GWASs of global methylation in the brain tissues of PD patients and healthy individuals conducted over the past 10 years revealed only one gene, whose methylation level changed – the cytochrome P450 gene (CYP2E1) [80]. Low reproducibility of the results may be due to influence of environmental factors that might affect both the risk of developing PD and differential methylation, which should be taken into account when conducting methylation analysis. These factors include smoking, coffee consumption, and exposure to pesticides and heavy metals [80]. It is also important to conduct comprehensive studies that include not only analysis of methylation profiles, but also identification of differentially expressed genes and their annotation. For example, Henderson et al. [81] identified a number of genome regions differentially methylated in peripheral blood lymphocytes in PD patients and found differentially expressed genes associated with these genome regions, although in most cases, differential methylation and differential expression correlated poorly with each other.
Therefore, it is currently difficult to draw a definitive conclusion about the contribution of DNA methylation to the risk of developing PD, which requires additional large-scale, long-term studies in large cohorts of patients that should be characterized in detail both clinically and in terms of lifestyle, with a focus on patients at the early stages of PD. The same is true for another actively analyzed epigenetic marker – the level of microRNA (miRNA) expression.
The first attempts at analysis of changes in miRNA expression in the nervous tissue in PD were carried out in 2015-2016 and yielded very conflicting results. The sets of identified differentially expressed miRNAs did not overlap, which may be due to the use of samples from different brain regions (substance nigra and prefrontal cortex), different miRNA analysis methods (sequencing, real-time PCR, NanoString technology), or different clinical and morphological characteristics of patients. At the same time, it should be noted that the reported characteristics of individuals in these studies were extremely insufficient – for example, none of the articles provided information on the stage and clinical form of PD [73, 82, 83]. A total of 99 miRNAs differentially expressed in the substantia nigra in PD have been identified to date, with 60 miRNAs being upregulated and 39 miRNAs – downregulated [84]. These studies have shown that the miRNA sets expressed in the brain in PD change significantly, and such changes can cause certain abnormalities in the expression of genes involved in the metabolic pathways associated with the PD pathogenesis.
Of particular interest are miRNAs whose targets are genes for the monogenic forms of PD; such miRNAs can be considered as a link between the idiopathic and familial forms of the disease. For example, miR-7 was found to have a binding site in the 3′-untranslated region of the alpha-synuclein gene; it blocked translation of alpha-synuclein mRNA and reduced the level of this protein both in vitro and in vivo. Blocking miR-7 activity in mice resulted in the death of dopaminergic neurons in the substantia nigra, decrease in the dopamine levels, and accumulation of alpha-synuclein in the nervous tissue [74]. However, the general picture is more complicated, as expression of alpha-synuclein is controlled by several miRNAs, such as miR-153, miR-203a-3p, miR-203a-3p, miR-30b, miR-34b/c, miR-214, and miR-433. A similar situation is observed for other genes for the familial forms of PD. Thus, expression of parkin gene is regulated by at least four miRNAs (miR-103a-3p, miR-146a, miR-181a, miR-218), dardarin gene – two miRNAs (miR-205, miR-599), and DJ-1 gene – two miRNAs (miR-494, miR-4639). As a result, miRNAs are involved in the regulation (both positive and negative) of a large number of metabolic processes associated with the PD development, such as mitochondrial dysfunction and oxidative stress, autophagy, apoptosis, inflammation, and neurotrophin expression [84, 85].
miRNAs may also be a link between the monogenic forms of PD, idiopathic cases, and exposure to environmental factors, e.g., such known risk factors as pesticides [86]. It has been shown that pesticides cause changes in the miRNA profiles in various body tissues, and these changes largely overlap for different pesticides. For example, rotenone, paraquat, organophosphates, and atrazine alter expression of miR-34 [87] involved in the regulation of alpha-synuclein expression. Another relationship between genetic factors and miRNAs may be based on the regulation of miRNA gene expression at the level of transcription and transcript stability. Very little is known about this process, but it has been shown that individual polymorphic sites in the human genome can affect the binding of miRNAs to them and thereby modulate expression of the target genes. Thus, the rs10024743 single-nucleotide polymorphism (minor allele) in the miR-34-binding site located in the 3′-untranslated region of the alpha-synuclein gene reduced the expression of alpha-synuclein more than 2 times [88]. The rs66737902 polymorphism identified in the miR-138-2-3p-binding site in the 3′-untranslated region of the dardarin gene (LRRK2) was also found to be associated with the risk of developing PD. The authors believe that disruption of the miR-138-2-3p binding to the dardarin mRNA leads to changes in its level and development of the pathological process [89].
An important role in the formation of regulatory miRNA networks is likely played by long noncoding RNAs (lncRNAs) – RNAs over 200 nucleotides in length that do not contain open reading frames and are not translated into proteins. These RNAs can be synthesized on the sense and antisense DNA strands and can be encoded in gene introns and intergenic regions. The exact number of such RNA transcripts and genes encoding them is unknown; however, it is believed to be comparable to, and possibly, to exceed the number of protein-coding genes [90].
Several lncRNAs associated with biological processes leading to PD, have been described. Thus, in mice with the drug (6-hydroxydopamine)-induced damage to the substantia nigra, lncRNA H19 activates expression of the tyrosine hydroxylase gene and Wnt/beta-catenin signaling pathway through the binding of miR-301b-3p, and thereby improves survival of dopaminergic neurons in the substantia nigra. On the contrary, a decrease in the H19 RNA level promotes neurodegeneration [91]. A number of lncRNAs (MALAT1, UCA1, SNGH14) regulate alpha-synuclein metabolism and formation of its aggregates [92, 93]. Many lncRNAs influence various neurodegeneration-associated processes. Thus, MALAT1, in addition to regulating alpha-synuclein metabolism, is involved in the modulation of neuroinflammation and activates expression of dardarin (LRKK2), thereby promoting apoptosis and autophagy in dopaminergic neurons. Blocking expression of MALAT1 improves survival of dopaminergic neurons [92]. The activity of dardarin can be controlled by another lncRNA, NEAT1. It forms intranuclear inclusions (paraspeckles), that contain a number of cellular proteins, including dardarin. This reduces the level of active dardarin in the cell and thereby increases cell resistance to oxidative stress [94].
Epigenetic regulatory interactions are not limited to the three mechanisms described above, but other options are far less studied. Still, a possible role in the pathogenesis of PD has been shown for histone modifications [95], circular RNAs (circRNAs), and competing endogenous RNAs (ceRNA) [96]. Analysis of the epigenetic profile in PD should be expanded to include new models of the disease, both cellular and organismal.
CONCLUSION
Over the past 25 years, identification of genetic and epigenetic factors associated with PD pathogenesis has created the basis to start the construction of the “Cologne Cathedral” or “Sagrada Familia” of PD concept. More likely, the second – after all, both in the case of Sagrada Familia and PD, the building has not yet been fully constructed, but the main contours are already visible.
The foundation for the PD “building” is formed, first of all, by the genes for the main monogenic PD forms. SNCA, PRKN, and LRRK2 are the cornerstones, on which the first mechanisms of disease etiopathogenesis were proposed based on the formation of alpha-synuclein aggregates, mitochondrial dysfunction, and disruption of proteasomal protein degradation processes. Further construction involved identification of new genes for the monogenic PD forms and added lysosomal dysfunction and disruption of vesicular transport processes to the main pathological mechanisms of PD. The concept of a single continuum of interacting cellular processes leading to the selective death of dopaminergic neurons if any of these functional elements is disrupted, has been formulated. It remains relevant for the idiopathic form of the disease as well: GWASs revealed the SNCA and LRKK2 genes as the main PD-associated loci. At the same time, it is obvious that the “walls of the cathedral” have not been completed, as not all genes associated with the familial and sporadic forms of the disease have been identified. However, the major construction has been finished, and identification of new gene(s) will not crumble this already erected building.
However, for any building, connection between its parts is very important, which in the case of an organism is achieved through epigenetic interactions ensuring the functioning of an entire ensemble of genes performing a given biological function. The research community has taken the first and successful steps in this direction, but there is still a lot to be done in the epigenetic “finishing” of the building. This will require the development of fundamentally new approaches both in studying molecular events associated with the epigenome and data analysis using machine learning and artificial intelligence methods [97, 98].
Abbreviations
- GWAS:
-
genome-wide association study
- PD:
-
Parkinson’s disease
References
Bandres-Ciga, S., Ahmed, S., Sabir, M. S., Blauwendraat, C., Adarmes-Gomez, A. D., Bernal-Bernal, I., Bonilla-Toribio, M., Buiza-Rueda, D., Carrillo, F., Carrion-Claro, M., Gomez-Garre, P., Jesus, S., Labrador-Espinosa, M. A., Macias, D., Mendez-Del-Barrio, C., Perinan-Tocino, T., Tejera-Parrado, C., Vargas-Gonzalez, L., Diez-Fairen, M., Alvarez, I., Tartari, J. P., Buongiorno, M., Aguilar, M., Gorostidi, A., Bergareche, J. A., Mondragon, E., Vinagre-Aragon, A., Croitoru, I., Ruiz-Martinez, J., Dols-Icardo, O., Kulisevsky, J., Marin-Lahoz, J., Pagonabarraga, J., Pascual-Sedano, B., Ezquerra, M., Camara, A., Compta, Y., Fernandez, M., Fernandez-Santiago, R., Munoz, E., Tolosa, E., Valldeoriola, F., Gonzalez-Aramburu, I., Sanchez Rodriguez, A., Sierra, M., Menendez-Gonzalez, M., Blazquez, M., Garcia, C., Suarez-San Martin, E., Garcia-Ruiz, P., Martinez-Castrillo, J. C., Vela-Desojo, L., Ruz, C., Barrero, F. J., Escamilla-Sevilla, F., Minguez-Castellanos, A., Cerdan, D., Tabernero, C., Gomez Heredia, M. J., Perez Errazquin, F., Romero-Acebal, M., Feliz, C., Lopez-Sendon, J. L., Mata, M., Martinez Torres, I., Kim, J. J., Dalgard, C. L., The American Genome Center, Brooks, J., Saez-Atienzar, S., Gibbs, J. R., Jorda, R., Botia, J. A., Bonet-Ponce, L., Morrison, K. E., Clarke, C., Tan, M., Morris, H., Edsall, C., Hernandez, D., Simon-Sanchez, J., Nalls, M. A., Scholz, S. W., Jimenez-Escrig, A., Duarte, J., Vives, F., Duran, R., Hoenicka, J., Alvarez, V., Infante, J., Marti, M. J., Clarimon, J., Lopez de Munain, A., Pastor, P., Mir, P., Singleton, A., and International Parkinson Disease Genomics Consortium (2019) The genetic architecture of Parkinson’s disease in Spain: characterizing population-specific risk, differential haplotype structures, and providing etiologic insight, Mov. Disord. Offic. J. Mov. Disord. Soc., 34, 1851-1863, https://doi.org/10.1002/mds.27864.
Savitt, J. M., Dawson, V. L., and Dawson, T. M. (2006) Diagnosis and treatment of Parkinson’s disease: molecules to medicine, J. Clin. Invest., 116, 1744-1754, https://doi.org/10.1172/JCI29178.
Vázquez-Vélez, G. E., and Zoghbi, H. Y. (2021) Parkinson’s disease genetics and pathophysiology, Annu. Rev. Neurosci., 44, 87-108, https://doi.org/10.1146/annurev-neuro-100720-034518.
Del Tredici, K., and Braak, H. (2016) Review: sporadic Parkinson’s disease: development and distribution of α-synuclein pathology, Neuropathol. Appl. Neurobiol., 42, 33-50, https://doi.org/10.1111/nan.12298.
Connolly, B. S., and Lang, A. E. (2014) Pharmacological treatment of Parkinson disease: a review, JAMA, 311, 1670-1683, https://doi.org/10.1001/jama.2014.3654.
Polymeropoulos, M. H., Lavedan, C., Leroy, E., Ide, S. E., Dehejia, A., Dutra, A., Pike, B., Root, H., Rubenstein, J., Boyer, R., Stenroos, E. S., Chandrasekharappa, S., Athanassiadou, A., Papapetropoulos, T., Johnson, W. G., Lazzarini, A. M., Duvoisin, R. C., Di Iorio, G., Golbe, L. I., and Nussbaum, R. L. (1997) Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease, Science, 276, 2045-2047, https://doi.org/10.1126/science.276.5321.2045.
Lunati, A., Lesage, S., and Brice, A. (2018) The genetic landscape of Parkinson’s disease, Rev. Neurolog., 174, 628-643, https://doi.org/10.1016/j.neurol.2018.08.004.
Lesage, S., and Brice, A. (2012) Role of mendelian genes in “sporadic” Parkinson’s disease, Parkinson. Rel. Disord., 18 Suppl 1, S66-S70, https://doi.org/10.1016/s1353-8020(11)70022-0.
Leroy, E., Boyer, R., Auburger, G., Leube, B., Ulm, G., Mezey, E., Harta, G., Brownstein, M. J., Jonnalagada, S., Chernova, T., Dehejia, A., Lavedan, C., Gasser, T., Steinbach, P. J., Wilkinson, K. D., and Polymeropoulos, M. H. (1998) The ubiquitin pathway in Parkinson’s disease, Nature, 395, 451-452, https://doi.org/10.1038/26652.
Lee, Y. C., and Hsu, S. D. (2017) Familial mutations and post-translational modifications of UCH-L1 in Parkinson’s disease and neurodegenerative disorders, Curr. Prot. Pept. Sci., 18, 733-745, https://doi.org/10.2174/1389203717666160217143721.
Wintermeyer, P., Kruger, R., Kuhn, W., Muller, T., Woitalla, D., Berg, D., Becker, G., Leroy, E., Polymeropoulos, M., Berger, K., Przuntek, H., Schols, L., Epplen, J. T., and Riess, O. (2000) Mutation analysis and association studies of the UCHL1 gene in German Parkinson’s disease patients, Neuroreport, 11, 2079-2082, https://doi.org/10.1097/00001756-200007140-00004.
Healy, D. G., Abou-Sleiman, P. M., Casas, J. P., Ahmadi, K. R., Lynch, T., Gandhi, S., Muqit, M. M., Foltynie, T., Barker, R., Bhatia, K. P., Quinn, N. P., Lees, A. J., Gibson, J. M., Holton, J. L., Revesz, T., Goldstein, D. B., and Wood, N. W. (2006) UCHL-1 is not a Parkinson’s disease susceptibility gene, Ann. Neurol., 59, 627-633, https://doi.org/10.1002/ana.20757.
Miyake, Y., Tanaka, K., Fukushima, W., Kiyohara, C., Sasaki, S., Tsuboi, Y., Yamada, T., Oeda, T., Shimada, H., Kawamura, N., Sakae, N., Fukuyama, H., Hirota, Y., and Nagai, M. (2012) UCHL1 S18Y variant is a risk factor for Parkinson’s disease in Japan, BMC Neurol., 12, 62, https://doi.org/10.1186/1471-2377-12-62.
Sun, S., Zhao, Y., Jin, G., and Kang, H. (2014) Lack of association between UCHL1 S18Y gene polymorphism and Parkinson’s disease in the Asian population: a meta-analysis, Neurol. Sci., 35, 1867-1876, https://doi.org/10.1007/s10072-014-1973-4.
Saigoh, K., Wang, Y. L., Suh, J. G., Yamanishi, T., Sakai, Y., Kiyosawa, H., Harada, T., Ichihara, N., Wakana, S., Kikuchi, T., and Wada, K. (1999) Intragenic deletion in the gene encoding ubiquitin carboxy-terminal hydrolase in gad mice, Nat. Genet., 23, 47-51, https://doi.org/10.1038/12647.
Setsuie, R., Wang, Y. L., Mochizuki, H., Osaka, H., Hayakawa, H., Ichihara, N., Li, H., Furuta, A., Sano, Y., Sun, Y. J., Kwon, J., Kabuta, T., Yoshimi, K., Aoki, S., Mizuno, Y., Noda, M., and Wada, K. (2007) Dopaminergic neuronal loss in transgenic mice expressing the Parkinson’s disease-associated UCH-L1 I93M mutant, Neurochem. Int., 50, 119-129, https://doi.org/10.1016/j.neuint.2006.07.015.
Yasuda, T., Nihira, T., Ren, Y. R., Cao, X. Q., Wada, K., Setsuie, R., Kabuta, T., Wada, K., Hattori, N., Mizuno, Y., and Mochizuki, H. (2009) Effects of UCH-L1 on alpha-synuclein over-expression mouse model of Parkinson’s disease, J. Neurochem., 108, 932-944, https://doi.org/10.1111/j.1471-4159.2008.05827.x.
Kyratzi, E., Pavlaki, M., and Stefanis, L. (2008) The S18Y polymorphic variant of UCH-L1 confers an antioxidant function to neuronal cells, Hum. Mol. Genet., 17, 2160-2171, https://doi.org/10.1093/hmg/ddn115.
Xilouri, M., Kyratzi, E., Pitychoutis, P. M., Papadopoulou-Daifoti, Z., Perier, C., Vila, M., Maniati, M., Ulusoy, A., Kirik, D., Park, D. S., Wada, K., and Stefanis, L. (2012) Selective neuroprotective effects of the S18Y polymorphic variant of UCH-L1 in the dopaminergic system, Hum. Mol. Genet., 21, 874-889, https://doi.org/10.1093/hmg/ddr521.
Riboldi, G. M., and Di Fonzo, A. B. (2019) GBA, Gaucher disease, and Parkinson’s disease: from genetic to clinic to new therapeutic approaches, Cells, 8, 364, https://doi.org/10.3390/cells8040364.
Thaler, A., Bregman, N., Gurevich, T., Shiner, T., Dror, Y., Zmira, O., Gan-Or, Z., Bar-Shira, A., Gana-Weisz, M., Orr-Urtreger, A., Giladi, N., and Mirelman, A. (2018) Parkinson’s disease phenotype is influenced by the severity of the mutations in the GBA gene, Parkinsonism Rel. Disord., 55, 45-49, https://doi.org/10.1016/j.parkreldis.2018.05.009.
Thaler, A., Gurevich, T., Bar Shira, A., Gana Weisz, M., Ash, E., Shiner, T., Orr-Urtreger, A., Giladi, N., and Mirelman, A. (2017) A “dose” effect of mutations in the GBA gene on Parkinson’s disease phenotype, Parkinsonism Rel. Disord., 36, 47-51, https://doi.org/10.1016/j.parkreldis.2016.12.014.
Gan-Or, Z., Liong, C., and Alcalay, R. N. (2018) GBA-associated Parkinson’s disease and other synucleinopathies, Curr. Neurol. Neurosci. Rep., 18, 44, https://doi.org/10.1007/s11910-018-0860-4.
Vieira, S. R. L., and Schapira, A. H. V. (2022) Glucocerebrosidase mutations and Parkinson’s disease, J. Neural Transmiss., 129, 1105-1117, https://doi.org/10.1007/s00702-022-02531-3.
Kluss, J. H., Mamais, A., and Cookson, M. R. (2019) LRRK2 links genetic and sporadic Parkinson’s disease, Biochem. Soc. Transact., 47, 651-661, https://doi.org/10.1042/bst20180462.
Lee, A. J., Wang, Y., Alcalay, R. N., Mejia-Santana, H., Saunders-Pullman, R., Bressman, S., Corvol, J. C., Brice, A., Lesage, S., Mangone, G., Tolosa, E., Pont-Sunyer, C., Vilas, D., Schüle, B., Kausar, F., Foroud, T., Berg, D., Brockmann, K., Goldwurm, S., Siri, C., Asselta, R., Ruiz-Martinez, J., Mondragón, E., Marras, C., Ghate, T., Giladi, N., Mirelman, A., and Marder, K. (2017) Penetrance estimate of LRRK2 p.G2019S mutation in individuals of non-Ashkenazi Jewish ancestry, Mov. Disord., 32, 1432-1438, https://doi.org/10.1002/mds.27059.
Trinh, J., Guella, I., and Farrer, M. J. (2014) Disease penetrance of late-onset parkinsonism: a meta-analysis, JAMA Neurol., 71, 1535-1539, https://doi.org/10.1001/jamaneurol.2014.1909.
Kestenbaum, M., and Alcalay, R. N. (2017) Clinical features of LRRK2 carriers with Parkinson’s disease, Adv. Neurobiol., 14, 31-48, https://doi.org/10.1007/978-3-319-49969-7_2.
Guadagnolo, D., Piane, M., Torrisi, M. R., Pizzuti, A., and Petrucci, S. (2021) Genotype-phenotype correlations in monogenic Parkinson’s disease: a review on clinical and molecular findings, Front. Neurol., 12, 648588, https://doi.org/10.3389/fneur.2021.648588.
Milanowski, Ł. M., Lindemann, J. A., Hoffman-Zacharska, D., Soto-Beasley, A. I., Barcikowska, M., Boczarska-Jedynak, M., Deutschlander, A., Kłodowska, G., Dulski, J., Fedoryshyn, L., Friedman, A., Jamrozik, Z., Janik, P., Karpinsky, K., Koziorowski, D., Krygowska-Wajs, A., Jasińska-Myga, B., Opala, G., Potulska-Chromik, A., Pulyk, A., Rektorova, I., Sanotsky, Y., Siuda, J., Sławek, J., Śmiłowska, K., Szczechowski, L., Rudzińska-Bar, M., Walton, R. L., Ross, O. A., and Wszolek, Z. K. (2021) Frequency of mutations in PRKN, PINK1, and DJ1 in patients with early-onset Parkinson’s disease from neighboring countries in Central Europe, Parkinsonism Rel. Disord., 86, 48-51, https://doi.org/10.1016/j.parkreldis.2021.03.026.
Olszewska, D. A., McCarthy, A., Soto-Beasley, A. I., Walton, R. L., Ross, O. A., and Lynch, T. (2022) PARKIN, PINK1, and DJ1 analysis in early-onset Parkinson’s disease in Ireland, Irish J. Med. Sci., 191, 901-907, https://doi.org/10.1007/s11845-021-02563-w.
Erer, S., Egeli, U., Zarifoglu, M., Tezcan, G., Cecener, G., Tunca, B., Ak, S., Demirdogen, E., Kenangil, G., Kaleagası, H., Dogu, O., Saka, E., and Elibol, B. (2016) Mutation analysis of the PARKIN, PINK1, DJ1, and SNCA genes in Turkish early-onset Parkinson’s patients and genotype-phenotype correlations, Clin. Neurol. Neurosurg., 148, 147-153, https://doi.org/10.1016/j.clineuro.2016.07.005.
Shulskaya, M. V., Shadrina, M. I., Fedotova, E. Y., Abramycheva, N. Y., Limborska, S. A., Illarioshkin, S. N., and Slominsky, P. A. (2017) Second mutation in PARK2 is absent in patients with sporadic Parkinson’s disease and heterozygous exonic deletions/duplications in parkin gene, Int. J. Neurosci., 127, 781-784, https://doi.org/10.1080/00207454.2016.1255612.
Semenova, E. V., Shadrina, M. I., Slominsky, P. A., Ivanova-Smolenskaya, I. A., Bagyeva, G., Illarioshkin, S. N., and Limborska, S. A. (2012) Analysis of PARK2 gene exon rearrangements in Russian patients with sporadic Parkinson’s disease, Mov. Disord., 27, 139-142, https://doi.org/10.1002/mds.23901.
Valente, E. M., and Ferraris, A. (2007) Heterozygous mutations in genes causing parkinsonism: monogenic disorders go complex, Lancet Neurol., 6, 576-578, https://doi.org/10.1016/s1474-4422(07)70158-8.
Brüggemann, N., Mitterer, M., Lanthaler, A. J., Djarmati, A., Hagenah, J., Wiegers, K., Winkler, S., Pawlack, H., Lohnau, T., Pramstaller, P. P., Klein, C., and Lohmann, K. (2009) Frequency of heterozygous Parkin mutations in healthy subjects: need for careful prospective follow-up examination of mutation carriers, Parkinsonism Rel. Disord., 15, 425-429, https://doi.org/10.1016/j.parkreldis.2008.11.014.
Pavese, N., Khan, N. L., Scherfler, C., Cohen, L., Brooks, D. J., Wood, N. W., Bhatia, K. P., Quinn, N. P., Lees, A. J., and Piccini, P. (2009) Nigrostriatal dysfunction in homozygous and heterozygous parkin gene carriers: an 18F-dopa PET progression study, Mov. Disord., 24, 2260-2266, https://doi.org/10.1002/mds.22817.
Van Nuenen, B. F., van Eimeren, T., van der Vegt, J. P., Buhmann, C., Klein, C., Bloem, B. R., and Siebner, H. R. (2009) Mapping preclinical compensation in Parkinson’s disease: an imaging genomics approach, Mov. Disord., 24 Suppl 2, S703-S710, https://doi.org/10.1002/mds.22635.
Marongiu, R., Ferraris, A., Ialongo, T., Michiorri, S., Soleti, F., Ferrari, F., Elia, A. E., Ghezzi, D., Albanese, A., Altavista, M. C., Antonini, A., Barone, P., Brusa, L., Cortelli, P., Martinelli, P., Pellecchia, M. T., Pezzoli, G., Scaglione, C., Stanzione, P., Tinazzi, M., Zecchinelli, A., Zeviani, M., Cassetta, E., Garavaglia, B., Dallapiccola, B., Bentivoglio, A. R., and Valente, E. M. (2008) PINK1 heterozygous rare variants: prevalence, significance and phenotypic spectrum, Hum. Mutat., 29, 565, https://doi.org/10.1002/humu.20719.
Yu, E., Rudakou, U., Krohn, L., Mufti, K., Ruskey, J. A., Asayesh, F., Estiar, M. A., Spiegelman, D., Surface, M., Fahn, S., Waters, C. H., Greenbaum, L., Espay, A. J., Dauvilliers, Y., Dupré, N., Rouleau, G. A., Hassin-Baer, S., Fon, E. A., Alcalay, R. N., and Gan-Or, Z. (2021) Analysis of heterozygous PRKN variants and copy-number variations in Parkinson’s disease, Mov. Disord., 36, 178-187, https://doi.org/10.1002/mds.28299.
Jia, F., Fellner, A., and Kumar, K. R. (2022) Monogenic Parkinson’s disease: genotype, phenotype, pathophysiology, and genetic testing, Genes, 13, 471, https://doi.org/10.3390/genes13030471.
Santos-Lobato, B. L., Schumacher-Schuh, A., Mata, I. F., Letro, G. H., Braga-Neto, P., Brandão, P. R. P., Godeiro-Junior, C. O., Coletta, M. V. D., Camargos, S. T., Borges, V., Rieder, C. R. M., and Tumas, V. (2021) Genetics of Parkinson’s disease in Brazil: a systematic review of monogenic forms, Arq. Neuropsiquiatr., 79, 612-623, https://doi.org/10.1590/0004-282X-anp-2020-0409.
Correia Guedes, L., Ferreira, J. J., Rosa, M. M., Coelho, M., Bonifati, V., and Sampaio, C. (2010) Worldwide frequency of G2019S LRRK2 mutation in Parkinson’s disease: a systematic review, Parkinsonism Rel. Disord., 16, 237-242, https://doi.org/10.1016/j.parkreldis.2009.11.004.
Gialluisi, A., Reccia, M. G., Modugno, N., Nutile, T., Lombardi, A., Di Giovannantonio, L. G., Pietracupa, S., Ruggiero, D., Scala, S., Gambardella, S., Iacoviello, L., Gianfrancesco, F., Acampora, D., D’Esposito, M., Simeone, A., Ciullo, M., and Esposito, T. (2021) Identification of sixteen novel candidate genes for late onset Parkinson’s disease, Mol. Neurodegener., 16, 35, https://doi.org/10.1186/s13024-021-00455-2.
Shulskaya, M. V., Alieva, A. K., Vlasov, I. N., Zyrin, V. V., Fedotova, E. Y., Abramycheva, N. Y., Usenko, T. S., Yakimovsky, A. F., Emelyanov, A. K., Pchelina, S. N., Illarioshkin, S. N., Slominsky, P. A., and Shadrina, M. I. (2018) Whole-exome sequencing in searching for new variants associated with the development of Parkinson’s disease, Front. Aging Neurosci., 10, 136, https://doi.org/10.3389/fnagi.2018.00136.
Cook, L., Schulze, J., Verbrugge, J., Beck, J. C., Marder, K. S., Saunders-Pullman, R., Klein, C., Naito, A., and Alcalay, R. N. (2021) The commercial genetic testing landscape for Parkinson’s disease, Parkinsonism Rel. Disord., 92, 107-111, https://doi.org/10.1016/j.parkreldis.2021.10.001.
Li, B., Zhao, G., Zhou, Q., Xie, Y., Wang, Z., Fang, Z., Lu, B., Qin, L., Zhao, Y., Zhang, R., Jiang, L., Pan, H., He, Y., Wang, X., Luo, T., Zhang, Y., Wang, Y., Chen, Q., Liu, Z., Guo, J., Tang, B., and Li, J. (2021) Gene4PD: a comprehensive genetic database of Parkinson’s disease, Front. Neurosci., 15, 679568, https://doi.org/10.3389/fnins.2021.679568.
Day, J. O., and Mullin, S. (2021) The genetics of Parkinson’s disease and implications for clinical practice, Genes, 12, 1006, https://doi.org/10.3390/genes12071006.
Fung, H. C., Scholz, S., Matarin, M., Simón-Sánchez, J., Hernandez, D., Britton, A., Gibbs, J. R., Langefeld, C., Stiegert, M. L., Schymick, J., Okun, M. S., Mandel, R. J., Fernandez, H. H., Foote, K. D., Rodríguez, R. L., Peckham, E., De Vrieze, F. W., Gwinn-Hardy, K., Hardy, J. A., and Singleton, A. (2006) Genome-wide genotyping in Parkinson’s disease and neurologically normal controls: first stage analysis and public release of data, Lancet Neurol., 5, 911-916, https://doi.org/10.1016/s1474-4422(06)70578-6.
Maraganore, D. M., de Andrade, M., Lesnick, T. G., Strain, K. J., Farrer, M. J., Rocca, W. A., Pant, P. V., Frazer, K. A., Cox, D. R., and Ballinger, D. G. (2005) High-resolution whole-genome association study of Parkinson disease, Am. J. Hum. Genet., 77, 685-693, https://doi.org/10.1086/496902.
Evangelou, E., Maraganore, D. M., and Ioannidis, J. P. (2007) Meta-analysis in genome-wide association datasets: strategies and application in Parkinson’s disease, PLoS One, 2, e196, https://doi.org/10.1371/journal.pone.0000196.
Leveille, E., Ross, O. A., and Gan-Or, Z. (2021) Tau and MAPT genetics in tauopathies and synucleinopathies, Parkinsonism Rel. Disord., 90, 142-154, https://doi.org/10.1016/j.parkreldis.2021.09.008.
Clark, L. N., Gao, Y., Wang, G. T., Hernandez, N., Ashley-Koch, A., Jankovic, J., Ottman, R., Leal, S. M., Rodriguez, S. M. B., and Louis, E. D. (2022) Whole genome sequencing identifies candidate genes for familial essential tremor and reveals biological pathways implicated in essential tremor aetiology, EBioMedicine, 85, 104290, https://doi.org/10.1016/j.ebiom.2022.104290.
Drazich-Taylor, E. H. S., Todd, E., Convery, R., Bocchetta, M., Clarke, M., Warren, J. D., Fox, N. C., Revesz, T., and Rohrer, J. D. (2023) Q351R MAPT mutation is associated with a mixed 3R/4R tauopathy and a slowly progressive cognitive, behavioural and parkinsonian syndrome, J. Neurol. Neurosurg. Psychiatry, 94, 169-171, https://doi.org/10.1136/jnnp-2022-329330.
Hu, M., Li, P., Wang, C., Feng, X., Geng, Q., Chen, W., Marthi, M., Zhang, W., Gao, C., Reid, W., Swanson, J., Du, W., Hume, R. I., and Xu, H. (2022) Parkinson’s disease-risk protein TMEM175 is a proton-activated proton channel in lysosomes, Cell, 185, 2292-2308.e2220, https://doi.org/10.1016/j.cell.2022.05.021.
Jinn, S., Drolet, R. E., Cramer, P. E., Wong, A. H., Toolan, D. M., Gretzula, C. A., Voleti, B., Vassileva, G., Disa, J., Tadin-Strapps, M., and Stone, D. J. (2017) TMEM175 deficiency impairs lysosomal and mitochondrial function and increases α-synuclein aggregation, Proc. Natl. Acad. Sci. USA, 114, 2389-2394, https://doi.org/10.1073/pnas.1616332114.
Krohn, L., Öztürk, T. N., Vanderperre, B., Ouled Amar Bencheikh, B., Ruskey, J. A., Laurent, S. B., Spiegelman, D., Postuma, R. B., Arnulf, I., Hu, M. T. M., Dauvilliers, Y., Högl, B., Stefani, A., Monaca, C. C., Plazzi, G., Antelmi, E., Ferini-Strambi, L., Heidbreder, A., Rudakou, U., Cochen De Cock, V., Young, P., Wolf, P., Oliva, P., Zhang, X. K., Greenbaum, L., Liong, C., Gagnon, J. F., Desautels, A., Hassin-Baer, S., Montplaisir, J. Y., Dupré, N., Rouleau, G. A., Fon, E. A., Trempe, J. F., Lamoureux, G., Alcalay, R. N., and Gan-Or, Z. (2020) Genetic, structural, and functional evidence Link TMEM175 to synucleinopathies, Ann. Neurol., 87, 139-153, https://doi.org/10.1002/ana.25629.
Wie, J., Liu, Z., Song, H., Tropea, T. F., Yang, L., Wang, H., Liang, Y., Cang, C., Aranda, K., Lohmann, J., Yang, J., Lu, B., Chen-Plotkin, A. S., Luk, K. C., and Ren, D. (2021) A growth-factor-activated lysosomal K+ channel regulates Parkinson’s pathology, Nature, 591, 431-437, https://doi.org/10.1038/s41586-021-03185-z.
Lunding, L. P., Krause, D., Stichtenoth, G., Stamme, C., Lauterbach, N., Hegermann, J., Ochs, M., Schuster, B., Sedlacek, R., Saftig, P., Schwudke, D., Wegmann, M., and Damme, M. (2021) LAMP3 deficiency affects surfactant homeostasis in mice, PLoS Genet., 17, e1009619, https://doi.org/10.1371/journal.pgen.1009619.
Tanaka, T., Warner, B. M., Michael, D. G., Nakamura, H., Odani, T., Yin, H., Atsumi, T., Noguchi, M., and Chiorini, J. A. (2022) LAMP3 inhibits autophagy and contributes to cell death by lysosomal membrane permeabilization, Autophagy, 18, 1629-1647, https://doi.org/10.1080/15548627.2021.1995150.
Tanaka, T., Warner, B. M., Odani, T., Ji, Y., Mo, Y. Q., Nakamura, H., Jang, S. I., Yin, H., Michael, D. G., Hirata, N., Suizu, F., Ishigaki, S., Oliveira, F. R., Motta, A. C. F., Ribeiro-Silva, A., Rocha, E. M., Atsumi, T., Noguchi, M., and Chiorini, J. A. (2020) LAMP3 induces apoptosis and autoantigen release in Sjögren’s syndrome patients, Sci. Rep., 10, 15169, https://doi.org/10.1038/s41598-020-71669-5.
Chosa, N., and Ishisaki, A. (2018) Two novel mechanisms for maintenance of stemness in mesenchymal stem cells: SCRG1/BST1 axis and cell-cell adhesion through N-cadherin, Jpn. Dent. Sci. Rev., 54, 37-44, https://doi.org/10.1016/j.jdsr.2017.10.001.
Higashida, H., Hashii, M., Tanaka, Y., Matsukawa, S., Higuchi, Y., Gabata, R., Tsubomoto, M., Seishima, N., Teramachi, M., Kamijima, T., Hattori, T., Hori, O., Tsuji, C., Cherepanov, S. M., Shabalova, A. A., Gerasimenko, M., Minami, K., Yokoyama, S., Munesue, S. I., Harashima, A., Yamamoto, Y., Salmina, A. B., and Lopatina, O. (2019) CD38, CD157, and RAGE as molecular determinants for social behavior, Cells, 9, 62, https://doi.org/10.3390/cells9010062.
Ortolan, E., Augeri, S., Fissolo, G., Musso, I., and Funaro, A. (2019) CD157: From immunoregulatory protein to potential therapeutic target, Immunol. Lett., 205, 59-64, https://doi.org/10.1016/j.imlet.2018.06.007.
Blauwendraat, C., Nalls, M. A., and Singleton, A. B. (2020) The genetic architecture of Parkinson’s disease, Lancet Neurol., 19, 170-178, https://doi.org/10.1016/S1474-4422(19)30287-X.
Wand, H., Lambert, S. A., Tamburro, C., Iacocca, M. A., O'Sullivan, J. W., Sillari, C., Kullo, I. J., Rowley, R., Dron, J. S., Brockman, D., Venner, E., McCarthy, M. I., Antoniou, A. C., Easton, D. F., Hegele, R. A., Khera, A. V., Chatterjee, N., Kooperberg, C., Edwards, K., Vlessis, K., Kinnear, K., Danesh, J. N., Parkinson, H., Ramos, E. M., Roberts, M. C., Ormond, K. E., Khoury, M. J., Janssens, A., Goddard, K. A. B., Kraft, P., MacArthur, J. A. L., Inouye, M., and Wojcik, G. L. (2021) Improving reporting standards for polygenic scores in risk prediction studies, Nature, 591, 211-219, https://doi.org/10.1038/s41586-021-03243-6.
Pang, A. W., MacDonald, J. R., Pinto, D., Wei, J., Rafiq, M. A., Conrad, D. F., Park, H., Hurles, M. E., Lee, C., Venter, J. C., Kirkness, E. F., Levy, S., Feuk, L., and Scherer, S. W. (2010) Towards a comprehensive structural variation map of an individual human genome, Genome Biol., 11, R52, https://doi.org/10.1186/gb-2010-11-5-r52.
Ng, A. R., Jamora, R. D. G., and Rosales, R. L. (2021) X-linked dystonia Parkinsonism: crossing a new threshold, J. Neural Transm., 128, 567-573, https://doi.org/10.1007/s00702-021-02324-0.
Di Lazzaro, G., Magrinelli, F., Estevez-Fraga, C., Valente, E. M., Pisani, A., and Bhatia, K. P. (2021) X-linked Parkinsonism: phenotypic and genetic heterogeneity, Mov. Disord., 36, 1511-1525, https://doi.org/10.1002/mds.28565.
Bragg, D. C., Sharma, N., and Ozelius, L. J. (2019) X-Linked Dystonia-Parkinsonism: recent advances, Curr. Opin. Neurol., 32, 604-609, https://doi.org/10.1097/wco.0000000000000708.
Marques, S. C., Oliveira, C. R., Pereira, C. M., and Outeiro, T. F. (2011) Epigenetics in neurodegeneration: a new layer of complexity, Prog. Neuropsychopharmacol. Biol. Psychiatry., 35, 348-355, https://doi.org/10.1016/j.pnpbp.2010.08.008.
Mohd Murshid, N., Aminullah Lubis, F., and Makpol, S. (2022) Epigenetic changes and its intervention in age-related neurodegenerative diseases, Cell. Mol. Neurobiol., 42, 577-595, https://doi.org/10.1007/s10571-020-00979-z.
Briggs, C. E., Wang, Y., Kong, B., Woo, T. U., Iyer, L. K., and Sonntag, K. C. (2015) Midbrain dopamine neurons in Parkinson’s disease exhibit a dysregulated miRNA and target-gene network, Brain Res., 1618, 111-121, https://doi.org/10.1016/j.brainres.2015.05.021.
McMillan, K. J., Murray, T. K., Bengoa-Vergniory, N., Cordero-Llana, O., Cooper, J., Buckley, A., Wade-Martins, R., Uney, J. B., O’Neill, M. J., Wong, L. F., and Caldwell, M. A. (2017) Loss of microRNA-7 regulation leads to alpha-synuclein accumulation and dopaminergic neuronal loss in vivo, Mol. Ther., 25, 2404-2414, https://doi.org/10.1016/j.ymthe.2017.08.017.
Alieva, A., Filatova, E. V., Karabanov, A. V., Illarioshkin, S. N., Limborska, S. A., Shadrina, M. I., and Slominsky, P. A. (2015) miRNA expression is highly sensitive to a drug therapy in Parkinson’s disease, Parkinsonism Rel. Disord., 21, 72-74, https://doi.org/10.1016/j.parkreldis.2014.10.018.
Wang, C., Chen, L., Zhang, M., Yang, Y., and Wong, G. (2020) PDmethDB: A curated Parkinson’s disease associated methylation information database, Computat. Struct. Biotechnol. J., 18, 3745-3749, https://doi.org/10.1016/j.csbj.2020.11.015.
Desplats, P., Spencer, B., Coffee, E., Patel, P., Michael, S., Patrick, C., Adame, A., Rockenstein, E., and Masliah, E. (2011) Alpha-synuclein sequesters Dnmt1 from the nucleus: a novel mechanism for epigenetic alterations in Lewy body diseases, J. Biol. Chem., 286, 9031-9037, https://doi.org/10.1074/jbc.C110.212589.
Pezzi, J. C., de Bem, C. M., da Rocha, T. J., Schumacher-Schuh, A. F., Chaves, M. L., Rieder, C. R., Hutz, M. H., Fiegenbaum, M., and Camozzato, A. L. (2017) Association between DNA methyltransferase gene polymorphism and Parkinson’s disease, Neurosci. Lett., 639, 146-150, https://doi.org/10.1016/j.neulet.2016.12.058.
Angelopoulou, E., Paudel, Y. N., Papageorgiou, S. G., and Piperi, C. (2022) Environmental impact on the epigenetic mechanisms underlying Parkinson’s disease pathogenesis: a narrative review, Brain Sci., 12, 175, https://doi.org/10.3390/brainsci12020175.
Schaffner, S. L., and Kobor, M. S. (2022) DNA methylation as a mediator of genetic and environmental influences on Parkinson’s disease susceptibility: impacts of alpha-Synuclein, physical activity, and pesticide exposure on the epigenome, Front. Genet., 13, 971298, https://doi.org/10.3389/fgene.2022.971298.
Henderson, A. R., Wang, Q., Meechoovet, B., Siniard, A. L., Naymik, M., De Both, M., Huentelman, M. J., Caselli, R. J., Driver-Dunckley, E., and Dunckley, T. (2021) DNA methylation and expression profiles of whole blood in Parkinson’s disease, Front. Genet., 12, 640266, https://doi.org/10.3389/fgene.2021.640266.
Hoss, A. G., Labadorf, A., Beach, T. G., Latourelle, J. C., and Myers, R. H. (2016) MicroRNA profiles in Parkinson’s disease prefrontal cortex, Front. Aging Neurosci., 8, 36, https://doi.org/10.3389/fnagi.2016.00036.
Nair, V. D., and Ge, Y. (2016) Alterations of miRNAs reveal a dysregulated molecular regulatory network in Parkinson’s disease striatum, Neurosci. Lett., 629, 99-104, https://doi.org/10.1016/j.neulet.2016.06.061.
Santos-Lobato, B. L., Vidal, A. F., and Ribeiro-Dos-Santos, Â. (2021) Regulatory miRNA-mRNA networks in Parkinson’s disease, Cells, 10, 1410, https://doi.org/10.3390/cells10061410.
Saghazadeh, A., and Rezaei, N. (2022) MicroRNA machinery in Parkinson’s disease: a platform for neurodegenerative diseases, Exp. Rev. Neurother., 22, 427-453, https://doi.org/10.1586/14737175.2015.1114886.
Ahmed, H., Abushouk, A. I., Gabr, M., Negida, A., and Abdel-Daim, M. M. (2017) Parkinson’s disease and pesticides: a meta-analysis of disease connection and genetic alterations, Biomed. Pharmacother., 90, 638-649, https://doi.org/10.1016/j.biopha.2017.03.100.
Aloizou, A. M., Siokas, V., Sapouni, E. M., Sita, N., Liampas, I., Brotis, A. G., Rakitskii, V. N., Burykina, T. I., Aschner, M., Bogdanos, D. P., Tsatsakis, A., Hadjigeorgiou, G. M., and Dardiotis, E. (2020) Parkinson’s disease and pesticides: are microRNAs the missing link?, Sci. Total Environ., 744, 140591, https://doi.org/10.1016/j.scitotenv.2020.140591.
Kabaria, S., Choi, D. C., Chaudhuri, A. D., Mouradian, M. M., and Junn, E. (2015) Inhibition of miR-34b and miR-34c enhances α-synuclein expression in Parkinson’s disease, FEBS Lett., 589, 319-325, https://doi.org/10.1016/j.febslet.2014.12.014.
Cardo, L. F., Coto, E., Ribacoba, R., Mata, I. F., Moris, G., Menéndez, M., and Alvarez, V. (2014) The screening of the 3′-UTR sequence of LRRK2 identified an association between the rs66737902 polymorphism and Parkinson’s disease, J. Hum. Genet., 59, 346-348, https://doi.org/10.1038/jhg.2014.26.
Nadhan, R., Isidoro, C., Song, Y. S., and Dhanasekaran, D. N. (2022) Signaling by LncRNAs: structure, cellular homeostasis, and disease pathology, Cells, 11, 2517, https://doi.org/10.3390/cells11162517.
Jiang, J., Piao, X., Hu, S., Gao, J., and Bao, M. (2020) LncRNA H19 diminishes dopaminergic neuron loss by mediating microRNA-301b-3p in Parkinson’s disease via the HPRT1-mediated Wnt/β-catenin signaling pathway, Aging, 12, 8820-8836, https://doi.org/10.18632/aging.102877.
Abrishamdar, M., Jalali, M. S., and Rashno, M. (2022) MALAT1 lncRNA and Parkinson’s disease: the role in the pathophysiology and significance for diagnostic and therapeutic approaches, Mol. Neurobiol., 59, 5253-5262, https://doi.org/10.1007/s12035-022-02899-z.
Taghizadeh, E., Gheibihayat, S. M., Taheri, F., Afshani, S. M., Farahani, N., and Saberi, A. (2021) LncRNAs as putative biomarkers and therapeutic targets for Parkinson’s disease, Neurol. Sci., 42, 4007-4015, https://doi.org/10.1007/s10072-021-05408-7.
Simchovitz, A., Hanan, M., Niederhoffer, N., Madrer, N., Yayon, N., Bennett, E. R., Greenberg, D. S., Kadener, S., and Soreq, H. (2019) NEAT1 is overexpressed in Parkinson’s disease substantia nigra and confers drug-inducible neuroprotection from oxidative stress, FASEB J., 33, 11223-11234, https://doi.org/10.1096/fj.201900830R.
Li, Y., Gu, Z., Lin, S., Chen, L., Dzreyan, V., Eid, M., Demyanenko, S., and He, B. (2022) Histone deacetylases as epigenetic targets for treating Parkinson’s disease, Brain Sci., 12, 672, https://doi.org/10.3390/brainsci12050672.
Zhang, H., Yao, L., Zheng, Z., Koc, S., and Lu, G. (2022) The role of non-coding RNAs in the pathogenesis of Parkinson’s disease: recent advancement, Pharmaceuticals, 15, 811, https://doi.org/10.3390/ph15070811.
Helmy, M., Eldaydamony, E., Mekky, N., Elmogy, M., and Soliman, H. (2022) Predicting Parkinson’s disease related genes based on PyFeat and gradient boosted decision tree, Sci. Rep., 12, 10004, https://doi.org/10.1038/s41598-022-14127-8.
Farrow, S. L., Schierding, W., Gokuladhas, S., Golovina, E., Fadason, T., Cooper, A. A., and O’Sullivan, J. M. (2022) Establishing gene regulatory networks from Parkinson’s disease risk loci, Brain, 145, 2422-2435, https://doi.org/10.1093/brain/awac022.
Quinn, P. M. J., Moreira, P. I., Ambrósio, A. F., and Alves, C. H. (2020) PINK1/PARKIN signalling in neurodegeneration and neuroinflammation, Acta Neuropathol. Commun., 8, 189, https://doi.org/10.1186/s40478-020-01062-w.
Dolgacheva, L. P., Berezhnov, A. V., Fedotova, E. I., Zinchenko, V. P., and Abramov, A. Y. (2019) Role of DJ-1 in the mechanism of pathogenesis of Parkinson’s disease, J. Bioenerg. Biomembr., 51, 175-188, https://doi.org/10.1007/s10863-019-09798-4.
Usmani, A., Shavarebi, F., and Hiniker, A. (2021) The cell biology of LRRK2 in Parkinson’s disease, Mol. Cell. Biol., 41, 1-16, https://doi.org/10.1128/mcb.00660-20.
Tanaka, K. (2020) The PINK1-Parkin axis: an overview, Neurosci. Res., 159, 9-15, https://doi.org/10.1016/j.neures.2020.01.006.
Cohen, B. H., Chinnery, P. F., and Copeland, W. C. (1993) POLG-related disorders, in GeneReviews (Adam, M. P., Everman, D. B., Mirzaa, G. M., Pagon, R. A., Wallace, S. E., Bean, L. J. H., Gripp, K. W., and Amemiya, A., eds) University of Washington, Seattle.
Van Veen, S., Martin, S., Van den Haute, C., Benoy, V., Lyons, J., Vanhoutte, R., Kahler, J. P., Decuypere, J. P., Gelders, G., Lambie, E., Zielich, J., Swinnen, J. V., Annaert, W., Agostinis, P., Ghesquière, B., Verhelst, S., Baekelandt, V., Eggermont, J., and Vangheluwe, P. (2020) ATP13A2 deficiency disrupts lysosomal polyamine export, Nature, 578, 419-424, https://doi.org/10.1038/s41586-020-1968-7.
Joseph, S., Schulz, J. B., and Stegmüller, J. (2018) Mechanistic contributions of FBXO7 to Parkinson’s disease, J. Neurochem., 144, 118-127, https://doi.org/10.1111/jnc.14253.
Zhou, Z. D., Lee, J. C. T., and Tan, E. K. (2018) Pathophysiological mechanisms linking F-box only protein 7 (FBXO7) and Parkinson’s disease (PD), Mutat. Res. Rev. Mutat. Res., 778, 72-78, https://doi.org/10.1016/j.mrrev.2018.10.001.
Lin, G., Lee, P. T., Chen, K., Mao, D., Tan, K. L., Zuo, Z., Lin, W. W., Wang, L., and Bellen, H. J. (2018) Phospholipase PLA2G6, a Parkinsonism-associated gene, affects Vps26 and Vps35, retromer function, and ceramide levels, similar to α-synuclein gain, Cell Metab., 28, 605-618.e606, https://doi.org/10.1016/j.cmet.2018.05.019.
Sassone, J., Reale, C., Dati, G., Regoni, M., Pellecchia, M. T., and Garavaglia, B. (2021) The role of VPS35 in the pathobiology of Parkinson’s disease, Cell. Mol. Neurobiol., 41, 199-227, https://doi.org/10.1007/s10571-020-00849-8.
Roosen, D. A., Blauwendraat, C., Cookson, M. R., and Lewis, P. A. (2019) DNAJC proteins and pathways to parkinsonism, FEBS J., 286, 3080-3094, https://doi.org/10.1111/febs.14936.
Choudhry, H., Aggarwal, M., and Pan, P. Y. (2021) Mini-review: synaptojanin 1 and its implications in membrane trafficking, Neurosci. Lett., 765, 136288, https://doi.org/10.1016/j.neulet.2021.136288.
Monfrini, E., Spagnolo, F., Canesi, M., Seresini, A., Rini, A., Passarella, B., Percetti, M., Seia, M., Goldwurm, S., Cereda, V., Comi, G. P., Pezzoli, G., and Di Fonzo, A. (2022) VPS13C-associated Parkinson’s disease: two novel cases and review of the literature, Parkinsonism Rel. Disord., 94, 37-39, https://doi.org/10.1016/j.parkreldis.2021.11.031.
Chang, D., Nalls, M. A., Hallgrímsdóttir, I. B., Hunkapiller, J., van der Brug, M., Cai, F., Kerchner, G. A., Ayalon, G., Bingol, B., Sheng, M., Hinds, D., Behrens, T. W., Singleton, A. B., Bhangale, T. R., and Graham, R. R. (2017) A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci, Nat. Genet., 49, 1511-1516, https://doi.org/10.1038/ng.3955.
Nalls, M. A., Pankratz, N., Lill, C. M., Do, C. B., Hernandez, D. G., Saad, M., DeStefano, A. L., Kara, E., Bras, J., Sharma, M., Schulte, C., Keller, M. F., Arepalli, S., Letson, C., Edsall, C., Stefansson, H., Liu, X., Pliner, H., Lee, J. H., Cheng, R., Ikram, M. A., Ioannidis, J. P., Hadjigeorgiou, G. M., Bis, J. C., Martinez, M., Perlmutter, J. S., Goate, A., Marder, K., Fiske, B., Sutherland, M., Xiromerisiou, G., Myers, R. H., Clark, L. N., Stefansson, K., Hardy, J. A., Heutink, P., Chen, H., Wood, N. W., Houlden, H., Payami, H., Brice, A., Scott, W. K., Gasser, T., Bertram, L., Eriksson, N., Foroud, T., and Singleton, A. B. (2014) Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease, Nat. Genet., 46, 989-993, https://doi.org/10.1038/ng.3043.
Nalls, M. A., Plagnol, V., Hernandez, D. G., Sharma, M., Sheerin, U. M., Saad, M., Simón-Sánchez, J., Schulte, C., Lesage, S., Sveinbjörnsdóttir, S., Stefánsson, K., Martinez, M., Hardy, J., Heutink, P., Brice, A., Gasser, T., Singleton, A. B., and Wood, N. W. (2011) Imputation of sequence variants for identification of genetic risks for Parkinson’s disease: a meta-analysis of genome-wide association studies, Lancet, 377, 641-649, https://doi.org/10.1016/s0140-6736(10)62345-8.
Pickrell, J. K., Berisa, T., Liu, J. Z., Ségurel, L., Tung, J. Y., and Hinds, D. A. (2016) Detection and interpretation of shared genetic influences on 42 human traits, Nat. Genet., 48, 709-717, https://doi.org/10.1038/ng.3570.
Rodrigo, L. M., and Nyholt, D. R. (2021) Imputation and reanalysis of ExomeChip Data identifies novel, conditional and joint genetic effects on Parkinson’s disease risk, Genes, 12, 689, https://doi.org/10.3390/genes12050689.
Lill, C. M., Roehr, J. T., McQueen, M. B., Kavvoura, F. K., Bagade, S., Schjeide, B. M., Schjeide, L. M., Meissner, E., Zauft, U., Allen, N. C., Liu, T., Schilling, M., Anderson, K. J., Beecham, G., Berg, D., Biernacka, J. M., Brice, A., DeStefano, A. L., Do, C. B., Eriksson, N., Factor, S. A., Farrer, M. J., Foroud, T., Gasser, T., Hamza, T., Hardy, J. A., Heutink, P., Hill-Burns, E. M., Klein, C., Latourelle, J. C., Maraganore, D. M., Martin, E. R., Martinez, M., Myers, R. H., Nalls, M. A., Pankratz, N., Payami, H., Satake, W., Scott, W. K., Sharma, M., Singleton, A. B., Stefansson, K., Toda, T., Tung, J. Y., Vance, J., Wood, N. W., Zabetian, C. P., Young, P., Tanzi, R. E., Khoury, M. J., Zipp, F., Lehrach, H., Ioannidis, J. P., and Bertram, L. (2012) Comprehensive research synopsis and systematic meta-analyses in Parkinson’s disease genetics: the PDGene database, PLoS Genet., 8, e1002548, https://doi.org/10.1371/journal.pgen.1002548.
Do, C. B., Tung, J. Y., Dorfman, E., Kiefer, A. K., Drabant, E. M., Francke, U., Mountain, J. L., Goldman, S. M., Tanner, C. M., Langston, J. W., Wojcicki, A., and Eriksson, N. (2011) Web-based genome-wide association study identifies two novel loci and a substantial genetic component for Parkinson’s disease, PLoS Genet., 7, e1002141, https://doi.org/10.1371/journal.pgen.1002141.
Spencer, C. C., Plagnol, V., Strange, A., Gardner, M., Paisan-Ruiz, C., Band, G., Barker, R. A., Bellenguez, C., Bhatia, K., Blackburn, H., Blackwell, J. M., Bramon, E., Brown, M. A., Brown, M. A., Burn, D., Casas, J. P., Chinnery, P. F., Clarke, C. E., Corvin, A., Craddock, N., Deloukas, P., Edkins, S., Evans, J., Freeman, C., Gray, E., Hardy, J., Hudson, G., Hunt, S., Jankowski, J., Langford, C., Lees, A. J., Markus, H. S., Mathew, C. G., McCarthy, M. I., Morrison, K. E., Palmer, C. N., Pearson, J. P., Peltonen, L., Pirinen, M., Plomin, R., Potter, S., Rautanen, A., Sawcer, S. J., Su, Z., Trembath, R. C., Viswanathan, A. C., Williams, N. W., Morris, H. R., Donnelly, P., and Wood, N. W. (2011) Dissection of the genetics of Parkinson’s disease identifies an additional association 5′ of SNCA and multiple associated haplotypes at 17q21, Hum. Mol. Genet., 20, 345-353, https://doi.org/10.1093/hmg/ddq469.
Funding
The review was supported by the Russian Science Foundation (project no. 20-15-00262, M.I. Shadrina) and by the Ministry of Science and Higher Education of the Russian Federation (agreement no. 075-15-2021-1357, P.A. Slominsky).
Author information
Authors and Affiliations
Contributions
M.I. Shadrina – writing and editing the paper; P.A. Slominsky – concept and writing the paper.
Corresponding author
Ethics declarations
The authors declare no conflict of interest. This review does not describe any studies involving humans or animals as tested subjects.
Rights and permissions
Open access. This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit https://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shadrina, M.I., Slominsky, P.A. Genetic Architecture of Parkinson’s Disease. Biochemistry Moscow 88, 417–433 (2023). https://doi.org/10.1134/S0006297923030100
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0006297923030100