
Abstract
The effects of cardiomyopathic mutations E56G, M149V, and E177G in the MYL3 gene encoding essential light chain of human ventricular myosin (ELCv), on the functional properties of cardiac myosin and its isolated head (myosin subfragment 1, S1) were investigated. Only the M149V mutation upregulated the actin-activated ATPase activity of S1. All mutations significantly increased the Ca2+-sensitivity of the sliding velocity of thin filaments on the surface with immobilized myosin in the in vitro motility assay, while mutations E56G and M149V (but not E177G) reduced the sliding velocity of regulated thin filaments and F-actin filaments almost twice. Therefore, despite the fact that all studied mutations in ELCv are involved in the development of hypertrophic cardiomyopathy, the mechanisms of their influence on the actin–myosin interaction are different.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
The molecular mechanism of muscle contraction is based on the cyclic interaction of myosin heads with actin filaments coupled with ATP hydrolysis. The molecule of muscle myosin consists of two heavy and four light chains; the N-terminal regions of heavy chains form globular heads, each containing the actin-binding sites and the active site of myosin ATPase [1]. Isolated myosin head, called myosin subfragment 1 (S1), consists of two main structural domains known as the motor (or catalytic) and regulatory domains, respectively. The motor domain is responsible for ATP hydrolysis and actin binding, whereas the regulatory domain is a long α-helix stabilized by the noncovalent interactions with two light chains – essential light chain (ELC) and regulatory light chain (RLC) [2]. The functioning of the myosin head as a molecular motor involves the rotation of the regulatory domain relative to the motor domain, caused by global conformational changes occurring in the motor domain during the ATPase cycle, in which the regulatory domain acts as a lever arm [1, 3]. An important role in this process belongs to the ELC, which stabilizes the α-helix of the regulatory domain, with which it is associated through its C-terminal part. There are some evidences that when the regulatory domain is rotated during the ATPase cycle, the C-terminal part of the ELC is able to interact with the motor domain of the head [4-7], and its unique N-terminal part is able to interact with both the motor domain [8] and actin [9].
ELCs play an important role in the functioning of cardiac myosin. Changes in the expression of ELC isoforms affect the contractile function of the myocardium [10]. Disorders in the primary structure of ELC of the human heart ventricle (ventricular ELC, ELCv) caused by mutations in the MYL3 gene are associated with the development of hypertrophic cardiomyopathy (HCM), a severe hereditary heart disease characterized by thickening of the left ventricle walls, fibrosis and diastolic myocardial dysfunction, which can lead to the heart rhythm abnormalities up to sudden cardiac arrest. To date, 13 amino acid substitutions in human ELCv are known caused by mutations in the MYL3 gene that are associated with the HCM development: E56G, A57G, R63C, V79I, R81H, G128C, E143K, M149V, E152K, R154H, H155D, M173V, and E177G [11]. It is important to note that all these substitutions are located in the ELCv region that interacts with the α-helix of the heavy chain of the regulatory domain of the myosin head [11]. At the same time, no HCM-associated mutations have been detected in the 52-amino acid N-terminal region of the ELCv containing functionally important sites responsible for the interaction with actin [9, 11] (except E56G and A57G located close to the N-terminal segment of ELCv).
For most HCM-associated mutations in the MYL3 gene, almost nothing is known about their effect on the properties of cardiac myosin. The data from such experiments, although in small numbers, are available only for a few mutations (E56G, A57G, M149V, and M173V). For example, it has been shown that E56G mutation dramatically decreases the affinity of ELCv to its binding site in the myosin heavy chain of the myosin head regulatory domain [12] and reduces the rigidity of the myosin molecule, the isometric strength of muscle fibers, and the sliding velocity of actin filaments along myosin in the in vitro motility assay [13]. Interesting data were obtained by the time-resolved fluorescence energy transfer (TR-FRET) method which allows to quickly measure the distances between the myosin head and actin directly in the course of the actomyosin ATPase cycle during transitions of the myosin head from the weak to strong binding with actin and back. At saturating ATP concentrations, the E56G mutation in ELCv markedly shifted the equilibrium between the weak and strong binding states during the ATPase cycle toward strong binding without affecting the actin-activated ATPase activity of cardiac S1 and its affinity for actin. The authors suggested that this shift in the equilibrium may lead to the development of a phenotype with an increased heart contractility in people with the cardiomyopathic E56G mutation in the MYL3 gene [14].
Interesting data were also obtained when comparing the effects caused by the A57G and M173V mutations in the myosin ELCv in transgenic mice. It has been shown that the A57G mutation increased the Ca2+-sensitivity of the muscle fiber strength development much more than M173V. The M173V mutation significantly reduced the actin-activated ATPase activity of cardiac myosin, whereas A57G had no effect on it [15]. Finally, we have to mention the properties of cardiac myosin with the M149V mutation in ELCv, which was the first to be identified among all the above mutations as one leading to the HCM development [16]. In the elegant experiments conducted using plasmon resonance, this mutation was shown to significantly reduce the affinity of ELCv to the recombinant fragment of the cardiac myosin heavy chain containing the IQ motif for the ELCv binding [12]. In addition, it was found that the M149V mutation in ELCv increased the sliding velocity of actin filaments on the surface with immobilized cardiac myosin in the in vitro motility assay [16].
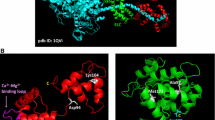
In this paper, we compared the properties of cardiac myosin and its isolated head (S1) with cardiomyopathic mutations E56G, M149V, or E177G in ELCv. The choice of these mutations was not accidental. Firstly, the substituted residues are highly conserved and located in different regions of the ELCv sequence: Glu56 (E56) is in the N-terminal part of the protein, near the unique N-terminal sequence of ELCv, while Met149 (M149) and Glu177 (E177) are in the C-terminal part (Fig. 1a). In the three-dimensional structure, M149 and E177 are located far from E56, in different parts of the ELCv molecule located at different sides of the α-helix of the heavy chain of the myosin head regulatory domain (Fig. 1b). Secondly, we tried to compare mutations, the effect of which on the properties of myosin has been studied to a different extent. Thus, the E56G mutation is one of the most well studied [9, 11-14], somewhat less is known about the functional effects of the M149V mutation [11, 12, 16], and almost nothing is known about the E177G mutation except that it was detected in a 3-month-old infant with severe progressive HCM, which led to the infant’s death at the age of 6 months [17].

Positions of cardiomyopathic mutations E56G, M149V, and E177G in the amino acid sequence (a) and spatial structure (shown in light gray) of ELCv associated with the α-helix of the myosin heavy chain (myosin heavy chain fragment including residues 773-810 is shown in dark gray) in the myosin head regulatory domain (PDB entry 5TBY) (b). Side chains of residues E56, M149, and E177 in panel (b) are shown in black; flexible N-terminal segment of ELCv, including 52 N-terminal residues is shaded in Fig. 1a and not shown in Fig. 1b.
MATERIALS AND METHODS
Protein preparations. ELCv preparations used in this work were recombinant proteins, products of the human MYL3 gene (UniProtKB P08590; MYL3_HUMAN), containing a 6-mer His-tag at the N-terminus for their purification by affinity chromatography. For this, the sequences encoding the His-tag and the proteolytic site for its subsequent removal by Factor Xa protease were added to the MYL3 nucleotide sequence. The modified MYL3 gene was cloned into the pET-9a vector (Merck, Sigma-Aldrich, Germany) at the NdeI and BamHI sites. Genetic constructs with the E56G, M149V, and E177G mutations were obtained by site-directed mutagenesis using the wild-type (WT) MYL3 cDNA and overlapping forward and reverse primers (Table 1). The primers were synthesized by Evrogen (Moscow, Russia).
The obtained constructs were used for bacterial expression of recombinant proteins (ELCv WT, ELCv E56G, ELCv M149V, and ELCv E177G) in E. coli BL21(DE3) cells. The proteins were purified by affinity chromatography on a 5-ml HisTrap HP column (GE Healthcare, USA). The concentration of ELCv preparations was determined spectrophotometrically using the extinction coefficient A1% = 0.2 cm–1 at 280 nm.
Cardiac myosin and troponin (Tn) were obtained by standard methods [18, 19] from the pig heart left ventricle. Recombinant tropomyosin (Tpm, cardiac isoform of Tpm 1.1) was prepared as described in detail earlier [20, 21]. Actin was obtained by the standard method from rabbit skeletal muscles [22]. Actin filaments (F-actin) were polymerized by adding 4 mM MgCl2 and 100 mM KCl to a solution of monomeric G-actin. For the in vitro motility assay, F-actin was fluorescently labeled by adding a twofold molar excess of TRITC-phalloidin (Sigma-Aldrich, USA). Isolated myosin heads (S1) were obtained by digesting cardiac myosin filaments with TLCK-treated α-chymotrypsin (Sigma-Aldrich) [23]. The concentration of S1 was determined spectrophotometrically using the extinction coefficient A1% = 7.5 cm–1 at 280 nm.
Replacement of native ELCs in cardiac myosin and S1 with recombinant ELCv. The reassociation technique, i.e., replacement of ELCs in the isolated myosin head (S1), has been repeatedly used by us earlier in the studies on S1 preparations obtained from the rabbit skeletal muscle myosin [7, 8]. We used the same technique to replace native ELCs with recombinant ELCs in the cardiac myosin S1. In short, S1 preparations were incubated for 30 min at 37°C with an 8-fold molar excess of recombinant ELCv in 50 mM imidazole-HCl (pH 7.0) containing 5 mM DTT, 10 mM MgATP, and 100 mM NaCl. The reaction was stopped by cooling on ice, after which the S1 preparation was purified by chromatography on a SP-Trisacryl column to separate S1 from the excess of free ELCv. However, this technique had to be significantly modified to substitute native ELCs with recombinant ELCs in the entire molecule of cardiac myosin, which is insoluble at low ionic strength. For this, cardiac myosin (2 mg/ml, 4 µM) was mixed with a 10-fold molar excess of recombinant ELCv (40 µM) in 50 mM imidazole-HCl buffer (pH 7.0) containing 5 mM DTT, 10 mM MgATP, and 0.7 M KCl, and incubated for 1 h at 37°C. Next, the mixture of myosin with ELCs was dialyzed against 50 mM Tris-HCl (pH 8.0) for the myosin aggregation and then centrifuged at 8000g for 10 min at 4°C, after which the myosin precipitate was resuspended and dialyzed against the same buffer containing 1 M NaCl.
The last stage of myosin or S1 purification was metal-affinity chromatography on a 1-ml His-Trap HP column (GE Healthcare) that retained only myosin heads containing the His-tag-modified ELCv. Chromatography was performed in 50 mM Tris-HCl buffer (pH 8.0) containing 300 mM NaCl (in the case of S1) or 1 M NaCl (for whole myosin) using a linear imidazole concentration gradient (15 mM to 500 mM). It is important to note that myosin molecules in which native ELCs were substituted with the recombinant ELCs in both heads were eluted from the column at higher imidazole concentrations than molecules containing recombinant ELCs in only one of the two heads. For further experiments, we collected only myosin fractions in which their own ELCs were completely replaced in both heads of the molecule by recombinant ELCv containing the N-terminal His-tag whose electrophoretic mobility was lower than that of native unmodified ELCs. The N-terminal His-tag was subsequently removed from the ELCv by treatment with Factor Xa protease (New England Biolabs, UK) at 4°C overnight; the reaction was stopped by the addition of PMSF. As a result, in the obtained cardiac myosin and S1 preparations, the native ELCs were completely replaced by the recombinant ELCs (either ELCv WT or ELCv with mutations E56G, M149V, or E177G).
In vitro motility assay allows to study the sliding velocity of F-actin or reconstructed thin filaments (i.e., actin filaments containing Tpm and Tn) on a glass surface with immobilized myosin, and in the case of reconstructed thin filaments, to determine the dependence of the sliding velocity on the concentration of calcium ions. Previously, we have repeatedly used this approach to study the functional properties of Tpm with cardiomyopathic mutations in various regions of the molecule [20, 21]. In this work, we used this method to study the properties of myosin containing recombinant ELCs in both heads, including ELCs with cardiomyopathic mutations E56G, M149V, or E177G. The measurements were carried out at 30°C as described earlier [20, 21]. In short, cardiac myosin (300 µg/ml) with recombinant ELCs was loaded into an experimental flow cell with the nitrocellulose-coated inner surface. Regulated thin filaments were obtained by adding cardiac Tpm and Tn at the final concentrations of 100 nM to a solution of 10 nM F-actin labeled with TRITC-phalloidin and placed in a flow cell with immobilized myosin. The sliding velocity of filaments was measured in the presence of 2 mM ATP using the GMimPro program [24] at various Ca2+ ion concentrations. The sliding velocity of F-actin was measured under the same conditions, but in the absence of Tpm, Tn, and Ca2+ ions. Experiments with myosin containing recombinant ELCs in both heads (either WT ELCv or ELCv with E56G, M149V, or E177G mutations) were repeated three times (each time with newly prepared myosin); the obtained values of the sliding velocity of thin filaments or F-actin were expressed as mean ±S.D. The calcium dependence of the sliding velocity of thin filaments was approximated by the Hill equation as described earlier [20, 25].
Measurements of ATPase activity. The concentration of myosin containing recombinant ELCs and its amount after reassociation and following purification were too low, which prevented us from using the standard method for determining myosin ATPase activity by measuring the amount of released inorganic phosphate, as it requires significantly higher protein concentrations. Therefore, to determine the ATPase activity, we used a much more sensitive technique that allowed us to measure with a high accuracy the decrease in the amount of ATP in the sample directly during the ATPase reaction from the reduction in the luminescence intensity measured using the luciferin–luciferase system. In our experiments, we used a luciferase/luciferin commercial kit (Sigma-Aldrich, #L-0633) for detecting picomolar concentrations of ATP in the samples. However, even this kit was not sensitive enough to measure the ATPase activity of the whole myosin with recombinant ELCs. Therefore, all experiments on the study of actin-activated Mg2+-dependent ATPase activity were carried out on the preparations of isolated cardiac myosin heads (S1) containing recombinant ELCs. The reaction mixture contained 5 mM imidazole (pH 7.0), 10 mM MgCl2, 87 nM S1, and 4.7 µM F-actin. The ATPase reaction was initiated by adding 10 mM ATP. Since luciferase itself is capable of hydrolyzing ATP (albeit at a low rate) and thus to affect the reaction kinetics, we used the time of complete luminescence quenching as the main parameter of the reaction rate. The reduction in the luminescence intensity was measured at 560 nm at 20°C with a PerkinElmer Multimode Plate Reader (PerkinElmer, USA).
RESULTS AND DISCUSSION
The functional properties of myosin containing recombinant ELCs in both heads were studied using the in vitro motility assay. First, we measured the sliding velocity of actin filaments (F-actin without Tpm and Tn) on the surface with immobilized myosin and found that the E177G mutation in ELCv had no effect on the sliding velocity of F-actin, whereas the E56G and M149V mutations reduced it almost twofold (Table 2). In the case of E56G mutation, our data correlate with the earlier published report showing that this mutation in ELCv noticeably (by ~25%) reduced the sliding velocity of F-actin on the surface with immobilized myosin [13]. However, in the case of M149V mutation in ELCv, Poetter et al. demonstrated that this mutation increased by almost 40% (and not reduced) the sliding velocity of F-actin on the surface with immobilized myosin [16]. Such discrepancies in the results can be explained by different methods employed for the preparation of myosin samples: in our experiments, the myosin samples were obtained from the pig heart and differed only in the presence of substituted recombinant ELCv, whereas Poetter et al. [16] used cardiac myosin isolated from the biopsies of patients with and without the M149V mutation in ELCv. The myosin preparations used in our experiments and studies by Potter et al. [16] not only had different composition of the cardiac myosin heavy chains, but could also differ in other parameters (such as, for example, the extent of ELCv phosphorylation in the myosin obtained from human biopsies vs. fully dephosphorylated recombinant ELCv).
We used the in vitro motility assay not only to investigate the effect of mutations in the cardiac myosin ELCv on the sliding velocity of F-actin, which has been done before [13, 16]. We were also the first to study the effect of these mutations on the sliding of reconstructed thin filaments (actin filaments containing Tpm and Tn) and to describe the dependence of their sliding velocity on the concentration of calcium ions, i.e., to reveal the influence of the studied mutations on the calcium regulation of actin–myosin interaction, which is a possible mechanism of the HCM development. The results of these studies are presented in Fig. 2 and Table 2. It should be noted that the maximum sliding velocity of thin filaments (Vmax), measured at high Ca2+ concentrations (at pCa 4.5) did not differ noticeably from the sliding velocity of F-actin in the absence of regulatory proteins (Table 2). At the same time, all mutations in the ELCv markedly increased the Ca2+-sensitivity of the sliding velocity of thin filaments, shifting the curves of the velocity dependence on pCa towards lower Ca2+ concentrations (Fig. 2) and thus reducing the concentration of Ca2+, at which the half-maximal sliding velocity (pCa50) was achieved. The most significant increase in the Ca2+-sensitivity was observed for the ELCv with the E177G mutation (Table 2). It is important to note that an increase in the Ca2+-sensitivity of the myosin interaction with actin is one of the characteristic features of HCM.

The effect of cardiomyopathic mutations E56G, M149V, and E177G in the myosin ELCv on the calcium dependence of the sliding velocity of regulated thin filaments on cardiac myosin in the in vitro motility assay. The experimental values of the sliding velocity (mean ±SD) were approximated by the Hill equation. The values of the Hill equation parameters are presented in Table 2.
We also investigated the effect of cardiomyopathic mutations E56G, M149V, and E177G in myosin ELCv on the ATPase activity of actomyosin. For the reasons described above (see “Materials and methods” section), we could not use traditional methods of measuring ATPase activity or use whole-myosin preparations with mutations in the ELCv in both heads of the molecule. Therefore, we used a technique that allowed us to measure the decrease in the amount of ATP in the sample directly in the course of the S1-catalyzed ATPase reaction from a decrease in the luminescence intensity measured using the luciferin–luciferase system. The results of the experiments indicated that among the studied mutations, only M149V significantly increased the ATPase activity of acto-S1, thus reducing almost twice the time required for complete luminescence quenching (15.3 ± 1.9 min for S1 with mutation M149V in ELCv vs. 26.0 ± 1.2 min for S1 with WT ELCv; data from seven independent measurements are presented as mean ±SD). Two other mutations, E56G and E177G, had no significant effect on the ATPase activity of acto-S1; with regard to the time required for complete luminescence quenching (26.5 ± 2.3 min), S1 preparations with these mutations in ELCv did not noticeably differ from S1 preparations with WT ELCv. In the case of the E56G, these results were consistent with the earlier published data indicating that this mutation in ELCv does not affect the actin-activated ATPase activity of S1 [14].
The differences between the effects caused by the E56G and M149V depended on the assay used. Thus, both mutations similarly reduced the sliding velocity of F-actin and thin filaments in the in vitro motility assay (Fig. 2, Table 2), whereas only M149V (but not E56G) increased the ATPase activity of acto-S1. This discrepancy can be explained by the fact that the in vitro motility assay was carried out with the whole myosin with mutations in ELCv molecules associated with both myosin heads, while the ATPase activity was measured in isolated myosin heads (S1). At the same time, there is evidence that the properties of whole myosin and S1 are quite different. This is due, firstly, to the cooperative interaction between the two heads in the myosin molecule [26-28], and secondly, to the possible interaction between the ELC and RLC in the heads of the whole myosin [29] (but not in S1, which does not contain the RLC).
It should be particularly noted that the E56G and M149V mutations in ELCv decreased twice the sliding velocity of F-actin and thin filaments in the in vitro motility assay (Fig. 2, Table 2). This effect was most likely due to the fact that both mutations reduce the affinity of ELCv to the myosin heavy chain in the regulatory domain of the myosin head [12], thus disrupting normal functioning of this domain, which is important for the operation of the head as a molecular motor. Interestingly, no such effect was found for the E177G mutation (Fig. 2, Table 2), the effect of which on the properties of cardiac myosin has not been studied before. It is possible that the influence of this mutation on the HCM development is due not to the decrease in the rate of interaction of myosin with actin in the cardiac muscle, but to a significant increase in the Ca2+-sensitivity of this interaction (Table 2). It is also worth mentioning that the amino acid residue 177 (E177G mutation) is located in an unordered mobile loop of the ELCv structure, whereas amino acid residues 56 and 149 (mutations E56G and M149V) are located in the α-helix and in a short loop connecting the β-sheet and the α-helix, respectively [11] (see Fig. 1b). This can explain, at least in part, the fact that the E177G mutation does not affect the rate of myosin interaction with actin, while increasing its Ca2+ sensitivity, which is determined by the presence of regulatory proteins, Tpm and Tn, in the reconstructed thin filaments.
CONCLUSIONS
Summing up, we were able to obtain new information on the effect of cardiomyopathic mutations E56G, M149V, and E177G in the MYL3 gene encoding ELCv on the functional properties of cardiac myosin and its isolated heads (S1). In particular, comparative analysis of the effects caused by these mutations showed that the E177G substitution, whose influence on the myosin properties has not been studied before, differed markedly from the other two mutations, E56G and M149V, in its effect on the sliding velocity of F-actin or reconstructed thin filaments in the in vitro motility assay. The E177G mutation had no effect on the sliding velocity of F-actin and on the maximum sliding velocity of thin filaments at a high concentration of calcium ions, whereas mutations E56G and M149V reduced it almost twofold. On the other hand, the E177G mutation increased the Ca2+-sensitivity of the sliding velocity of thin filaments more noticeably than the E56G and M149V mutations. These data suggest that the mechanism of the HCM development caused by the E177G mutation in ELCv differs from those resulting from the cardiomyopathic mutations E56G and M149V.
Abbreviations
- ELC:
-
myosin essential light chain
- ELCv:
-
ventricular ELC
- HCM:
-
hypertrophic cardiomyopathy
- S1:
-
myosin subfragment 1
- Tn:
-
troponin
- Tpm:
-
tropomyosin
References
Levitsky, D. I. (2004) Actomyosin systems of biological motility, Biochemistry (Moscow), 69, 1177-1189, https://doi.org/10.1007/s10541-005-0063-x.
Rayment, I., Rypniewski, W., Schmidt-Base, K., Smith, R., Tomchick, D., et al. (1993) Three-dimentional structure of myosin subfragment 1: a molecular motor, Science, 261, 50-58, https://doi.org/10.1126/science.8316857.
Rayment, I. (1996) The structural basis of the myosin ATPase activity, J. Biol. Chem., 271, 15850-15853, https://doi.org/10.1074/jbc.271.27.15850.
Milligan, R. A. (1996) Protein–protein interactions in the rigor actomyosin complex, Proc. Natl. Acad. Sci. USA, 93, 21-26, https://doi.org/10.1073/pnas.93.1.21.
Dominguez, R., Freyzon, Y., Trybus, K. M., and Cohen, C. (1998) Crystal structure of a vertebrate smooth muscle myosin motor domain and its complex with the essential light chain: visualization of the pre-power stroke state, Cell, 94, 559-571, https://doi.org/10.1016/s0092-8674(00)81598-6.
Borejdo, J., Ushakov, D. S., Moreland, R., Akopova, I., Reshetnyak, Y., et al. (2001) The power stroke causes changes in the orientation and mobility of the termini of essential light chain 1 of myosin, Biochemistry, 40, 3796-3803, https://doi.org/10.1021/bi002527u.
Logvinova, D. S., Markov, D. I., Nikolaeva, O. P., Sluchanko, N. N., Ushakov, D. S., et al. (2015) Does interaction between the motor and regulatory domains of the myosin head occur during ATPase cycle? Evidence from thermal unfolding studies on myosin subfragment 1, PLoS One, 10, e0137517, https://doi.org/10.1371/journal.pone.0137517.
Logvinova, D. S., Matyushenko, A. M., Nikolaeva, O. P., and Levitsky, D. I. (2018) Transient interaction between the N-terminal extension of the essential light chain-1 and motor domain of the myosin head during the ATPase cycle, Biochem. Biophys. Res.Commun., 495, 163-167, https://doi.org/10.1016/j.bbrc.2017.10.172.
Logvinova, D. S., and Levitsky, D. I. (2018) Essential light chains of myosin and their role in functioning of the myosin motor, Biochemistry (Moscow), 83, 944-960, https://doi.org/10.1134/S0006297918080060.
Schaub, M.C., Hefti, M. A., Zuellig, R. A., and Morano, I. (1998) Modulation of contractility in human cardiac hypertrophy by myosin essential light chain isoforms, Cardiovasc. Res., 37, 381-404, https://doi.org/10.1016/s0008-6363(97)00258-7.
Yadav, S., Sitbon, Y. H., Kazmierczak, K., and Szczesna-Cordary, D. (2019) Hereditary heart disease: pathophysiology, clinical presentation, and animal models of HCM, RCM, and DCM associated with mutations in cardiac myosin light chains, Pflügers Arch. Eur. J. Physiol., 471, 683-699, https://doi.org/10.1007/s00424-019-02257-4.
Lossie, J., Ushakov, D. S., Ferenczi, M. A., Werner, S., Keller, S., et al. (2012) Mutations of ventricular essential myosin light chain disturb myosin binding and sarcomeric sorting, Cardiovasc. Res., 93, 390-396, https://doi.org/10.1093/cvr/cvr320.
Lossie, J., Köhncke, C., Mahmoodzadeh, S., Steffen, W., Canepari, M., et al. (2014) Molecular mechanism regulating myosin and cardiac functions by ELC, Biochem. Biophys. Res. Commun., 450, 464-469, https://doi.org/10.1016/j.bbrc.2014.05.142.
Guhathakurta, P., Prochniewicz, E., Roopnarine, O., Rohde, J. A., and Thomas, D. D. (2017) A cardiomyopathy mutation in the myosin essential light chain alters actomyosin structure, Biophys. J., 113, 91-100, https://doi.org/10.1016/j.bpj.2017.05.027.
Huang, W., and Szczesna-Cordary, D. (2015) Molecular mechanisms of cardiomyopathy phenotypes associated with myosin light chain mutations, J. Muscle Res. Cell Motil., 36, 433-445, https://doi.org/10.1007/s10974-015-9423-3.
Poetter, K., Jiang, H., Hassanzadeh, S., Master, S. R., Chang, A., et al. (1996) Mutations in either the essential or regulatory light chains of myosin are associated with a rare myopathy in human heart and skeletal muscle, Nat. Genet., 13, 63-69, https://doi.org/10.1038/ng0596-63.
Jay, A., Chikarmane, R., Poulik, J., and Misra, V. K. (2013) Infantile hypertrophic cardiomyopathy associated with a novel MYL3 mutation, Cardiology, 124, 248-251, https://doi.org/10.1159/000347138.
Margossian, S. S., and Lowey, S. (1982) Preparation of myosin and its subfragments from rabbit skeletal muscle, Methods Enzymol., 85, 55-71, https://doi.org/10.1016/0076-6879(82)85009-x.
Potter, J. D. (1982) Preparation of troponin and its subunits, Methods Enzymol., 85 (Part B), 241-263, https://doi.org/10.1016/0076-6879(82)85024-6.
Matyushenko, A. M., Shchepkin, D. V., Kopylova, G. V., Popruga, K. E., Artemova, N. V., et al. (2017) Structural and functional effects of cardiomyopathy-causing mutations in the troponin T-binding region of cardiac tropomyosin, Biochemistry, 56, 250-259, https://doi.org/10.1021/acs.biochem.6b00994.
Matyushenko, A. M., Koubassova, N. A., Shchepkin, D. V., Kopylova, G. V., Nabiev, S. R., et al. (2019) The effects of cardiomyopathy-associated mutations in the head-to-tail overlap junction of α-tropomyosin on its properties and interaction with actin, Int. J. Biol. Macromol., 125, 1266-1274, https://doi.org/10.1016/j.ijbiomac.2018.09.105.
Pardee, J. D., and Spudich, J. A. (1982) Purification of muscle actin, Methods Enzymol., 85, 164-181, https://doi.org/10.1016/0076-6879(82)85020-9.
Weeds, A. G., and Taylor, R. S. (1975) Separation of subfragment-1 isoenzymes from rabbit skeletal muscle myosin, Nature, 257, 54-56, https://doi.org/10.1038/257054a0.
Mashanov, G. I., and Molloy, J. E. (2007) Automatic detection of single fluorophores in live cells, Biophys. J., 92, 2199-2211, https://doi.org/10.1529/biophysj.106.081117.
Matyushenko, A. M., Artemova, N. V., Shchepkin, D. V., Kopylova, G. V., Bershitsky, S. Y., et al. (2014) Structural and functional effects of two stabilizing substitutions, D137L and G126R, in the middle part of α-tropomyosin molecule, FEBS J., 281, 2004-2016, https://doi.org/10.1111/febs.12756.
Schaub, M. C., Watterson, J. G., and Waser, P. G. (1977) Evidence for head-head interactions in myosin from cardiac and skeletal muscles, Basic Res. Cardiol., 72, 124-132, https://doi.org/10.1007/BF01906350.
Tyska, M. J., Dupuis, D. E., Guilford, W. H., Patlak, J. B., Waller, G. S., et al. (1999) Two heads of myosin are better than one for generating force and motion, Proc. Natl. Acad. Sci. USA, 96, 4402-4407, https://doi.org/10.1073/pnas.96.8.4402.
Albet-Torres, N., Bloemink, M. J., Barman, T., Candau, R., Froölander, K., et al. (2009) Drug effect unveils inter-head cooperativity and strain-dependent ADP release in fast skeletal actomyosin, J. Biol. Chem., 284, 22926-22937, https://doi.org/10.1074/jbc.M109.019232.
Houdusse, A., and Cohen, C. (1996) Structure of the regulatory domain of scallop myosin at 2 Å resolution: implications for regulation, Structure, 4, 21-32, https://doi.org/10.1016/s0969-2126(96)00006-8.
Acknowledgments
The authors express their deep gratitude to Valentina Yurievna Berg for her help in conducting experiments on the in vitro motility assay.
Funding
The work was supported by the Russian Science Foundation (project no. 22-14-00059 to D.I.L.).
Author information
Authors and Affiliations
Contributions
D. S. Yampolskaya, A. M. Matyushenko, and D. I. Levitsky – developed the concept and supervised the study; G. V. Kopylova, D. V. Shchepkin, and S. Y. Bershitsky – conducted in vitro motility assay; D. S. Yampolskaya – conducted measurements of the ATPase activity; D. S. Yampolskaya and D. I. Levitsky – wrote the text of the article. All the authors took part in the discussion of the research results and editing of the final version of the manuscript.
Corresponding author
Ethics declarations
The authors declare no conflict of interest. This article does not describe any studies involving humans or animals performed by any of the authors.
Rights and permissions
Open access. This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit https://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yampolskaya, D.S., Kopylova, G.V., Shchepkin, D.V. et al. Properties of Cardiac Myosin with Cardiomyopathic Mutations in Essential Light Chains. Biochemistry Moscow 87, 1260–1267 (2022). https://doi.org/10.1134/S0006297922110050
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0006297922110050