
Abstract
Cigarette smoking is a leading cause of preventable death throughout the world. Nicotine, the primary addictive compound in tobacco, plays a vital role in the initiation and maintenance of its use. Nicotine exerts its pharmacological roles through nicotinic acetylcholine receptors (nAChRs), which are ligand-gated ion channels consisting of five membrane-spanning subunits. Besides the CHRNA4, CHRNB2 and CHRNA5/A3/B4 cluster on chromosome 15, which has been investigated intensively, recent evidence from both genome-wide association studies and candidate gene-based association studies has revealed the crucial roles of the CHRNB3–CHRNA6 gene cluster on chromosome 8 in nicotine dependence (ND). These studies demonstrate two distinct loci within this region. The first one is tagged by rs13277254, upstream of the CHRNB3 gene, and the other is tagged by rs4952, a coding single nucleotide polymorphism in exon 5 of that gene. Functional studies by genetic manipulation in mice have shown that α6*-nAChRs, located in the ventral tegmental area (VTA), are of great importance in controlling nicotine self-administration. However, when the α6 subunit is selectively re-expressed in the VTA of the α6−/− mouse by a lentiviral vector, the reinforcing property of nicotine is restored. To further determine the role of α6*-nAChRs in the process of nicotine-induced reward and withdrawal, genetic knock-in strains have been examined, which showed that replacement of Leu with Ser in the 9′ residue in the M2 domain of α6 produces nicotine-hypersensitive mice (α6 L9′S) with enhanced dopamine release. Moreover, nicotine-induced upregulation may be another ingredient in the pathology of nicotine addiction although the effect of chronic nicotine exposure on the expression of α6-containing receptors is controversial. To gain a better understanding of the pathological processes underlying ND and ND-related behaviors and to promote the development of effective smoking cessation therapies, we here present the most recent studies concerning the genetic effects of the CHRNB3–CHRNA6 gene cluster in ND.
Similar content being viewed by others


Introduction
Cigarette smoking is one of the leading causes of preventable morbidity and death worldwide,1 being responsible for approximately five million deaths annually, mostly caused by smoking-related cancers and cardiovascular and respiratory diseases.2 Because of the continuous effort of legislation against tobacco smoking and public realization of its health consequences, the prevalence of cigarette smoking is lower than in the past but has been relatively stable since the mid-1990 s;3 however, the smoking cessation rate still is relatively low. In contrast to developed countries, the prevalence of smoking is still increasing in developing countries,4 further highlighting the urgency of developing new medicines for smoking cessation.
Tobacco use, including smoking initiation (SI) and nicotine dependence (ND), is a complex and multifactorial behavior determined by both genetic and environmental factors, as well as their interactions.5 Findings from twin studies have clearly demonstrated that genetics contributes to various smoking-related behaviors.6, 7, 8 After reviewing the reported genetic epidemiological studies on smoking behaviors, Sullivan and Kendler9 concluded that the heritability of SI and ND was 0.56 and 0.67, respectively, and Li et al.10 estimated the mean heritability of ND to be 0.59 in male smokers and 0.46 in female smokers.
There are >4000 ingredients in cigarette smoke,11 but the pharmacological effects of dependence are produced primarily by nicotine, which exerts its physiological roles through neuronal nicotinic acetylcholine receptors (nAChRs).12 The nAChRs, widely distributed in the central and peripheral nervous systems, are ligand-gated ion channels consisting of five membrane-spanning subunits13 that can modulate the release of neurotransmitters14 and mediate fast signal transmission at synapses.15 Binding of nicotine to nAChRs forms the molecular basis for the reward of nicotine and, eventually, the development of ND.
Different approaches have been employed, such as genetic, pharmacologic, and in vitro or in vivo functional studies, to link ND to one or more specific nAChR subunits.16 Because of the wide distribution of α4β2* (where ‘*’ indicates additional subunits) nicotinic receptors in the brain and their high affinity for nicotine, a large body of research has focused primarily on these subunits.17 Recently, several genetic variants located in nAChR subunit encoding genes other than CHRNA4 or CHRNB2 were detected by genome-wide association studies (GWAS)18 and various candidate gene-based association and functional studies.19, 20, 21 For example, the most compelling SNP rs16969968 in CHRNA5,22 leading to an amino acid change in the position 398 (D398N) of the α5 subunit protein, has been consistently demonstrated to be a major biological contributor to ND.20, 23 For details on this part of the research, please refer to recent reviews.22, 24, 25, 26 It is thus believed that additional subunits or receptor subtypes are involved in the determination of different ND behaviors.
In this report, we review the evidence of genetic association between variants in the CHRNB3–CHRNA6 gene cluster on chromosome 8 and ND or ND-related phenotypes. Further, we include the functional studies of α6 and β3 nAChR subunits in the etiologies of ND using genetically engineered knock out (KO) and knock-in (KI) mice.
GWAS of the CHRNB3–CHRNA6 gene cluster and ND
GWAS is an effective approach when one does not have prior knowledge of gene function(s) and pathological process of the disease of interest as a means to identify common susceptibility genetic variants for ND or other complex phenotypes of interest. Bierut et al.27 reported the first high-density association study on ND with the aim of identifying common genetic variants that contribute to the transition from occasional cigarette smoking to ND. The sample in this report consisted of 1050 heavy smokers, with a Fagerström Test for Nicotine Dependence (FTND) score of >4.0, and 879 light smokers, who showed no symptoms of ND. Among 2.4 million single nucleotide polymorphisms (SNPs) examined, multiple risk SNPs in the CHRNB3–CHRNA6 cluster were identified, with the most compelling evidence for rs13277254 in CHRNB3 (P=6.54 × 10−5). In addition, another SNP, rs6474413, in complete linkage disequilibrium with rs13277254 (r2=1) in the same gene, was identified, with a P-value of 9.36 × 10−5. These nominal associations (according to the current genome-wide significant threshold of 10−8) were subsequently replicated in a GWAS meta-analysis21 using the number of cigarettes smoked per day (CPD) to measure ND. Furthermore, Rice et al.21, 28 reported that CHRNB3 was more strongly associated with FTND than with CPD, pointing out the importance of selecting an appropriate phenotype for association analysis. These authors carried out an independent GWAS with 1294 ND cases (defined by FTND score) and 2071 non-ND controls who had smoked at least 1 cigarette, revealing that the genetic locus most strongly associated with ND was rs1451240 in CHRNB3 (odds ratio (OR) 0.65; P=2.4 × 10−8). This association was strengthened when combined in a subsequent meta-analysis with a previously published dataset19 (combined P=6.7 × 10−16; total N=4200). However, when CPD was used as an alternate phenotype, the association no longer reached genome-wide significance (P=0.0007). This result highlights that phenotype selection is important in genetic association study of ND. Although CPD is the most commonly used phenotype of smoking because of its easy measurement, available evidence supports the view that CPD is related to culture and ethnicity.29 Contrarily, the FTND score appeared to be a relatively invariant measure of ND.
Candidate gene-based association studies of the CHRNB3–CHRNA6 gene cluster with ND
Besides the latest application of GWAS, significant efforts have been made to identify susceptibility loci for ND and ND-related phenotypes through a candidate gene approach with both case–control and family-based designs. Because a set of common, highly correlated variants (r2=1) tagged by rs6474413 and rs13277254 in the CHRNB3–CHRNA6 gene cluster has been associated with ND at genome-wide significance,21, 27 more attention has been paid to this region. So far, many candidate gene-based association studies have implicated various SNPs in the CHRNB3–CHRNA6 cluster as having a significant effect on ND and ND-related phenotypes in multiple ethnic populations (Tables 1 and 2).
Nicotine dependence
After analyzing 3713 SNPs in >300 candidate genes for their association with ND, Saccone et al.19 reported that rs6474413 (P=9.36 × 10−5) and rs10958726 (P=1.33 × 10−4) in CHRNB3 are significantly associated with ND. Both SNPs are located in the putative 5′ promoter region of the gene, with rs6474413 being 2 kb away from the start codon and 15 kb from rs10958726. Because of the high linkage disequilibrium between the two SNPs, they may contribute to a single association signal. Using a sample of 1050 ND cases and 879 non-ND controls of European descent, the same population as used in the study of Saccone et al.,19 another study30 revealed a significant locus, tagged by rs13277254 at the 5′ end of CHRNB3–CHRNA6, that influences the transition from smoking to ND. This finding was replicated in the follow-up study,38 which considered peer smoking as a social environmental risk factor for smoking behavior.
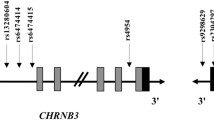
On the basis of the previous association results of a high-density study covering the complete family of 16 CHRN genes in a population of European ancestry, Saccone et al.30 extended their research to determine whether variants in the CHRNB3–CHRNA6 gene cluster also are associated with ND in African-Americans (AAs).33 They did not detect any associated SNP in their AA sample with a sample size of 710. It was suggested that there might exist at least two distinct loci in the CHRNB3–CHRNA6 gene cluster that are associated with ND in European Americans (EAs). The first one was tagged by rs13277254, upstream of the gene cluster, together with additional associated SNPs in this region constituted Signal 1. Signal 2 was tagged by rs4952, the only known coding SNP in the exon 5 of CHRNB3, which had a low correlation with rs13277254 (Figure 1).

Schematic diagram of the human CHRNB3–CHRNA6 gene cluster. Horizontal black arrows indicate the direction of transcription of each gene. Gray and black rectangles indicate exons and untranslated regions, respectively, while horizontal black lines represent introns (not drawn to scale). The genetic variants significantly associated with ND in EAs are shown by vertical arrows, which represent two distinct signals. EA, European-American; ND, nicotine dependence.
There also exist many other common variants in the CHRNB3–CHRNA6 gene cluster that show a significant association with ND in multiple ethnic populations, including Han Chinese,40 AAs,39 EAs,43, 44 and Israelis.46 We performed a meta-analysis of variants in CHRNB3 in relation to ND by combining data from the studies of subjects of different ethnicities.34 Although allele frequencies in AAs were different from those in EAs and subjects of Asian ancestry, where the last two ethnic samples appeared to be similar, we found that the genetic effect of seven SNPs in CHRNB3 are in the same direction among the three populations. More importantly, all these SNPs showed a significant association with ND. However, because of the different genetic structures of various ancestries, inconsistent results were found at the SNP level. We detected only four of seven SNPs in the samples of African origin, whereas the associations of all SNPs in the samples of European and Asian ancestry were significant.34 In contrast, none of these SNPs was reported to be associated with ND among three other studies in Finnish,47 Swiss48 and Czech49 populations. Hubacek49 attributed this discrepancy partly to socioeconomic status in that the prevalence of smoking was higher in post-Communist countries than in western European countries, and this fact could mask the real effect of each SNP. Thus, further replication studies in additional independent samples of different origins are warranted.
ND-related phenotypes
The early-subjective response to tobacco smoking is a subphenotype of SI, which can predict later persistence of smoking and addiction. DiFranza et al.50 reported that greater sensitivity to nicotine during early-smoking attempts, as manifested by relaxation, dizziness or nausea, was a determinant of later ND. Pomerleau et al.51 found that smokers who felt a pleasurable buzz during early smoking smoked much later than those who did not. Thus, it was reasonable to assume that genes, especially CHRN, associated with ND might also play a role in early-subjective responses to tobacco.
The first report concerning the association between the variants in CHRNB3–CHRNA6 and subjective responses to tobacco was published by Zeiger et al.31 using as subjects 1056 ethnically diverse adolescents and a separate community sample of 1524 families. The most significant associations were found between two CHRNB3 SNPs (that is, rs4950 and rs13280604) and three subjective response factors to initial tobacco use (adverse, negative physical and positive). Since then, several studies36, 42, 52 have examined the association between variants in the CHRNB3–CHRNA6 gene cluster and dizziness at first inhalation of cigarette smoke. Although both Ehringer et al.36 and Pedneault et al.42 have detected associations with several SNPs in the putative promoter region of CHRNB3 and CHRNA6, Hoft et al.52 did not, which might be attributable to the small sample and the discrepancy of the phenotypic assessment tools used in these studies.
Apart from early-subjective responses to tobacco, there exist many other ND-related phenotypes where the CHRNB3–CHRNA6 gene cluster may play an important role, such as smoking status (never smoking versus ever smoking),35 smoking trajectories from early adolescence to adulthood,45 and various ND endophenotypes such as ‘novelty seeking’53 or ‘drive.’37 In addition, smoking cessation is of great interest, because it is the ultimate goal of studying tobacco addiction and any other smoking-related phenotypes. Hoft et al.32 examined the association of SNPs in the CHRNB3–CHRNA6 cluster with quit attempts in a nationally representative sample of households, which revealed that three SNPs upstream of CHRNB3 (that is, rs7004381, rs4950, rs13280604) and an SNP (rs2304297) in the 3′-region of CHRNA6 were significantly associated with the number of unsuccessful quit attempts in Caucasians. Further, Fletcher et al.41 provided novel evidence of the importance of genetics in explaining different responses to tobacco taxation policy. These investigators found that individuals with the protective G/G polymorphism of rs2304297 in CHRNA6 responded to high tobacco taxation, which may help with abstention, whereas others had no response. The inability of this tobacco control policy (high taxation) to reduce the use of cigarettes in individuals with the C/C genotype suggests that alternative methods might be needed to increase smoking cessation in this population.
Analysis of rare variants in the CHRNB3–CHRNA6 gene cluster
Both GWAS and candidate gene-based association studies have identified multiple common variants in the CHRNB3–CHRNA6 gene cluster that contribute to ND and ND-related phenotypes. However, the role of rare variants of this cluster in ND has rarely been studied, largely because the extremely low minor allele frequency (MAF) poses great difficulties in ensuring adequate statistical power. The only study of this topic was carried out by Haller et al.,54 in which a DNA-pooling approach was used to sequence the coding and flanking regions of CHRNA6 and CHRNB3 in AA and EA ND smokers or smokers without any ND symptom (for the AAs, two case pools and two control pools; for the EAs, one case pool and one control pool). In contrast to another study performed by the same group,55 which showed that rare missense variants in CHRNB3 were associated with a risk of alcohol and cocaine dependence, there was no evidence supporting the role of the same variants in ND.54, 55
Despite the absence of genetic association data for most SNPs, functional studies conducted by us56 indicated that rare variants in the hα6 subunit gene play a vital role in the etiology of ND. Although missense variations such as Asp57Asn (rs149966755) and Ser156Arg (rs373147726), Asn171Lys (rs79945499) compromises the function of hα6*-nAChRs heterologously expressed in Xenopus oocytes, the nicotine sensitivity of these receptors is marginally or significantly increased by introducing Arg96His (rs188620180), Ala184Asp (rs200745568), Asp199Tyr (rs372469952) or Ser233Cys (rs369966241) variations into the hα6 subunit gene. Greater sensitivity to activation by agonists (nicotine or ACh) may result in a lower risk of ND, whereas reduced sensitivity increases the risk.57 Individuals displaying altered α6*-nAChR pharmacology as a result of rare variants in CHRNA6 are expected to exhibit different responses to cigarette smoking.
Because rare variants (defined as those having an MAF of <1%), together with copy-number variants and small insertion/deletion polymorphisms (indels) constitute the majority of human genetic variations, they might contribute, at least partly, to the missing heritability of ND. Thus, we need to take rare variants into consideration when studying ND-related phenotypes, especially rare missense functional variants.
Functional studies of the β3 and α6 subunits by genetic manipulation in rodents
As described above, numerous genetic studies have revealed a highly significant association between variants in the CHRNB3–CHRNA6 gene cluster and increased vulnerability to ND,21, 27, 28 which generates a need to explore the underlying mechanisms. However, to date, few pharmacologic ligands have been developed that can selectively target specific nAChR subtypes. Therefore, to understand the contribution of α6 and β3 subunits to ND susceptibility in vivo, and to circumvent the problem mentioned above, together with the difficulty associated with α6*-nAChRs in vitro expression genetic manipulation in mice becomes highly valuable. These manipulations generally include preventing the expression of the α6 or β3-subunit (KO) and replacing it with hyperactive derivatives (KI).
More attention has been paid to α6*- and β3*-nAChRs since the demonstration that these subunits exhibit an expression pattern restricted mainly to catecholaminergic and visual system neurons.58, 59, 60, 61 By using transgenic mice expressing the α6 subunit fused with green fluorescent protein, the α6 subunit was found to be highly and selectively expressed in the ventral tegmental area (VTA) and substantia nigra pars compacta, important regions for reinforcement of nicotine use,62, 63 with functional expression also in the locus coeruleus and retinal ganglion cells.64, 65 Immunoprecipitation and high-affinity [125I]α-conotoxinMII (αCtxMII)-binding studies showed that α6β2β3* and α6α4β2β3* pentamers are the predominant α6*-nAChRs in the striatum.66, 67 Furthermore, the gene encoding the β3-subunit, which is adjacent to CHRNA6 (Figure 1), usually is co-expressed with α6. Because of the accessory role of the β3-subunit, it cannot form an acetylcholine-binding site, although it has an essential role in α6*-nAChR biogenesis and function.68, 69 Gotti et al.69 discovered that β3-subunit deletion dramatically reduced, but did not eliminate, α6*-nAChRs expression in the DA cell body (VTA) and terminal region (striatum), suggesting the importance of β3 for the correct assembly, stability and transport of α6-containing receptors in dopaminergic neurons. In addition, a study conducted by Cui et al.68 demonstrated that disruption of the β3 gene does not affect expression of mRNA for α6 and other subunits in the same brain areas. They also found that β3-KO mice have altered locomotor activity and prepulse inhibition of acoustic startle responses, behaviors that are regulated in part by nigrostriatal and mesolimbic dopaminergic neurotransmission. Knowledge of these alterations is supported by the evidence that a population of β3-dependent nAChRs, which are sensitive to inhibition by αCTxMII, modulate striatal dopamine release.68 In addition, Kamens et al.70 showed that the protective variant rs6474413 from human studies reduced expression of the CHRNB3 subunit, and decreased β3 gene expression resulted in reduced nicotine intake in mice.
The α6-null mice grow normally and show no obvious developmental, neurologic or behavior deficits.66, 71 By using autoradiography, Champtiaux et al.71 found complete disappearance of [125I] α-CtxMII binding in both midbrain dopamergic neurons and the visual system after deleting the α6 subunit, indicating that α6 is an essential component of the native-binding site of this toxin. Another study72 has shown the central role of α6 in the VTA in acute nicotine reinforcement.
There are two primary strategies for measuring the reinforcing effect of nicotine: one is intravenous or intracranial nicotine self-administration (SA)73, 74 and the other is nicotine-induced conditioned place preference (CPP).75 The SA paradigm is usually conducted in 30 min with matched animal pairs placed in the experimental boxes, with one animal defined as active and the other as passive. Each nose-poke (NP) by the active mouse activates the computer-operated syringe pump delivering either nicotine or saline to both the active and passive animals, while NPs of the passive mouse are recoded with no scheduled consequences. By calculating the ratio between the number of responses (NPs) of the active and passive mouse, the reinforcing effects of nicotine can be determined. When tested in this way, α6-WT mice self-administered nicotine in a unit dose of 26.3 μg kg−1 per infusion (inf), whereas their α6-KO drug-naive littermates did not. The α6-KO animals did not self-administer nicotine even in an extensive range of lower (8.7-17.5 μg kg−1 per inf) and higher (35-52.6 μg kg−1 per inf) doses. Importantly, when the α6 subunit was selectively re-expressed in the VTA of α6−/− mice using a lentiviral vector, the reinforcement property of nicotine was restored (Figure 2).72 In intracranial SA experiments where learning is required, α6-KO mice showed a trend (although it was not significant) toward reduced nicotine SA compared with wild-type (WT) control mice.76 These findings demonstrate that the α6 subunit in the VTA is necessary for maintaining nicotine SA. By employing the latter model, Sanjakdar et al.77 showed that nicotine displayed a typical inverted U-shaped CPP response curve in the WT mice. Although the dose of 0.5 mg kg−1 nicotine led to a significant CPP in the WT mice, it failed to produce a CPP response in α6-KO mice. In contrast, the higher nicotine dose of 1.0 mg kg−1 resulted in preference scores in α6-KO mice, which were significantly higher than in α6-WT littermates (Figure 3). The α6-KO mice exhibit a rightward shift in the nicotine dose–response curve compared with WT mice, indicating that the rewarding effect of nicotine is mediated by α6*-nAChRs. Pharmacologic blockade of the α6 subunit by selective antagonists (for example, α-contoxinMII) attenuates nicotine-induced CPP,77, 78 further supporting the vital role of α6 in the nicotine reinforcement.

Nicotine intravenous self-administration in WT and α6-VEC-VTA mice, but not in α6-KO mice. Data are presented as mean (±s.e.m.) reinforcement index (that is, ratio of the cumulative nose pokes (NPS) of the active mice with respect to yoked control passive mice over the 30- min session in each group). The dose of nicotine was 26.3 μg kg−1 per inf. P<0.01 indicates statistically significant differences between nicotine-treated and saline control groups. The data used in the figure are adapted from the study by Pons et al.72 KO, knock out; VTA, ventral tegmental area; WT, wild type.

Crucial role of α6* nAChRs in the rewarding effects of nicotine based on conditioned place preference (CPP). The α6-KO mice exhibited a rightward shift in the nicotine dose–response curve compared with WT littermates. Data are presented as mean (±s.e.m.) preference score (sec). ***P<0.001. The data are adapted from the study by Sanjakdar et al.77 KO, knock out; nAChR, nicotinic acetylcholine receptor; WT, wild type.
Although the KO mice model is an essential research tool for understanding the mechanisms of ND, it typically allows addressing only questions of necessity, not sufficiency. To fully understand the diverse roles of different subunits or subtypes in the process of nicotine-induced reward and withdrawal, genetic KI strains have been developed. Replacement of ‘Leu’ with ‘Ser’ in the 9′ residue in the M2 domain of the α6 subunit produces nicotine-hypersensitive mice. These α6 L9′S strains show hyperactive locomotion and fail to habituate to a home cage, a novel environment or reduced wheel rotations,79, 80, 81, 82 which is consistent with enhanced dopamine neuron firing and release.79, 80, 82, 83 In addition, by crossing the α4-KO mice with α6L9′S strains, it was found that the hyperactive effects caused by the gain-of-function mutation are mediated by α6α4* pentamers, because α6L9′S mice lacking the α4 subunit display essentially normal behavior.80 Together, these studies demonstrate that α6L9′S mice are valuable in investigating the role of the α6 subunit in ND-related behaviors.
Effect of chronic nicotine exposure on the expression of α6-containing nAChRs
Nicotine, like other substances of abuse, enhances dopamine transmission in the mesolimbic dopamine pathway, which is thought to play a critical role in the reinforcing effects that maintain smoking behaviors. Many studies on the rewarding effects of nicotine employed an acute administration approach. However, because smoking is a chronic behavior leading to long-term adaptive changes in the brain, knowledge of these chronic changes is essential for understanding ND and implementing measures that cause smoking cessation. Therefore, if genetic manipulation of nAChR genes in mouse KO or KI models represents a powerful research tool for identification of the particular contribution of specific receptor subunits to ND susceptibility, chronic nicotine treatment in vivo or in vitro, which mimics the process of smoking in humans, is a valuable strategy.
After long-term nicotine exposure, high-affinity agonist binding to nAChRs in the central nervous system increases in both animal84, 85 and human86 brains. This process, termed ‘nicotine-induced upregulation,’ 87 may be involved in the pathology of nicotine addiction. An increase in [3H]Ach-binding sites was reported in the brains of smokers compared with non-smokers.88 The essence of nAChRs upregulation is more related to greater receptor numbers than to augmentation of receptor affinity for nicotine.89 Furthermore, a hypothesis that nicotine acts as a pharmacologic chaperone to enhance a critical step inside the cell during the maturation of nAChRs has gained support recently.90 Specifically, nicotine binding to partially assembled nAChRs induces conformations that assemble more efficiently. This could be a compensatory response following desensitization of neuronal AChRs after chronic nicotine exposure.91, 92
Accumulating studies have consistently observed upregulation by radiolabeled epibatidine, which identified several nAChR subtypes in numerous brain regions after various nicotine treatments, including injection by osmotic minipumps or jugular cannula and infusion in drinking water.85, 93, 94, 95, 96 Using [125I]epibatidine, A-85380, and cytosine, Nguten et al.97 demonstrated that chronic exposure to nicotine upregulates α4β2-containing receptors while having little effect on other nAChR subtypes. Nevertheless, α4β2*-nAChRs, with a wide distribution in the brain and high affinity for nicotine, clearly become desensitized at an early stage of smoking behavior and thus do not function for most of the day in smokers. Despite the clarity of α4β2*-nAChR upregulation, it is not sufficient to explain continued smoking throughout the day.16, 98 On the other hand, nAChRs with low affinity for nicotine (for example, α7, α6) are not susceptible to rapid saturation and might play an important role in continued smoking. Besides α4β2-containing receptors, other diverse populations of nAChRs, such as α6β2* and α7*, have been identified in the mesolimbic dopamine pathway. These findings shed light on the vital importance of research on the upregulation of other nAChRs.
Unlike the situation with α4β2*-nAChRs, upregulation of α6-containing receptors in response to chronic nicotine exposure is controversial.99 There have been reports of upregulation, downregulation and no change in in vitro and in vivo experiments (Table 3). Upregulation of α6β2*- or α6β2β3*-nAChRs by incubation with nicotine was observed in cultured cell lines,100, 101, 102 although functional expression of α6-containing receptors in a heterologous expression system proved to be difficult until some specific strategies were used, such as chimeras, gain-of-function mutagenesis and so on. Unfortunately, in rodents, although Nguyen et al.97 and Parker et al.103 suggested upregulation of α6*-nAChRs in the nucleus accumbens, several other research groups104, 105, 106, 107, 108 observed downregulation in the striatum. Interestingly, Perez et al.107 showed, by using the novel α-CtxMII analog E11A in α4-KO mice, that nicotine administration in the drinking water for 2 weeks increased the α6 (non-α4) β2*-nAChR population in the striatum, contrary to the reduction of total α6β2* subtypes in WT littermates. This leads us to hypothesize that α6α4β2* contributes to the downregulation in the striatum. Furthermore, in non-human primates such as the squirrel monkey, nicotine in the drinking water with a final concentration of 650 μg ml−1 for >6 months did not significantly change the α6β2*-nAChR-binding site109, 110, 111 except in the study conducted by McCallum et al.112 This effect might be caused by region-specific actions, because earlier studies concentrated mainly on the nucleus accumbens, whereas the later ones focused on the striatum. Analyses in other reward-related regions of the brain also were performed, but this work has yielded no clear results or conclusions.102, 103, 113
Several factors may account for these disparate findings. First, different nicotine treatment regimens with different concentrations of nicotine and exposure time were used. The importance of such changes is supported by evidence that α6β2β3*-nAChR showed upregulation after 50 nm nicotine treatment but downregulation with 500 nm nicotine.102 Second, different species/cell lines, brain regions and α6-containing subtypes may play a role in the inconsistent results. Last but not least, heterogeneity of the detection methods is an influencing factor, implying the urgency of developing more subunit-specific agonists and antibodies.
Conclusions and future research
In this report, we have summarized several lines of evidence that support the involvement of the CHRNB3–CHRNA6 gene cluster in ND. A multitude of genetic studies (GWAS and candidate gene-based association studies) analyzing various ND phenotypes have implicated variants in this gene cluster in the development of ND. The most compelling evidence is for SNPs rs13277254 and rs6474413 in CHRNB3, as well as rs10958726 and rs1955186 within this same signal. However, not much has been found specifically for the CHRNA6 subunit gene, contrary to its vital role in maintaining ND as demonstrated with functional studies. These findings reveal only a small fraction of variants, that is, these known polymorphisms have small effects and can explain only a small proportion of the heritability of smoking-related behaviors. Therefore, additional loci (especially rare variants) need to be identified. Furthermore, despite the inconsistent results, it is important to study the genetics of ND in diverse populations. Differences in genetic architectures and allele frequencies in different ethnic populations can help assign statistically significant signals to potentially causal variants.
Genetic modification of the CHRNB3 and CHRNA6 in mice is a valuable approach to evaluate the contribution of each subunit to ND susceptibility. The use of KO mice has displayed various behavioral phenotypes related to ND. For example, α6-KO mice do not self-administer nicotine, unlike their WT counterparts. In addition, studies in α6-hypersensitive mice (KI mice) are powerful in identifying compounds that activate or antagonize α6*-nAChRs as a means to improve the development of drugs for smoking cessation. Nevertheless, this approach is limited in the in vivo or in vitro studies focusing on elucidating the functional consequences of different SNPs. This level of investigation will provide significant insights into how genetic variations in humans underlie individual differences in the reinforcement, aversion and withdrawal of nicotine. There exist significant differences in the pharmacologic properties of the α6 and β3 subunits, such as receptor upregulation after chronic nicotine treatment and differences among subtypes and brain regions. It remains to be determined how nicotine regulates the expression of α6*-nAChRs. Inconsistent results from different studies were likely a consequence of the unpredictable behavior in heterologous expression systems. Functional expression of WT α6*-nAChRs is difficult to achieve unless some modifications have been adopted, for instance, subunit chimeras, concatameric subunits and point mutagenesis of the α6 or β3 subunits. In spite of the significant progress, there still are many obstacles to be overcome. That may be why conflicting results concerning upregulation of α6-containing receptors occur in relatively few studies. Thus, advancing the heterologous expression of α6* receptors should be another focus of future research.
References
CDC. Current cigarette smoking among adults --- United States, 2011. MMWR Morb Mortal Wkly Rep 2012; 61: 889–894.
Li MD . Identifying susceptibility loci for nicotine dependence: 2008 update based on recent genome-wide linkage analyses. Hum Genet 2008; 123: 119–131.
Centers for Disease Control and Prevention (CDC). State-specific prevalence of current cigarette smoking among adults and secondhand smoke rules and policies in homes and workplaces. MMWR Morb Mortal Wkly Rep 2006; 55: 1148–1151.
Benowitz NL . Clinical pharmacology of nicotine: implications for understanding, preventing, and treating tobacco addiction. Clin Pharmacol Ther 2008; 83: 531–541.
Batra V, Patkar AA, Berrettini WH, Weinstein SP, Leone FT . The genetic determinants of smoking. Chest 2003; 123: 1730–1739.
Lessov CN, Martin NG, Statham DJ, Todorov AA, Slutske WS, Bucholz KK et al. Defining nicotine dependence for genetic research: evidence from Australian twins. Psychol Med 2004; 34: 865–879.
Lessov-Schlaggar CN, Pang Z, Swan GE, Guo Q, Wang S, Cao W et al. Heritability of cigarette smoking and alcohol use in Chinese male twins: the Qingdao twin registry. Int J Epidemiol 2006; 35: 1278–1285.
Maes HH, Sullivan PF, Bulik CM, Neale MC, Prescott CA, Eaves LJ et al. A twin study of genetic and environmental influences on tobacco initiation, regular tobacco use and nicotine dependence. Psychol Med 2004; 34: 1251–1261.
Sullivan PF, Kendler K . The genetic epidemiology of smoking. Nicotine Tob Res 1999; 1: S51–S57.
Li MD, Cheng R, Ma JZ, Swan GE . A meta-analysis of estimated genetic and environmental effects on smoking behavior in male and female adult twins. Addiction 2003; 98: 23–31.
Baker RR, Pereira da Silva JR, Smith G . The effect of tobacco ingredients on smoke chemistry. Part I: flavourings and additives. Food Chem Toxicol 2004; 42: S3–37.
Dani JA, Harris RA . Nicotine addiction and comorbidity with alcohol abuse and mental illness. Nat Neurosci 2005; 8: 1465–1470.
Le Novere N, Corringer PJ, Changeux JP . The diversity of subunit composition in nAChRs: evolutionary origins, physiologic and pharmacologic consequences. J Neurobiol 2002; 53: 447–456.
Dani JA, Bertrand D . Nicotinic acetylcholine receptors and nicotinic cholinergic mechanisms of the central nervous system. Annu Rev Pharmacol Toxicol 2007; 47: 699–729.
Gotti C, Zoli M, Clementi F . Brain nicotinic acetylcholine receptors: native subtypes and their relevance. Trends Pharmacol Sci 2006; 27: 482–491.
Rose JE . Multiple brain pathways and receptors underlying tobacco addiction. Biochem Pharmacol 2007; 74: 1263–1270.
Kalamida D, Poulas K, Avramopoulou V, Fostieri E, Lagoumintzis G, Lazaridis K et al. Muscle and neuronal nicotinic acetylcholine receptors. Structure, function and pathogenicity. FEBS J 2007; 274: 3799–3845.
Waters AJ, Shiffman S, Sayette MA, Paty JA, Gwaltney CJ, Balabanis MH . Attentional bias predicts outcome in smoking cessation. Health Psychol 2003; 22: 378–387.
Saccone SF, Hinrichs AL, Saccone NL, Chase GA, Konvicka K, Madden PA et al. Cholinergic nicotinic receptor genes implicated in a nicotine dependence association study targeting 348 candidate genes with 3713 SNPs. Hum Mol Genet 2007; 16: 36–49.
Bierut LJ, Stitzel JA, Wang JC, Hinrichs AL, Grucza RA, Xuei X et al. Variants in nicotinic receptors and risk for nicotine dependence. Am J Psychiatry 2008; 165: 1163–1171.
Thorgeirsson TE, Gudbjartsson DF, Surakka I, Vink JM, Amin N, Geller F et al. Sequence variants at CHRNB3-CHRNA6 and CYP2A6 affect smoking behavior. Nat Genet 2010; 42: 448–453.
Bierut LJ . Genetic vulnerability and susceptibility to substance dependence. Neuron 2011; 69: 618–627.
Morel C, Fattore L, Pons S, Hay YA, Marti F, Lambolez B et al. Nicotine consumption is regulated by a human polymorphism in dopamine neurons. Mol Psychiatr 2014; 19: 930–936.
Wen L, Jiang K, Yuan W, Cui W, Li MD . Contribution of variants in CHRNA5/A3/B4 gene cluster on chromosome 15 to tobacco smoking: from genetic association to mechanism. Mol Neurobiol 2014; 53: 472–484.
Berrettini WH, Doyle GA . The CHRNA5-A3-B4 gene cluster in nicotine addiction. Mol Psychiatry 2012; 17: 856–866.
Melroy-Greif WE, Stitzel JA, Ehringer MA . Nicotinic acetylcholine receptors: upregulation, age-related effects and associations with drug use. Genes Brain Behav 2016; 15: 89–107.
Bierut LJ, Madden PA, Breslau N, Johnson EO, Hatsukami D, Pomerleau OF et al. Novel genes identified in a high-density genome wide association study for nicotine dependence. Hum Mol Genet 2007; 16: 24–35.
Rice JP, Hartz SM, Agrawal A, Almasy L, Bennett S, Breslau N et al. CHRNB3 is more strongly associated with Fagerstrom test for cigarette dependence-based nicotine dependence than cigarettes per day: phenotype definition changes genome-wide association studies results. Addiction 2012; 107: 2019–2028.
Johnson EO, Morgan-Lopez AA, Breslau N, Hatsukami DK, Bierut LJ . Test of measurement invariance of the FTND across demographic groups: assessment, effect size, and prediction of cessation. Drug Alcohol Depend 2008; 93: 260–270.
Saccone NL, Saccone SF, Hinrichs AL, Stitzel JA, Duan W, Pergadia ML et al. Multiple distinct risk loci for nicotine dependence identified by dense coverage of the complete family of nicotinic receptor subunit (CHRN) genes. Am J Med Genet B Neuropsychiatr Genet 2009; 150B: 453–466.
Zeiger JS, Haberstick BC, Schlaepfer I, Collins AC, Corley RP, Crowley TJ et al. The neuronal nicotinic receptor subunit genes (CHRNA6 and CHRNB3) are associated with subjective responses to tobacco. Hum Mol Genet 2008; 17: 724–734.
Hoft NR, Corley RP, McQueen MB, Schlaepfer IR, Huizinga D, Ehringer MA . Genetic association of the CHRNA6 and CHRNB3 genes with tobacco dependence in a nationally representative sample. Neuropsychopharmacology 2009; 34: 698–706.
Saccone NL, Schwantes-An TH, Wang JC, Grucza RA, Breslau N, Hatsukami D et al. Multiple cholinergic nicotinic receptor genes affect nicotine dependence risk in African and European Americans. Genes Brain Behav 2010; 9: 741–750.
Cui WY, Wang S, Yang J, Yi SG, Yoon D, Kim YJ et al. Significant association of CHRNB3 variants with nicotine dependence in multiple ethnic populations. Mol Psychiatry 2013; 18: 1149–1151.
Bar-Shira A, Gana-Weisz M, Gan-Or Z, Giladi E, Giladi N, Orr-Urtreger A . CHRNB3 c.-57 A>G functional promoter change affects Parkinson's disease and smoking. Neurobiol Aging 2014; 35: 2179 e2171–2176.
Ehringer MA, McQueen MB, Hoft NR, Saccone NL, Stitzel JA, Wang JC et al. Association of CHRN genes with ‘dizziness’ to tobacco. Am J Med Genet B Neuropsychiatr Genet 2010; 153B: 600–609.
Won WY, Park B, Choi SW, Kim L, Kwon M, Kim JH et al. Genetic association of CHRNB3 and CHRNA6 gene polymorphisms with nicotine dependence syndrome scale in Korean population. Psychiatr Invest 2014; 11: 307–312.
Johnson EO, Chen LS, Breslau N, Hatsukami D, Robbins T, Saccone NL et al. Peer smoking and the nicotinic receptor genes: an examination of genetic and environmental risks for nicotine dependence. Addiction 2010; 105: 2014–2022.
Culverhouse RC, Johnson EO, Breslau N, Hatsukami DK, Sadler B, Brooks AI et al. Multiple distinct CHRNB3-CHRNA6 variants are genetic risk factors for nicotine dependence in African Americans and European Americans. Addiction 2014; 109: 814–822.
Wei J, Chu C, Wang Y, Yang Y, Wang Q, Li T et al. Association study of 45 candidate genes in nicotine dependence in Han Chinese. Addict Behav 2012; 37: 622–626.
Fletcher JM . Why have tobacco control policies stalled? Using genetic moderation to examine policy impacts. PLoS One 2012; 7: e50576.
Pedneault M, Labbe A, Roy-Gagnon MH, Low NC, Dugas E, Engert JC et al. The association between CHRN genetic variants and dizziness at first inhalation of cigarette smoke. Addict Behav 2014; 39: 316–320.
Stevens VL, Bierut LJ, Talbot JT, Wang JC, Sun J, Hinrichs AL et al. Nicotinic receptor gene variants influence susceptibility to heavy smoking. Cancer Epidemiol Biomarkers Prev 2008; 17: 3517–3525.
Wang S, DvdV A, Xu Q, Seneviratne C, Pomerleau OF, Pomerleau CS et al. Significant associations of CHRNA2 and CHRNA6 with nicotine dependence in European American and African American populations. Hum Genet 2014; 133: 575–586.
Lee CT, Fuemmeler BF, McClernon FJ, Ashley-Koch A, Kollins SH . Nicotinic receptor gene variants interact with attention deficient hyperactive disorder symptoms to predict smoking trajectories from early adolescence to adulthood. Addict Behav 2013; 38: 2683–2689.
Greenbaum L, Kanyas K, Karni O, Merbl Y, Olender T, Horowitz A et al. Why do young women smoke? I. Direct and interactive effects of environment, psychological characteristics and nicotinic cholinergic receptor genes. Mol Psychiatry 2006; 11: 312–322.
Keskitalo-Vuokko K, Pitkaniemi J, Broms U, Heliovaara M, Aromaa A, Perola M et al. Associations of nicotine intake measures with CHRN genes in Finnish smokers. Nicotine Tob Res 2011; 13: 686–690.
Etter JF, Hoda JC, Perroud N, Munafo M, Buresi C, Duret C et al. Association of genes coding for the alpha-4, alpha-5, beta-2 and beta-3 subunits of nicotinic receptors with cigarette smoking and nicotine dependence. Addict Behav 2009; 34: 772–775.
Hubacek JA, Lanska V, Adamkova V . Lack of an association between SNPs within the cholinergic receptor genes and smoking behavior in a Czech post-MONICA study. Genet Mol Biol 2014; 37: 625–630.
DiFranza JR, Savageau JA, Fletcher K, Ockene JK, Rigotti NA, McNeill AD et al. Recollections and repercussions of the first inhaled cigarette. Addict Behav 2004; 29: 261–272.
Pomerleau OF, Pomerleau CS, Mehringer AM, Snedecor SM, Cameron OG . Validation of retrospective reports of early experiences with smoking. Addict Behav 2005; 30: 607–611.
Hoft NR, Stitzel JA, Hutchison KE, Ehringer MA . CHRNB2 promoter region: association with subjective effects to nicotine and gene expression differences. Genes Brain Behav 2011; 10: 176–185.
Landgren S, Berglund K, Jerlhag E, Fahlke C, Balldin J, Berggren U et al. Reward-related genes and personality traits in alcohol-dependent individuals: a pilot case control study. Neuropsychobiology 2011; 64: 38–46.
Haller G, Druley T, Vallania FL, Mitra RD, Li P, Akk G et al. Rare missense variants in CHRNB4 are associated with reduced risk of nicotine dependence. Hum Mol Genet 2012; 21: 647–655.
Haller G, Kapoor M, Budde J, Xuei X, Edenberg H, Nurnberger J et al. Rare missense variants in CHRNB3 and CHRNA3 are associated with risk of alcohol and cocaine dependence. Hum Mol Genet 2014; 23: 810–819.
Dash B, Li MD . Analysis of rare variations reveals roles of amino acid residues in the N-terminal extracellular domain of nicotinic acetylcholine receptor (nAChR) alpha6 subunit in the functional expression of human alpha6*-nAChRs. Mol Brain 2014; 7: 35.
Haller G, Li P, Esch C, Hsu S, Goate AM, Steinbach JH . Functional characterization improves associations between rare non-synonymous variants in CHRNB4 and smoking behavior. PLoS One 2014; 9: e96753.
Deneris ES, Boulter J, Swanson LW, Patrick J, Heinemann S . Beta 3: a new member of nicotinic acetylcholine receptor gene family is expressed in brain. J Biol Chem 1989; 264: 6268–6272.
Forsayeth JR, Kobrin E . Formation of oligomers containing the beta3 and beta4 subunits of the rat nicotinic receptor. J Neurosci 1997; 17: 1531–1538.
Vailati S, Moretti M, Balestra B, McIntosh M, Clementi F, Gotti C . beta3 subunit is present in different nicotinic receptor subtypes in chick retina. Eu J Pharmacol 2000; 393: 23–30.
Le Novere N, Zoli M, Changeux JP . Neuronal nicotinic receptor alpha 6 subunit mRNA is selectively concentrated in catecholaminergic nuclei of the rat brain. Eur J Neurosci 1996; 8: 2428–2439.
Mackey ED, Engle SE, Kim MR, O'Neill HC, Wageman CR, Patzlaff NE et al. alpha6* nicotinic acetylcholine receptor expression and function in a visual salience circuit. J Neurosci 2012; 32: 10226–10237.
Powers MS, Broderick HJ, Drenan RM, Chester JA . Nicotinic acetylcholine receptors containing alpha6 subunits contribute to alcohol reward-related behaviours. Genes Brain Behav 2013; 12: 543–553.
Azam L, Winzer-Serhan UH, Chen Y, Leslie FM . Expression of neuronal nicotinic acetylcholine receptor subunit mRNAs within midbrain dopamine neurons. J Comp Neurol 2002; 444: 260–274.
Azam L, McIntosh JM . Characterization of nicotinic acetylcholine receptors that modulate nicotine-evoked [3H]norepinephrine release from mouse hippocampal synaptosomes. Mol Pharmacol 2006; 70: 967–976.
Champtiaux N, Gotti C, Cordero-Erausquin M, David DJ, Przybylski C, Lena C et al. Subunit composition of functional nicotinic receptors in dopaminergic neurons investigated with knock-out mice. J Neurosci 2003; 23: 7820–7829.
Zoli M, Moretti M, Zanardi A, McIntosh JM, Clementi F, Gotti C . Identification of the nicotinic receptor subtypes expressed on dopaminergic terminals in the rat striatum. J Neurosci 2002; 22: 8785–8789.
Cui C, Booker TK, Allen RS, Grady SR, Whiteaker P, Marks MJ et al. The beta3 nicotinic receptor subunit: a component of alpha-conotoxin MII-binding nicotinic acetylcholine receptors that modulate dopamine release and related behaviors. J Neurosci 2003; 23: 11045–11053.
Gotti C, Moretti M, Clementi F, Riganti L, McIntosh JM, Collins AC et al. Expression of nigrostriatal alpha 6-containing nicotinic acetylcholine receptors is selectively reduced, but not eliminated, by beta 3 subunit gene deletion. Mol Pharmacol 2005; 67: 2007–2015.
Kamens HM, Miyamoto J, Powers MS, Ro K, Soto M, Cox R et al. The beta3 subunit of the nicotinic acetylcholine receptor: modulation of gene expression and nicotine consumption. Neuropharmacology 2015; 99: 639–649.
Champtiaux N, Han ZY, Bessis A, Rossi FM, Zoli M, Marubio L et al. Distribution and pharmacology of alpha 6-containing nicotinic acetylcholine receptors analyzed with mutant mice. J Neurosci 2002; 22: 1208–1217.
Pons S, Fattore L, Cossu G, Tolu S, Porcu E, McIntosh JM et al. Crucial role of alpha4 and alpha6 nicotinic acetylcholine receptor subunits from ventral tegmental area in systemic nicotine self-administration. J Neurosci 2008; 28: 12318–12327.
Orejarena MJ, Herrera-Solis A, Pons S, Maskos U, Maldonado R, Robledo P . Selective re-expression of beta2 nicotinic acetylcholine receptor subunits in the ventral tegmental area of the mouse restores intravenous nicotine self-administration. Neuropharmacology 2012; 63: 235–241.
Besson M, David V, Baudonnat M, Cazala P, Guilloux JP, Reperant C et al. Alpha7-nicotinic receptors modulate nicotine-induced reinforcement and extracellular dopamine outflow in the mesolimbic system in mice. Psychopharmacology 2012; 220: 1–14.
Grabus SD, Martin BR, Brown SE, Damaj MI . Nicotine place preference in the mouse: influences of prior handling, dose and strain and attenuation by nicotinic receptor antagonists. Psychopharmacology 2006; 184: 456–463.
Exley R, Maubourguet N, David V, Eddine R, Evrard A, Pons S et al. Distinct contributions of nicotinic acetylcholine receptor subunit alpha4 and subunit alpha6 to the reinforcing effects of nicotine. Proc Natl Acad Sci USA 2011; 108: 7577–7582.
Sanjakdar SS, Maldoon PP, Marks MJ, Brunzell DH, Maskos U, McIntosh JM et al. Differential roles of alpha6beta2* and alpha4beta2* neuronal nicotinic receptors in nicotine- and cocaine-conditioned reward in mice. Neuropsychopharmacology 2015; 40: 350–360.
Jackson KJ, McIntosh JM, Brunzell DH, Sanjakdar SS, Damaj MI . The role of alpha6-containing nicotinic acetylcholine receptors in nicotine reward and withdrawal. J Pharmacol Exp Ther 2009; 331: 547–554.
Drenan RM, Grady SR, Whiteaker P, McClure-Begley T, McKinney S, Miwa JM et al. In vivo activation of midbrain dopamine neurons via sensitized, high-affinity alpha 6 nicotinic acetylcholine receptors. Neuron 2008; 60: 123–136.
Drenan RM, Grady SR, Steele AD, McKinney S, Patzlaff NE, McIntosh JM et al. Cholinergic modulation of locomotion and striatal dopamine release is mediated by alpha6alpha4* nicotinic acetylcholine receptors. J Neurosci 2010; 30: 9877–9889.
Grady SR, Drenan RM, Breining SR, Yohannes D, Wageman CR, Fedorov NB et al. Structural differences determine the relative selectivity of nicotinic compounds for native alpha 4 beta 2*-, alpha 6 beta 2*-, alpha 3 beta 4*- and alpha 7-nicotine acetylcholine receptors. Neuropharmacology 2010; 58: 1054–1066.
Cohen BN, Mackey ED, Grady SR, McKinney S, Patzlaff NE, Wageman CR et al. Nicotinic cholinergic mechanisms causing elevated dopamine release and abnormal locomotor behavior. Neuroscience 2012; 200: 31–41.
Wang Y, Lee JW, Oh G, Grady SR, McIntosh JM, Brunzell DH et al. Enhanced synthesis and release of dopamine in transgenic mice with gain-of-function alpha6* nAChRs. J Neurochem 2014; 129: 315–327.
Webster JC, Francis MM, Porter JK, Robinson G, Stokes C, Horenstein B et al. Antagonist activities of mecamylamine and nicotine show reciprocal dependence on beta subunit sequence in the second transmembrane domain. Br J Pharmacol 1999; 127: 1337–1348.
Marks MJ, Pauly JR, Gross SD, Deneris ES, Hermans-Borgmeyer I, Heinemann SF et al. Nicotine binding and nicotinic receptor subunit RNA after chronic nicotine treatment. J Neurosci 1992; 12: 2765–2784.
Perry DC, Davila-Garcia MI, Stockmeier CA, Kellar KJ . Increased nicotinic receptors in brains from smokers: membrane binding and autoradiography studies. J Pharmacol Exp Ther 1999; 289: 1545–1552.
Hogg RC, Raggenbass M, Bertrand D . Nicotinic acetylcholine receptors: from structure to brain function. Rev Physiol Biochem Pharmacol 2003; 147: 1–46.
Breese CR, Marks MJ, Logel J, Adams CE, Sullivan B, Collins AC et al. Effect of smoking history on [3H]nicotine binding in human postmortem brain. J Pharmacol Exp Ther 1997; 282: 7–13.
Buisson B, Bertrand D . Chronic exposure to nicotine upregulates the human (alpha)4((beta)2 nicotinic acetylcholine receptor function. J Neurosci 2001; 21: 1819–1829.
Lester HA, Xiao C, Srinivasan R, Son CD, Miwa J, Pantoja R et al. Nicotine is a selective pharmacological chaperone of acetylcholine receptor number and stoichiometry. Implications for drug discovery. AAPS J 2009; 11: 167–177.
Picciotto MR, Addy NA, Mineur YS, Brunzell DH . It is not ‘either/or’: activation and desensitization of nicotinic acetylcholine receptors both contribute to behaviors related to nicotine addiction and mood. Prog Neurobiol 2008; 84: 329–342.
Fenster CP, Hicks JH, Beckman ML, Covernton PJ, Quick MW, Lester RA . Desensitization of nicotinic receptors in the central nervous system. Ann NY Acad Sci 1999; 868: 620–623.
Rogers SW, Gahring LC, Collins AC, Marks M . Age-related changes in neuronal nicotinic acetylcholine receptor subunit alpha4 expression are modified by long-term nicotine administration. J Neurosci 1998; 18: 4825–4832.
Ryan RE, Ross SA, Drago J, Loiacono RE . Dose-related neuroprotective effects of chronic nicotine in 6-hydroxydopamine treated rats, and loss of neuroprotection in alpha4 nicotinic receptor subunit knockout mice. Br J Pharmacol 2001; 132: 1650–1656.
Sparks JA, Pauly JR . Effects of continuous oral nicotine administration on brain nicotinic receptors and responsiveness to nicotine in C57Bl/6 mice. Psychopharmacology (Berl) 1999; 141: 145–153.
Flores CM, Davila-Garcia MI, Ulrich YM, Kellar KJ . Differential regulation of neuronal nicotinic receptor binding sites following chronic nicotine administration. J Neurochem 1997; 69: 2216–2219.
Nguyen HN, Rasmussen BA, Perry DC . Subtype-selective up-regulation by chronic nicotine of high-affinity nicotinic receptors in rat brain demonstrated by receptor autoradiography. J Pharmacol Exp Ther 2003; 307: 1090–1097.
Wooltorton JR, Pidoplichko VI, Broide RS, Dani JA . Differential desensitization and distribution of nicotinic acetylcholine receptor subtypes in midbrain dopamine areas. J Neurosci 2003; 23: 3176–3185.
Srinivasan R, Henderson BJ, Lester HA, Richards CI . Pharmacological chaperoning of nAChRs: a therapeutic target for Parkinson's disease. Pharmacol Res 2014; 83: 20–29.
Tumkosit P, Kuryatov A, Luo J, Lindstrom J . Beta3 subunits promote expression and nicotine-induced up-regulation of human nicotinic alpha6* nicotinic acetylcholine receptors expressed in transfected cell lines. Mol Pharmacol 2006; 70: 1358–1368.
Walsh H, Govind AP, Mastro R, Hoda JC, Bertrand D, Vallejo Y et al. Up-regulation of nicotinic receptors by nicotine varies with receptor subtype. J Biol Chem 2008; 283: 6022–6032.
Henderson BJ, Srinivasan R, Nichols WA, Dilworth CN, Gutierrez DF, Mackey ED et al. Nicotine exploits a COPI-mediated process for chaperone-mediated up-regulation of its receptors. J Gen Physiol 2014; 143: 51–66.
Parker SL, Fu Y, McAllen K, Luo J, McIntosh JM, Lindstrom JM et al. Up-regulation of brain nicotinic acetylcholine receptors in the rat during long-term self-administration of nicotine: disproportionate increase of the alpha6 subunit. Mol Pharmacol 2004; 65: 611–622.
Lai A, Parameswaran N, Khwaja M, Whiteaker P, Lindstrom JM, Fan H et al. Long-term nicotine treatment decreases striatal alpha 6* nicotinic acetylcholine receptor sites and function in mice. Mol Pharmacol 2005; 67: 1639–1647.
Perry DC, Mao D, Gold AB, McIntosh JM, Pezzullo JC, Kellar KJ . Chronic nicotine differentially regulates alpha6- and beta3-containing nicotinic cholinergic receptors in rat brain. J Pharmacol Exp Ther 2007; 322: 306–315.
Doura MB, Gold AB, Keller AB, Perry DC . Adult and periadolescent rats differ in expression of nicotinic cholinergic receptor subtypes and in the response of these subtypes to chronic nicotine exposure. Brain Res 2008; 1215: 40–52.
Perez XA, Bordia T, McIntosh JM, Grady SR, Quik M . Long-term nicotine treatment differentially regulates striatal alpha6alpha4beta2* and alpha6(nonalpha4)beta2* nAChR expression and function. Mol Pharmacol 2008; 74: 844–853.
Marks MJ, Grady SR, Salminen O, Paley MA, Wageman CR, McIntosh JM et al. alpha6beta2*-subtype nicotinic acetylcholine receptors are more sensitive than alpha4beta2*-subtype receptors to regulation by chronic nicotine administration. J Neurochem 2014; 130: 185–198.
McCallum SE, Parameswaran N, Bordia T, Fan H, McIntosh JM, Quik M . Differential regulation of mesolimbic alpha 3/alpha 6 beta 2 and alpha 4 beta 2 nicotinic acetylcholine receptor sites and function after long-term oral nicotine to monkeys. J Pharmacol Exp Ther 2006; 318: 381–388.
Perez XA, O'Leary KT, Parameswaran N, McIntosh JM, Quik M . Prominent role of alpha3/alpha6beta2* nAChRs in regulating evoked dopamine release in primate putamen: effect of long-term nicotine treatment. Mol Pharmacol 2009; 75: 938–946.
Perez XA, Ly J, McIntosh JM, Quik M . Long-term nicotine exposure depresses dopamine release in nonhuman primate nucleus accumbens. J Pharmacol Exp Ther 2012; 342: 335–344.
McCallum SE, Parameswaran N, Bordia T, McIntosh JM, Grady SR, Quik M . Decrease in alpha3*/alpha6* nicotinic receptors but not nicotine-evoked dopamine release in monkey brain after nigrostriatal damage. Mol Pharmacol 2005; 68: 737–746.
Mugnaini M, Garzotti M, Sartori I, Pilla M, Repeto P, Heidbreder CA et al. Selective down-regulation of [(125)I]Y0-alpha-conotoxin MII binding in rat mesostriatal dopamine pathway following continuous infusion of nicotine. Neuroscience 2006; 137: 565–572.
Perez XA, McIntosh JM, Quik M . Long-term nicotine treatment down-regulates alpha6beta2* nicotinic receptor expression and function in nucleus accumbens. J Neurochem 2013; 127: 762–771.
Acknowledgements
We thank Dr David L Bronson for excellent editing of this manuscript. This study was supported in part by the Research Center for Air Pollution and Health of Zhejiang University, Ministry of Science and Technology of China (2012AA020405), the National Natural Science Foundation of China grant 81273223, Young Scientists Fund of National Science Foundation of China (81301140) and NIH grant DA012844.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Wen, L., Yang, Z., Cui, W. et al. Crucial roles of the CHRNB3–CHRNA6 gene cluster on chromosome 8 in nicotine dependence: update and subjects for future research. Transl Psychiatry 6, e843 (2016). https://doi.org/10.1038/tp.2016.103
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/tp.2016.103
- Springer Nature Limited