
Abstract
A semi-comprehensive metabolomics was used to identify the candidate metabolites and genes to decipher mechanisms of resistance in wheat near-isogenic lines (NILs) containing QTL-2DL against Fusarium graminearum (Fg). Metabolites, with high fold-change in abundance, belonging to hydroxycinnamic acid amides (HCAAs): such as coumaroylagmatine, coumaroylputrescine and Fatty acids: phosphatidic acids (PAs) were identified as resistance related induced (RRI) metabolites in rachis of resistant NIL (NIL-R), inoculated with Fg. A WRKY like transcription factor (TF) was identified within the QTL-2DL region, along with three resistance genes that biosynthesized RRI metabolites. Sequencing and in-silico analysis of WRKY confirmed it to be wheat TaWRKY70. Quantitative real time-PCR studies showed a higher expression of TaWRKY70 in NIL-R as compared to NIL-S after Fg inoculation. Further, the functional validation of TaWRKY70 based on virus induced gene silencing (VIGS) in NIL-R, not only confirmed an increased fungal biomass but also decreased expressions of downstream resistance genes: TaACT, TaDGK and TaGLI1, along with decreased abundances of RRI metabolites biosynthesized by them. Among more than 200 FHB resistance QTL identified in wheat, this is the first QTL from which a TF was identified, and its downstream target genes as well as the FHB resistance functions were deciphered.
Similar content being viewed by others

Introduction
Fusarium head blight (FHB) is one of the major constraints in wheat and barley production. Several methods have been used to manage FHB in wheat, among which the use of FHB resistant cultivars is considered to be the most efficient, economic and environmental friendly method1. More than 200 QTL have been identified, including a total of 52 QTL associated with rachis resistance based on single floret inoculation2. Among these, the QTL-2DL is one of the major and the most stable QTL across different genetic backgrounds and various environments2. This was first identified from Wuhan-1, a Chinese genotype, in which it explained up to 28% of the total phenotypic variation3. However, the genetic determinants undelying these QTL still remain largely unknown. Thus, the identification and functional elucidation of genes from these QTL are very important for their use in breeding.
A recent transcriptomic study of NILs containing QTL-2DL attempted to identify candidate genes, but failed to identify any QTL specific genes for resistance to FHB4. Apart from this, plentiful transcriptomics and metabolomics studies reported numerous differentially expressed genes and accumulation of metabolites involved in FHB resistance but none of them were validated for gene functions5,6,7,8,9,10,11 except for TaACT gene in QTL-2DL12. Therefore, functional analysis of mapped QTL using alternative disciplines like metabolomics integrated with genomics is considered as one of the best tools to decipher the functions of underlying genes. Semi-comprehensive metabolite profiling of barley13,14 and wheat7,8,13,14,15,16 genotypes with varying levels of resistance to FHB has led to the identification of several RR metabolites and their role in resistance. Recently a semi-comprehensive metabolomics study of barley genotypes, resistant and susceptible to FHB identified a transcription factor HvWIN1 that regulated downstream resistance genes to biosynthesize fatty acids that were deposited to reinforce cuticle to contain Fg infection17. In potato, not only the RR metabolites against Pytophthora infestans but also their biosynthetic genes were identified18,19 and functionally validated20. Integrated transcriptomics and metabolomics have revealed induction of hierarchies of resistance genes and differential accumulation of defense related metabolites in potato against late blight21. The resistance in plants against biotic stress is considered to be due to hierarchies of resistance (R) genes with regulatory roles such as elicitor/effector recognition receptors (RELRR and/or RERR), phytohormone biosynthetic genes (RPHR), mitogen-activated protein kinase (RMAPK), and transcription factors (RTF) which regulate the metabolic pathway network genes that biosynthesize resistance related metabolites (RRRM) and/or RR proteins (RRRP) to suppress or contain the pathogen to initial infection22.
The WRKYs are one of the largest families of transcriptional regulators in plants and are involved in biotic and abiotic stress responses such as metabolite biosynthesis, cell wall formation, senescence, trichome development, and hormone responses23,24,25,26,27. WRKY proteins have either one or two WRKY DNA binding domains with a consensus amino acid sequence, WRKYGQK at N-terminal end and a zinc-finger motif at their C-terminal end25,28. The WRKY TFs regulate target genes by binding to the specific DNA sequence motif (T)TGAC(C/T), which is known as the W-box28. WRKYs may be positive or negative regulators of downstream defense mechanisms25. For example, WRKY TFs regulates the production of a variety of phenolic-based compounds including lignin23,29,30. Knocking down of StWRKY1 in potato compromised resistance to P. infestans due to reduced accumulation of hydroxycinnamic acid amides20. OsWRKY45 is a positive regulator of terpenes such as momilactone, phytocassane, and oryzalexin accumulation, which are involved in plant defense against pathogens and herbivores by activating biosynthetic gene expression31. Silencing of TaWRKY53 in wheat has confirmed its role in aphid defense32. Two WRKY genes, NaWRKY3 and NaWRKY6 coordinate defense response against herbivory in tobacco33. AtWRKY33 is known to regulate biosynthesis of camalexin, and a positive regulator of resistance against the necrotrophic fungi Botrytis cinerea and Alternaria brassicicola34. Contrastingly, AtWRKY38 and AtWRKY62 are negative regulators of basal resistance to Pseudomonas syringae35. Over expression of AtWRKY48, showed negative effects towards resistance to P. syringae36. Similarly, HvWRKY1 and HvWRKY2 are negative defense regulators of powdery mildew resistance in barley37. Gossypium hirsutum (Gh) WRKY25 negatively regulates B. cinerea infection in transgenic tobacco38. Over expression of GhWRKY27a reduced resistance to Rhizoctonia solani infection in tobacco39. AtWRKY50/51 involved in enhanced resistance to A. brassicicola but at the same time it also involved in increased susceptibility to B. cinerea by mediating salicylic acid and low oleic acid dependent repression of jasmonic acid signaling40. In rice, overexpression of OsWRKY13 up-regulated the phenylpropanoid pathway genes and at the same time down-regulated those involved in terpenoid biosynthesis, illustrating the important role of WRKYs in differential regulation of diverse metabolite biosynthetic genes involved in plant defense41. This shows that the WRKYs are of great interest to reveal diverse biotic stress responses in plants.
In wheat, despite huge efforts the elucidation of molecular functions of WRKY genes is still limited. There are at least 200 WRKY genes in wheat but unfortunately only a few of them were studied in detail42. Therefore, identification and functional analysis of WRKY TFs in wheat is very important. In this study, we identified and characterized TaWRKY70 TF from bread wheat. The TaWRKY70 gene was confirmed to be located within the QTL-2DL region, and imparted resistance against FHB by accumulating PAs and HCAAs through regulation of downstream biosynthetic resistance genes TaDGK, TaGLI1, and TaACT.
Materials and Methods
Plant production and experimental design
The near-isogenic lines (NILs) used here were derived from a cross BW301 X HC3743,43. The BW301 is FHB susceptible hard red spring wheat line from western Canada, and HC374 is resistant to FHB (derived from the cross Wuhan1 x Nyubai). The NILs were genotyped with microsatellite markers. Homozygous lines with susceptible background differing only in the alleles of the QTL-2DL locus and did not have any other known FHB resistance QTL located on chromosomes 3B, 4B, 5A and 6B were used to derive the NILs44. The seeds of NILs with FHB susceptible and resistant alleles of QTL-2DL were obtained from Dr. McCartney, AAFC, Winnipeg, Canada. The experiment was laid out in a randomized complete block design (RCBD) with two genotypes (resistant and susceptible NILs), two inoculations (pathogen and mock-solution) and five biological replications over time with nine plants in three pots as experimental units. The plants grown in greenhouse were maintained at temperature 23 ± 2 °C, photoperiod of 16 h, and relative humidity 70 ± 10%, throughout the growing period. A complex slow releasing fertilizer 14:14:14 (NPK) at the rate of 5 g per pot and 0.03% of trace elements was applied every 15 days to each pot.
Pathogen production and inoculation
The Fg isolate (GZ-3639, obtained from Dr. R. H. Proctor, USA) was grown on potato dextrose agar at 26 °C for four days. For spore production, Fg was further sub-cultured on Rye B agar media and kept inverted by exposing the plates to near UV light for three days. From a seven day old culture macroconidia were harvested and spore count was adjusted to 1 × 105 macroconidia ml−1 using a hemocytometer (American Scientific Products, USA)13. The experimental units consisted of at least 10 spikes per replication selected from three pots containing three plants in each. Three alternate pairs of wheat spikelets at 50% anthesis stage were point inoculated with 10 μl of either macroconidial suspension or mock-solution using a syringe (GASTIGHT 1750 DAD, Reno, USA). Plants were covered with transparent plastic bags sprayed with water to maintain high humidity and the bags were removed at 48 hours post inoculation (hpi).
Sample collection, metabolite analysis using liquid chromatography-high resolution mass spectrometry (LC-HRMS) and data processing
At 72 hpi, ten spikes for each replicate were harvested and the spike region with three inoculated and three alternate un-inoculated pairs of spikelets was retained. Spikelets (10 × 6 = 60 pairs) from rachis (10 pieces) were separated, and both the samples were frozen immediately in liquid nitrogen and separately stored at −80 °C until further use. Metabolites were extracted from rachis samples in 60% ice cold aqueous methanol. The 5 μl of clear sample extract was used for metabolite analysis based on LC-HRMS (at IRCM, Montreal, Canada) as previously described13.
The LC-HRMS output Xcalibur RAW files were converted into mzXML format. The data was analyzed using MZMine2, and the peaks were identified as metabolites based on monoisotopic mass and fragmentation match with databases and available literature7,13,19,20. The relative peak intensities of monoisotopic masses of metabolites were subjected to Students t-test (SAS v 9.3) in pair wise treatment combinations (RP vs RM, RM vs SM, SP vs SM and RP vs SP, where RP = resistant NIL inoculated with pathogen, RM = resistant NIL inoculated with mock-solution, SP = susceptible NIL inoculated with pathogen, SM = susceptible NIL inoculated with mock-solution) to identify treatment significant metabolites. The abundances of peaks significant at P < 0.0545, and false discovery rate threshold of 0.0546 were retained. False discovery rate of peaks depends mainly on the signal/noise (S/N) ratio; lower the ratio higher is the false discovery rate. Therefore, S/N ratio was kept high to avoid any false discovery17. The metabolites, significantly higher in abundance in resistant than susceptible NIL were considered as resistance related (RR) metabolites. Further, these metabolites were grouped into RR constitutive (RRC = RM > SM) and RR induced (RRI = (RP > RM) > (SP > SM)) metabolites. The fold change (FC) in abundance of metabolites in NIL-R was calculated relative to NIL-S (NIL-R/NIL-S)7. Only the highly significant and high FC RRI metabolites were prioritized to increase the probability to identify the most effective resistance candidate genes.
Disease severity and fungal biomass assessment
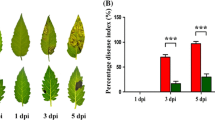
To evaluate rachis resistance in wheat genotypes, two NILs with resistant and susceptible alleles were planted in RCBD with three biological replications each with three pots sown at three day intervals. Ten spikes were selected, and in each one pair of spikelets in the mid region was inoculated with Fg to assess the spread of pathogen from the inoculated spikelet to other through rachis. Plants were covered with transparent plastic bags sprayed with water to maintain high moisture and the bags were removed at 48 hpi. Observations on the number of spikelets diseased were taken at three day intervals until 15 days post inoculation (dpi). Dark brown discolored and/or bleached spikelets were considered as diseased. Disease severity in NILs was quantified as proportion of spikelets diseased (PSD) in a spike, from which the area under the disease progress curve (AUDPC) was calculated8. Data was analyzed for significance based on ANOVA using SAS program (SAS v 9.3).
A separate experiment was conducted to assess resistance based on fungal biomass. The experiment was conducted as RCBD with two NILs with two inoculations (pathogen or mock) and three biological replications with two pots each containing three plants. At 50% anthesis stage, five spikes were selected and three alternate pairs of spikelets were point inoculated with 10 μl of either macroconidial suspension in water or mock-solution using a syringe (GASTIGHT 1750 DAD, Reno, USA). After inoculation, plants were covered with polyethylene bags sprayed with water and bags were removed at 48 hpi. The rachis regions containing six pairs of spikelets were harvested at six dpi and immediately frozen in liquid nitrogen and stored at −80 °C until further use. The genomic DNA was extracted and the fungal biomass was quantified using a real-time qPCR by measuring relative copy number of fungal housekeeping gene Tri6. The abundance of this gene was normalized with TaActin. The relative gene copy number of Tri6 based on real-time qPCR was used to estimate the amount of fungal biomass47. Statistical significance was calculated using Students t-test.
Candidate gene identification based on high fold-change RR metabolites and their physical localization within QTL-2DL
The RRI metabolites with high FC in abundance were mapped on to metabolic pathways to find their catalytic enzymes and the coding genes, which were identified using genomic databases (such as KEGG, MetaCyc, PlantCyc and Arabidopsis Acyl metabolic pathways) and available literature. Presence of SSR markers, wmc245, gpw8003, gwm539 and gwm608, were used to define the interval for QTL-2DL. Some flanking marker sequences available at GrainGenes database were retrieved, and if not available, they were sequenced in our lab (gpw8003, gwm539 and gwm608). Flanking marker sequences were subjected to BLAST48 search in IWGSC chromosome survey sequence repository (Wheat CSS genome reference v2) to physically localize the markers and to define the QTL-2DL interval temporarily. Later, the candidate genes identified based on high FC RRI metabolites were BLAST searched to check their co-localization within the temporarily mapped QTL-2DL region. Further, this was confirmed by gene prediction using the 2DL chromosome arm sequence from the IWGSC (Chromosome arm sequence assemblies) between the two flanking makers (wmc245 and gwm608). Contigs identified as the best hit for candidate genes were retrieved from database and the gene prediction was performed using SoftBery–FGENESH (http://linux1.softberry.com/berry.phtml?topic=fgenesh&group=programs&subgroup=gfind) program to study the gene structure. The identified gene was amplified using gene specific primers designed using NCBI Primer-BLAST tool (http://www.ncbi.nlm.nih.gov/tools/primer-blast/). Gene prediction and synteny mapping was also performed with rice and brachypodium to predict and locate other putative genes in the QTL-2DL region.
Gene cloning, sequencing and sequence analysis
The genomic DNA was isolated and the full length TaWRKY70 gene was amplified using primer pairs TaWRKY_F and TaWRKY_R from NILs. Gene amplification was conducted using a thermal cycler (Bio-Rad, Mississauga, ON, Canada) with the following steps: Initial denaturation at 95 °C for 5 min followed by 35 cycles of 94 °C for 30 s, 55 °C for 1 min, 72 °C for 2 min followed by a final extension at 72 °C for 10 min. PCR products were separated on a 1% agarose gel. A band size corresponding to ~1300 bp was then purified from the gel, cloned into the pGEM®-T Easy vector (Promega, USA), and sequenced using the ABI Automated DNA sequencer. DNA sequences were translated to amino acid sequences using the ExPASy Translate Tool (http://web.expasy.org/translate/). The MOTIF Search tool (http://www.genome.jp/tools/motif/) was used to search for functional domains present in deduced amino acid sequence. Further, these results were confirmed using PROSITE tool (http://www.expasy.ch/prosite) and NCBI Conserved Domain Database (NCBI CDD). The multiple sequence alignment was performed using MultAlin (http://multalin.toulouse.inra.fr/multalin/) and maximum-likelihood phylogenetic relationships were determined using Phylogey.fr (http://www.phylogeny.fr/) program.
RNA isolation and gene expression based on qRT-PCR
For relative quantification of transcript expression, the total RNA of rachis was isolated from five biological replicates using RNeasy plant mini kit (Qiagen Inc.). Purified total RNA (1–2 μg) was used to reverse transcribe RNA into cDNA using iScript cDNA synthesis kit (BioRad, ON, Canada). Using equal quantity of cDNA (20 ng) for each sample, real-time qRT-PCR was performed using Qi SYBR Green supermix (BioRad, Canada) in a CFX384TM Real-Time system (BioRad, Canada). The mRNA abundance of target gene was normalized with TaActin transcript level. PCR results were analyzed using comparative delta-delta Ct method (2−ΔΔCT)49. The statistical significance of observations was analyzed based on Students t-test.
Nuclear localization assay
The LocSigDB (http://genome.unmc.edu/LocSigDB/) was used for nuclear localization signal (NLS) prediction. The full-length coding region of TaWRKY70 was amplified and cloned into pCX-DG vector containing green florescence protein (GFP) and Cauliflower Mosaic Virus (CaMV) 35S promoter50. For subcellular localization study, TaWRKY70 + GFP fusion and GFP alone (as a control) were transfected into potato protoplasts using a polyethylene glycol-calcium method51. Transfected protoplasts were incubated at 23 °C for 16 h and analyzed for GFP florescence by florescence microscopy. This experiment was conducted three times.
Luciferase (LUC) transient expression assay
The coding region of the TaWRKY70 gene and the promoters of TaACT, TaDGK and TaGLI1 from the resistant genotype were amplified, cloned into pGEM®-T Easy vector (Promega, USA), and confirmed by sequencing. This was followed by sub-cloning into the in FU63 (CD3-1841) vector52. For LUC transient expression assays, reporter plasmids (ACTp-LUC or DGKp-LUC or GLI1p-LUC or vector control having 30 bp DNA fragment without w-box), effector constructs containing TaWRKY70, and 35 S::β-glucuronidase (GUS) internal control were co-transformed into potato protoplasts. The protoplasts were pelleted and re-suspended in 1× cell culture lysis reagent (Promega, USA). GUS fluorescence was measured using a Modulus luminometer/fluorometer with a UV fluorescence optical kit (Fluorescence Microplate Reader; BioTek, USA). The experiment was carried out in three replicates; each replicate contained 20 μl protoplast lysate and 100 μl LUC mix. LUC activity was detected with a luminescence kit using LUC assay substrate (Fluorescence Microplate Reader). The relative reporter gene expression levels were expressed as LUC/GUS ratios, which were used to discriminate treatments. The significance between treatments and vector control was analyzed using students t-test at P < 0.01.
Construction of BSMV vectors and virus induced gene silencing of TaWRKY70
For transient gene silencing, 283 bp fragment of TaWRKY70 gene was selected with efficient siRNA generation and no off-target genes into the modified viral genome using siRNA Scan tool (http://bioinfo2.noble.org/RNAiScan.htm), and a BLAST search of fragment against GenBank database. We chose the most divergent sequence containing (3′ UTR region) to increase the specificity (Fig. S1a). The fragment was amplified from cDNA using the primers listed in Table S1. Fragment was cloned into the pGEM®-T Easy Vector (Promega Corp., WI, USA) and sequence was confirmed. Plasmid DNA was digested using NotI (New England Biolabs, MA, USA), thereby generating NotI ends in DNA fragment. The cDNA fragment was subsequently ligated to pSL038-1 vector a plasmid encoding a modified BSMV γ genome segment with a NotI cloning site downstream of the γb gene53 (Fig. S1b and c). The pSL038-1 vector carrying either phytoene desaturase (PDS) or without any gene served as positive and negative controls respectively. The plasmids BSMV α, pγSL038-1 were linearized with Mlu1 restriction enzyme whereas; BSMV β was linearized by using Spe1 enzyme. Linearized plasmids were converted into capped in-vitro transcripts using mMessage Machine™ T7 in-vitro transcription kit (Ambion, Inc., Austin, TX, USA), following the manufacturer’s protocol.
The experiment was designed as a RCBD with one genotype the resistant NIL, two treatments of silenced or non-silenced with five biological replicates over time each with two pots. Plants produced as detailed earlier were rub-inoculated with all the three in-vitro transcript reactions (α, β and γ BSMV) in 1:1:1 ratio (1 μl of each) along with 22.5 μl inoculation buffer (1% sodium pyrophosphate, 1% bentonite, 1% celite in 0.1 M glycine, 0.06 M dipotassium phosphate)54 that facilitated viral entry and infection. To increase the silencing efficiency both the flag leaf and spikelets were rub-inoculated55. The experimental units consisted of five plants from two pots with a total of ten spikes per replicate which were separately rub-inoculated with test (BSMV + TaWRKY70) and negative control (BSMV:00). The flag leaves and spikelets rub inoculated with BSMV + PDS were served as positive controls.
Confirmation of gene silencing by qRT-PCR, estimation of fungal biomass and abundances of targeted metabolites
At 12 dpi with virus, three alternate spikelets were inoculated with 10 μl of Fg spore suspension and covered with water sprayed plastic bags. The bags were removed at 48 hpi and five out of ten spikes were collected at 72 hpi for qRT-PCR and metabolite analysis in rachis. Similarly, at six dpi, remaining spike samples were collected for the relative quantification of fungal biomass in rachis as relative gene copy number of the fungal housekeeping gene Tri6 over TaActin housekeeping gene.
Results
Disease severity and fungal biomass in spikelets and rachis of NIL
The disease severity in spikelets of NILs, with alternate alleles for resistance at QTL-2DL with one pair of mid spikelets inoculated with Fg was assessed as proportion of spikelets diseased in a spike (PSD), from which the AUDPC was calculated. The AUDPC was significantly higher in NIL-S (2.33) compared to NIL-R (1.48), with a FC = 1.57. The rachis resistance in NILs was assessed based on the amount of fungal biomass in the rachis regions containing six pairs of spilelets, where three alternate pairs were inoculated with Fg. The fungal biomass estimated at 6 dpi as relative gene copy number of Tri6 normalized to TaActin based on real-time qPCR was also significantly higher (5.8 FC) in NIL-S than in NIL-R (Kage et al.12). This clearly demonstrated a high level of rachis resistance associated with QTL-2DL against FHB.
Metabolite profiles of NILs
Semi-comprehensive metabolomics of rachis samples collected at 72 hpi identified several differentially accumulated RR metabolites in NILs with contrasting alleles at QTL-2DL for resistance against Fg. The significant metabolites were categorized into RRC and RRI. The RRI metabolites with high FC in abundance mainly belonged to two chemical groups: (i) phosphatidic acids and derivatives (PAs): [(1-heptadecanoyl,2-(5Z,8Z,11Z,14Z-eicosatetraenoyl)-sn-glycero-3-phosphat = PA(17:0/20:4(5Z,8Z,11Z,14Z) (FC = 54.3)), (1-pentadecanoyl-2-(8Z,11Z,14Z-eicosatrienoyl)-glycero-3-phosphate = (PA(15:0/20:3(8Z,11Z,14Z)(FC = 9.5)) and (1-(9Z-nonadecenoyl)-2-(13Z,16Z-docosadienoyl)-glycero-3-phosphate = (PA(19:1(9Z)/22:2 (13Z,16Z)(FC = 2.3))]; (ii) hydroxycinnamic acid amides (HCAAs): p-coumaroylagmatine (FC = 28.7), p-coumaroyputrescine (FC = 9.5) (Table 1).
Identification of candidate genes in QTL-2DL
Putatively identified high FC RRI metabolites were mapped on to metabolic pathways and the candidate genes (RRRM) corresponding to the enzymes that biosynthesized these RRI metabolites were identified using the public databases and available literature. The agmatinecoumaroyl transferase (ACT) is a rate limiting enzyme in the biosynthesis of HCAAs such as coumaroylagmatine and coumaroylputrescine56,57. Whereas, the diacylglycerol kinase (DGK) and glycerol kinase (GLI1) are important enzymes in the biosynthesis of PAs in plants58,59,60,61. BLAST analysis positioned the closest gene matches for these enzymes, ACT, DGK and GLI1 within the presumed interval of QTL-2DL (Fig. 1a). The genes (TaACT and TaDGK), including their promoters were sequenced and the sequences were deposited in NCBI database. Sequence comparison of TaACT and TaDGK in contrasting NILs revealed that only the TaACT was polymorphic but not the TaDGK. This led us to suspect a possible involvement of TF in regulating PAs pathway. While searching for other candidate genes, coincidentally a WRKY like TF was found in the QTL-2DL region based on gene prediction. A list of other predicted genes present in QTL-2DL identified based on synteny study with rice and brachypodium are given in Supplementary Data (Table S2), but their roles in FHB resistance are yet to be confirmed.

(a) Physical map of the QTL-2DL on long arm of wheat chromosome 2D. Location of the of candidate genes identified within the QTL-2DL region are shown on the right side and flanking markers on the left side of the 2D chromosome; (b) Schematic diagram depicting the TaWRKY70 gene structure containing exon, intron and coding regions; (c) Conserved domain predicted based on NCBI Conserved Domain Database Search, it shows presence of conserved WRKY domain; (d) Figure showing the characteristic features of Group III WRKY transcription factors, showing the DNA binding domain containing 69 amino acids underlined. It has WRKYGQK and C2HC, WRKY and zinc finger conserved motifs, respectively.
TaWRKY gene sequencing and sequence analysis
The WRKY TFs are known to be involved in regulating plant responses to biotic and abiotic stresses. Therefore, we sequenced the predicted full length WRKY gene from the genomic DNA of NILs. Sequence analysis based on FGENESH suggested that WRKY has three exons and two introns (Fig. 1b) and the intron-exon boundaries were confirmed to be AG and GT at the acceptor and donor sites respectively through FSPLICE (http://linux1.softberry.com/berry.phtml?topic=fsplice&group=programs&subgroup=gfind). The full length sequence of TaWRKY was 1288 bp in length containing an open reading frame (ORF) of 1165 bp, a 96 bp 3′ untranslated region (UTR) and 27 bp 5′ UTR (Fig. 1b). One of the plant canonical polyadenylation signals, a six-nucleotide near-upstream element (NUE - AAATAA) was found in the 3′ UTR at the position 1251 to 1257 bp (Fig. 1b)62. The complete genomic sequence was submitted to NCBI and was assigned a GenBank accession number KU562861. The putative protein encoded by TaWRKY consisted of 290 amino acids. It has conserved characteristic DNA-binding domain comprising a single WRKY domain and Cys2-His-Cys type zinc-binding motif spanning from position 98 to 166 amino acids (Fig. 1c and d). The Group III WRKYs differs from groups I and II in its altered C2-HC zinc finger motif C-X7-C-X23-HX-C63 (Fig. 1d). Multiple sequence alignment and phylogenetic analyses indicated that the TaWRKY belonged to Group III type of WRKY family (Fig. 2a). The TaWRKY showed 99% identity with Aegilops tauschi putative WRKY70 orthologues of AtWRKY70 and wheat WRKY45. Recent studies have placed several WRKY TFs, including AtWRKY70, downstream of NPR1 in the SA signaling pathway on the basis of transcriptional profiling using Arabidopsis npr1 mutant plants64. Whereas, rice WRKY45 which is orthologue to AtWRKY70 acts under SA pathway but independent of NH1 (orthologue of AtNPR1)65. Therefore, it was designated as OsWRKY45 but not OsWRKY7065. Based on this we putatively designated TaWRKY as TaWRKY70 like TF gene (or as TaWRKY70) since we are not sure about the position of wheat WRKY45/70 in SA pathway.

(a) Phylogenetic relationships of TaWRKY70 (in green box) with other plant WRKY sequences obtained from NCBI database for wheat (TaWRKY1A, TaWRKY19A, TaWRKY74a, TaWRKY53a, TaWRKY71, TaWRKY74b, and TaWRKY19b), rice (OsWRKY45 and OsWRKY32), arabidospsis (AtWRKY18, AtWRKY6, AtWRKY70 and AtWRKY4), barley (HvWRKY32), Aegilops tauschii (WRKY70), Triticum urartu (WRKY70). Neighbor-joining tree representing relationships among WRKY proteins from different plant species. Numbers in red color represents branch length; (b) Comparison of DNA sequence variation between NIL-R, NIL-S and Chinese spring TaWRKY70. It shows a single nucleotide polymorphism at 294 bp position; (c) Comparison of protein sequence variation between NIL-R, NIL-S and Chinese spring TaWRKY70. Box shows the predicted nuclear localization signal. It shows 14 amino acids deletion in NIL-S as a result of SNP at 294 bp in the DNA sequence causing shift in splice site.
Sequence variation of TaWRKY70 between NILs and differential gene expression during Fg infection
Multiple sequence alignment of TaWRKY70 gene between NILs and T. aestivum cv. Chinese Spring revealed single nucleotide polymorphisms (SNP) in NIL-S at 294 bp position (Fig. 2b), which is exactly at the first exon-intron junction. This resulted in 14 amino acid deletion in predicted protein sequence by shifting open reading frame (ORF) upstream to its normal, causing a lack of 42 bp sequence in the first exon of the TaWRKY70 transcript (Fig. 2c). Further the deleterious effect of these SNPs on protein functionality was confirmed based on in-silico analysis by Phyre 2 Investigator (data not shown). However, additional experimental proofs are needed to confirm the mutation and to know how it induces the truncated protein in-vivo and alters the transcriptional activity of TaWRKY70 ultimately affecting resistance against Fg.
The relative gene expression of TaWRKY70 following Fg inoculation was significantly (P < 0.05) higher (2.3 FC at 48 hpi and 2.0 FC at 72 hpi) in pathogen treated NIL-R compared to pathogen treated NIL-S. Similarly, its expression was higher in pathogen treated NIL-R compared to mock treated samples of both the NILs at both the time points (48 hpi and 72 hpi) though the expression levels were slightly lower at 72 hpi (Fig. 3a), suggesting TaWRKY70 has a potential role against Fg resistance in the early stages of defense through activation of downstream genes.

Here target gene expression is normalized to reference gene TaActin. (a) Relative transcriptional changes of TaWRKY70 at 48 and 72 hpi; (b) Gene expression of TaDGK and TaGLI1 and TaACT (which did not show any expression in mock treated samples) at 72 hpi. RP = Resistant pathogen, RM = Resistant mock, SP = Susceptible pathogen and SM = Susceptible mock. Significant differences in expression levels of RP as compared to SP using Students t-test: *P < 0.05; **P < 0.01.
Gene expression, promoter analysis of RR metabolite biosynthetic genes and their physical interaction with RTaWRKY70
At 72 hpi, the relative gene expression levels of downstream RRRM genes: TaDGK (2.4 FC), TaGLI (2.0 FC), and TaACT (3.3 FC) were significantly (P < 0.01) higher in NIL-R than in NIL-S after Fg inoculation; the trend was similar with TaWRKY70 (Fig. 3b). To further study the downstream RRRMtargets of TaWRKY70, we performed promoter analysis of RTaACT, RTaDGK and RTaGLI1. The promoter sequences from −1 bp to −1000 bp upstream of ATG start site were considered for analysis using a PLACE database (http://www.dna.affrc.go.jp/PLACE/) and manual search. The promoter analysis of these genes revealed the presence of putative W-box sequence in their promoters within −500 bp (Table 2). Further we confirmed the potential interaction of TaWRKY70 with these downstream RRRM genes using Arabidopsis as a search organism in GeneMANIA software (http://www.genemania.org/). Resulting networks showed a clear interaction between TaWRKY70 and the RRRM genes (TaACT, TaDGK and TaGLI1) (Fig. S2). The physical interaction in-vivo was confirmed based on luciferase assay. The reporter and effector constructs were transformed into potato protoplasts to check the expression of LUC reporter. We found drastic increase in the expression of LUC reporter in TaACT (34.7), TaDGK (32.9) and TaGLI1 (31.6) promoters as compared with vector alone (4.5) (Fig. 4a,b), suggesting TaWRKY70 regulates transcription of TaDGK, TaGLI1 and TaACT to biosynthesize PAs and HCAA, thus confirming their in-silico predicted interaction.

(a) Constructs used in the transient expression assay and (b) relative luciferase (LUC) reporter activity by TaWRKY70. The relative reporter gene expression levels were expressed as LUC/GUS ratios. Values are averages of three replicates. Significant differences in expression levels in promoters compared with vector based on Student’s t-test: **P < 0.01.
Nuclear localization of TaWRKY70 protein
To investigate the subcellular localization of TaWRKY70 protein, we used LocSigDB (http://genome.unmc.edu/LocSigDB/) with default setting. We found three conserved amino acids region (KRK) potentially acting as NLS for TaWRKY70 protein (Fig. 5a). Additionally, we used transient expression system in potato protoplasts to characterize the subcellular localization of TaWRKY70 protein. This demonstrated that TaWRKY70 + GFP fusion protein was localized in the nucleus, while the control vector (GFP alone) was expressed in the cytosol and nucleus (Fig. 5b). These results agreed with the subcellular localization prediction, suggesting that the TaWRKY70 is a nuclear protein.

(a) Nuclear localization signal (NLS) predicted. Red colored amino acid region in bold font and underlined is a NLS; (b) Nuclear localization analysis. Constructs consisting of either TaWRKY70-GFP fusion or GFP alone were used to transiently transform into potato protoplasts. Free GFP and TaWRKY70-GFP fusion proteins were transiently expressed in potato protoplast and observed with a fluorescence microscope. Here, the extreme left panel (GFP fluorescence), the middle panel (bright field) and the right panel (merged view of two images). Transient expression assays were conducted at least three times.
Response to Fg infection after knocking down of TaWRKY70 in wheat
Based on the changes in TaWRKY70 expression after Fg inoculation, the BSMV-VIGS system was employed to knock down the transcription of TaWRKY70 and to further investigate its function in response to Fg infection. The feasibility and silencing efficiency of the BSMV-VIGS system in NIL-R was tested using the wheat phytoene desaturase (TaPDS) as a positive control. At 12 dpi with BSMV:TaPDS, photo-bleaching symptoms started appearing on wheat spikes when TaPDS was silenced (Fig. S3). Therefore, the BSMV-VIGS system was used for assessing the potential roles of TaWRKY70 in wheat resistance against Fg infection. Under the same conditions, the BSMV:TaWRKY70 (test/silenced) and BSMV:00 (control/non-silenced) recombinant vectors were rub-inoculated onto the NIL-R.
To study the efficiency of silencing of TaWRKY70 in NIL-R plants that had been infected with recombinant BSMV vectors, the relative expression levels in rachis were detected by qRT-PCR. The relative expression of TaWRKY70 was significantly (P < 0.01) reduced by 86.04% in plants infected with BSMV:TaWRKY70 compared to BSMV:00 infected plants at 72 hpi with Fg confirming the silencing or down regulation of target gene in wheat rachis (Fig. 6a). To further determine whether silencing of TaWRKY70 in NIL-R compromised resistance to Fg infection, the fungal biomass of Fg was estimated by measuring the relative transcript levels of the Fg housekeeping gene Tri6 over TaActin. Fungal biomass was significantly higher (P < 0.01) in silenced plants as compared to nonsilenced control plants (Fig. 6b). These results suggest that the enhanced susceptible phenotypes observed in NIL-R inoculated with the Fg were due to the silencing of TaWRKY70.

(a) Confirmation of knocking down of TaWRKY70 by assaying relative transcript expression of TaWRKY70 normalized to reference gene TaActin in silenced plant (BSMV:TaWRKY70) compared to non-silenced (BSMV:00) at 3 dpi after Fg inoculation; (b) Fungal biomass in BSMV-infected plants at 6 dpi with Fg. Relative copy number of Tri6 fungal housekeeping gene (=fungal biomass) was quantified in TaWRKY70 knocked down (BSMV:TaWRKY70) plants and compared with control (BSMV:00). Here relative target gene copy number is normalized to reference gene TaActin; and (c) Relative transcript levels of TaDGK, TaACT and TaGLI1 assayed individually in TaWRKY70 knocked down (BSMV:TaWRKY70) plants compared to non-silenced (BSMV:00) at 3 dpi after Fg inoculation. Here target gene expression is normalized to reference gene TaActin; (d) Relative metabolite abundances of RRI metabolites in silenced (BSMV:TaWRKY70) and non-silenced (BSMV:00) NIL-R at 3 dpi after Fg inoculation. PA-1 – PA(17:0/20:4(5Z,8Z,11Z,14Z)), PA-2 – PA(15:0/20:3(8Z,11Z,14Z)), PA-3 – PA(19:1(9Z)/22:2(13Z,16Z)), Cou-Ag – p-coumaroylagmatine and Cou-put – p-coumaroylputrescine. Significant differences in expression levels as compared in silenced (BSMV:TaWRKY70) with non-silenced (BSMV:00) using Students t-test: *P < 0.05; **P < 0.01.
Silencing of TaWRKY70 affected transcriptional response of RRRM genes and RR metabolite accumulation
It is evident from the confirmation of the presence of W-Box in promoters based on GeneMANIA analysis, and luciferase assay that TaWRKY70 physically interacts with the downstream targets like TaACT, TaDGK and TaGLI1. Therefore, to check whether knocking down of TaWRKY70 has affected the transcriptional responses of downstream candidate genes, the relative expression levels of these genes were estimated using qRT-PCR in silenced samples. Coincidently, the expression levels of TaACT (FC = 3.44), TaDGK (FC = 1.36) and TaGLI1 (FC = 1.88) were significantly down regulated in silenced as compared to non-silenced samples, further confirming these genes as potential interacting targets of TaWRKY70 (Fig. 6c). Additionally, to confirm the biochemical and molecular mechanisms of the involvement of TaWRKY70 to resist FHB, metabolite profiling was performed in silenced and non-silenced NIL-R plants. In NIL-R silenced plants, the abundances of candidate RR metabolites such as, PAs: (PA(17:0/20:4(5Z,8Z,11Z,14Z)), PA(15:0/20:3(8Z,11Z,14Z)) and PA(19:1(9Z)/22:2(13Z,16Z)) with FC = 8.3, 2.5 & 1.6, respectively), and HCAAs: p-coumaroylagmatine (FC = 6.7) and p-coumarolyputrescine (FC = 3.5) were significantly (P < 0.01) reduced compared to non-silenced plants (Fig. 6d). This clearly implied the involvement of TaWRKY70 in the regulation of downstream RR metabolite biosynthetic genes in NIL-R, production of RRI metabolites and an eventual resistance against FHB.
Discussion
Resistance in plants against pathogen attack is controlled by several hierarchies of resistance genes that eventually biosynthesize resistance related metabolites and proteins that directly suppress and/or contain the pathogen to initial infection through their antimicrobial and/or cell wall reinforcement properties22,66. Plant RELRR genes recognize the pathogen produced elicitors and trigger downstream RMAPK and RTFgenes, which regulate the RRRMand RRRPgenes that biosynthesize RR metabolites and proteins. Thus it is crucial to map the network of plant genes involved in the hierarchy to resist the pathogen. Some of these genes have major or minor resistance effects. In this study, we report wheat TF as one of the candidate genes with significant FHB resistance effect, through regulation of several downstream RRRM genes that biosynthesize RRI metabolites that directly suppress and/or contain pathogen advancement.
Metabolites are the end products of genes, and thus they better represent the phenotype. Accordingly, metabolite profiling was used as a primary tool to explore the RRRM genes involved in NILs with contrasting levels of FHB resistance at QTL-2DL. Only the high FC RRI metabolites were considered to explore the RRRM genes. The PAs and HCAAs were the major RRI metabolites found in wheat rachis after pathogen invasion. Phosphotidic acid is the essential intermediate for the de-novo biosynthesis of all glycerolipids67. PAs and their derivatives are basically involved in signaling and structural fortification of the cell wall through deposition of glycerol 3-phosphates68,69. This also helps in the suppression of cell death induced by hydrogen peroxide70. The HCAAs are not only phytoalexins that suppress pathogens due to their antimicrobial activity but also are deposited in the secondary cell walls reinforcing them to contain the pathogen to initial infection71. Considering these roles of RRI metabolites identified here, we mapped these metabolites on to their metabolic pathways to identify their biosynthetic genes. Based on available information from databases and literature, the genes involved in biosynthesizing these RRI metabolites identified were: TaACT, TaDGK and TaGLI1. Several studies have reported the biotic stress resistance roles of ACT56,57,72, DGK73,74 and GLI159,75,76. While searching for other genes in the QTL-2DL region we found a gene encoding WRKY like protein. The WRKY proteins are regulatory in nature and have a role in plant biotic and abiotic stress resistance by controlling the transcription of downstream R genes by binding to W-Box cis-elements present in their promoters77. Further, sequence analysis of TaWRKY70 gene revealed polymorphism between NILs. The SNPs in NIL-S at the position of 294 bp, in the first exon-intron junction, led to a shift in the open reading frame, which resulted in truncated protein. This might affect the protein structure and function, proving which would require additional studies. The levels of gene expression of TaACT, TaDGK, TaGLI1 and TaWRKY70 were higher in NIL-R compared to its susceptible counterpart. Further, the disease severity and fungal biomass in NIL-R were significantly lower than in NIL-S, as confirmed in our previous study12. Taken together; these results demonstrate the potential roles of these candidate genes in FHB resistance in wheat.
Sequence comparison of TaDGK in NILs revealed the absence of sequence variation, both at the coding and promoter regions, and in spite of this the transcript expression levels were higher in NIL-R than in NIL-S. To answer this, we sequenced and analyzed the promoter regions of TaACT, TaDGK and TaGLI1, which revealed the presence of W-Box cis-element in their promoter, giving a clue on the involvement of TaWRKY70 protein in the regulation of these downstream RRRMgenes by binding to their promoter. Subcellular localization study showed TaWRKY70 to be localized in nucleus. Further, the bioinformatics analysis of protein-DNA interaction networks using GenMANIA software showed that TaWRKY70 interacts with all the three RRRMgenes (TaACT, TaDGK and TaGLI1) that biosynthesized the candidate RRI metabolites identified here. Further, their physical interaction was proved based on luciferase assay in-vivo. These results present compelling evidence on the involvement of TaWRKY70 in FHB disease resistance, by regulating downstream genes that produced RRI metabolites with signaling, antimicrobial and cell wall reinforcement properties.
Association of RRI metabolites with RRRMgenes alone is not enough to claim the role of TaWRKY70 gene in FHB resistance, and they need to be functionally validated. Among several tools available such as gene mutagenesis, insertional mutagenesis, RNAi and VIGS, the VIGS is considered to be the best tool for its easy and rapid knockdown ability of genes during plant development and also it enables assessment of the lack of resistance effect induction in plant by the pathogen78. There are several successful reports on the use of VIGS in functional genomics in tobacco, tomato, Arabidopsis, potato, wheat and barley, as these plants have well established vectors for gene silencing. Therefore, we used VIGS as a tool in functional characterization of TaWRKY70 in NIL-R. The knocking down of TaWRKY70 in NIL-R resulted in the reduction of transcript abundance of TaACT, TaDGK and TaGLI1 which in turn decreased the abundances of their biosynthetic RRI metabolites, resulting in increased fungal biomass. The silenced NIL-R phenotype was quite similar to NIL-S phenotype as determined based on the amount of fungal biomass. These results indicate that TaWRKY70 TF modulates the expression of several RRRM genes, of which TaACT, TaDGK and TaGLI1 may be a subset. PAs biosynthetic genes were confirmed to be regulated by TFs in Nannochloropsis spp., a group of oleaginous microalgae79. Knock-down of FcWRKY70 in kumquat down-regulated ADC (arginine decarboxylase) gene expression and decreased putrescine abundance level accompanied by compromised dehydration tolerance80. Late blight pathogen infection in potato induced HCAA biosynthetic genes regulated by StWRKY1, and the promoter region sequence analysis of 4-coumarate:CoA ligase (St4CL) and tyramine hydroxycinnamoyl transferase (StTHT) revealed the W-box sequence, demonstrating the WRKY binding activity20. Overexpression of TaWRKY45 has showed enhanced resistance to powdery mildew, leaf rust and fusarium head blight diseases in wheat but mechanisms of resistance were not known81. Whitefly infestation in Arabidopsis also induced AtWRKY and regulated At4Cl4 expression by binding to W-box present in its promoter82. Similarly, in wheat aphid infestation induced TaWRKY53, silencing of which significantly reduced the expression of PAL gene32 suggesting that the network of these genes are involved in imparting resistance to several biotic stresses.
In summary, we have identified and isolated Group III stress-responsive WRKY gene designated as TaWRKY70 from wheat, which acts as a positive regulator of resistance against Fg. The FHB resistant NIL containing TaWRKY70 accumulated high amounts of RRI metabolites, whereas the TaWRKY70 silenced plants had reduced amounts. The RRRM genes such as TaACT, TaDGK and TaGLI1 along with RTF gene TaWRKY70 were localized within the QTL-2DL region. Furthermore, the promoter analysis of the candidate RRRMgenes, TaACT, TaDGK and TaGLI1 revealed W-box elements and the luciferase assay confirmed their regulation by TaWRKY70. Collectively, these results indicated that the TaWRKY70 gene functioned in mediating FHB resistance by elevating the accumulation of PAs and HCAAs metabolites by regulating downstream TaACT, TaDGK and TaGLI genes (Fig. S4). A simplified model to explain the interaction of regulatory and RR metabolite biosynthetic genes can be expressed as: RTaWRKY70 * (RTaACT + RTaDGK + RTaGLI1) to provide a global view of the mode of action of TaWRKY70 in FHB resistance. In conclusion, taken together, the TaWRKY70 gene in the QTL-2DL governs major resistance effect against Fg. Following further validation, this gene can be used in wheat FHB resistance breeding programs or for genome editing in susceptible commercial cultivars, if these genes are found non-functional to enhance resistance in wheat against FHB (Kushalappa et al.66).
Additional Information
How to cite this article: Kage, U. et al. TaWRKY70 transcription factor in wheat QTL-2DL regulates downstream metabolite biosynthetic genes to resist Fusarium graminearum infection spread within spike. Sci. Rep. 7, 42596; doi: 10.1038/srep42596 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Bai, G. & Shaner, G. Management and resistance in wheat and barley to fusarium head blight. Annual review of phytopathology 42, 135–161, doi: 10.1146/annurev.phyto.42.040803.140340 (2004).
Buerstmayr, H., Ban, T. & Anderson, J. A. QTL mapping and marker‐assisted selection for Fusarium head blight resistance in wheat: a review. Plant breeding 128, 1–26 (2009).
Somers, D. J., Fedak, G. & Savard, M. Molecular mapping of novel genes controlling Fusarium head blight resistance and deoxynivalenol accumulation in spring wheat. Genome 46, 555–564 (2003).
Long, X. et al. Expression profiling identifies differentially expressed genes associated with the fusarium head blight resistance QTL 2DL from the wheat variety Wuhan-1. Physiological and Molecular Plant Pathology 90, 1–11 (2015).
Bernardo, A. N., Ma, H., Zhang, D. & Bai, G. Single nucleotide polymorphism in wheat chromosome region harboring Fhb1 for Fusarium head blight resistance. Molecular breeding 29, 477–488 (2012).
Dhokane, D., Karre, S., Kushalappa, A. C. & McCartney, C. Integrated Metabolo-Transcriptomics Reveals Fusarium Head Blight Candidate Resistance Genes in Wheat QTL-Fhb2. PloS one 11, e0155851 (2016).
Gunnaiah, R., Kushalappa, A. C., Duggavathi, R., Fox, S. & Somers, D. J. Integrated metabolo-proteomic approach to decipher the mechanisms by which wheat QTL (Fhb1) contributes to resistance against Fusarium graminearum . PloS one 7, e40695 (2012).
Hamzehzarghani, H. et al. Metabolic profiling to discriminate wheat near isogenic lines, with quantitative trait loci at chromosome 2DL, varying in resistance to fusarium head blight. Canadian Journal of Plant Science 88, 789–797 (2008).
Jia, H., Cho, S. & Muehlbauer, G. J. Transcriptome analysis of a wheat near-isogenic line pair carrying Fusarium head blight-resistant and-susceptible alleles. Molecular plant-microbe interactions 22, 1366–1378 (2009).
Schweiger, W. et al. Transcriptomic characterization of two major Fusarium resistance quantitative trait loci (QTLs), Fhb1 and Qfhs. ifa‐5A, identifies novel candidate genes. Molecular plant pathology 14, 772–785 (2013).
Xiao, J. et al. Transcriptome-based discovery of pathways and genes related to resistance against Fusarium head blight in wheat landrace Wangshuibai. BMC genomics 14, 197 (2013).
Kage, U., Karre, S., Kushalappa, A. C. & McCartney, C. Identification and characterization of a fusarium head blight resistance gene TaACT in wheat QTL‐2DL. Plant Biotechnology Journal, doi: 10.1111/pbi.12641 (2016).
Bollina, V. et al. Mass spectrometry‐based metabolomics application to identify quantitative resistance‐related metabolites in barley against Fusarium head blight. Molecular plant pathology 11, 769–782 (2010).
Kumaraswamy, G. K. et al. Metabolomics technology to phenotype resistance in barley against Gibberella zeae. European Journal of Plant Pathology 130, 29–43 (2011).
Gunnaiah, R. & Kushalappa, A. C. Metabolomics deciphers the host resistance mechanisms in wheat cultivar Sumai-3, against trichothecene producing and non-producing isolates of Fusarium graminearum . Plant Physiology and Biochemistry 83, 40–50 (2014).
Hamzehzarghani, H. et al. Metabolic profiling and factor analysis to discriminate quantitative resistance in wheat cultivars against fusarium head blight. Physiological and Molecular Plant Pathology 66, 119–133 (2005).
Kumar, A. et al. WAX INDUCER1 (HvWIN1) transcription factor regulates free fatty acid biosynthetic genes to reinforce cuticle to resist Fusarium head blight in barley spikelets. Journal of Experimental Botany, erw187 (2016).
Pushpa, D., Yogendra, K. N., Gunnaiah, R., Kushalappa, A. C. & Murphy, A. Identification of Late Blight Resistance-Related Metabolites and Genes in Potato through Nontargeted Metabolomics. Plant Molecular Biology Reporter 1–12 (2013).
Yogendra, K. N. et al. Quantitative resistance in potato leaves to late blight associated with induced hydroxycinnamic acid amides. Functional & integrative genomics 1–14 (2014).
Yogendra, K. N. et al. Transcription factor StWRKY1 regulates phenylpropanoid metabolites conferring late blight resistance in potato. Journal of experimental botany 66, 7377–7389 (2015).
Yogendra, K. N. & Kushalappa, A. C. Integrated transcriptomics and metabolomics reveal induction of hierarchies of resistance genes in potato against late blight. Functional Plant Biology (2016).
Kushalappa, A. C., Yogendra, K. N. & Karre, S. Plant Innate Immune Response: Qualitative and Quantitative Resistance. Critical Reviews in Plant Sciences 35, 38–55 (2016).
Guillaumie, S. et al. The grapevine transcription factor WRKY2 influences the lignin pathway and xylem development in tobacco. Plant molecular biology 72, 215–234 (2010).
Rushton, D. L. et al. WRKY transcription factors: key components in abscisic acid signalling. Plant biotechnology journal 10, 2–11 (2012).
Rushton, P. J., Somssich, I. E., Ringler, P. & Shen, Q. J. WRKY transcription factors. Trends in plant science 15, 247–258 (2010).
Yu, Y. et al. MlWRKY12, a novel Miscanthus transcription factor, participates in pith secondary cell wall formation and promotes flowering. Plant science 212, 1–9 (2013).
Zou, X., Seemann, J. R., Neuman, D. & Shen, Q. J. A WRKY gene from creosote bush encodes an activator of the abscisic acid signaling pathway. Journal of Biological Chemistry 279, 55770–55779 (2004).
Eulgem, T., Rushton, P. J., Schmelzer, E., Hahlbrock, K. & Somssich, I. E. Early nuclear events in plant defence signalling: rapid gene activation by WRKY transcription factors. The EMBO Journal 18, 4689–4699 (1999).
Naoumkina, M. A., He, X. & Dixon, R. A. Elicitor-induced transcription factors for metabolic reprogramming of secondary metabolism in Medicago truncatula. BMC plant biology 8, 1 (2008).
Wang, H. et al. Mutation of WRKY transcription factors initiates pith secondary wall formation and increases stem biomass in dicotyledonous plants. Proceedings of the National Academy of Sciences 107, 22338–22343 (2010).
Akagi, A. et al. WRKY45-dependent priming of diterpenoid phytoalexin biosynthesis in rice and the role of cytokinin in triggering the reaction. Plant molecular biology 86, 171–183 (2014).
Van Eck, L. et al. Virus‐induced gene silencing of WRKY53 and an inducible phenylalanine ammonia‐lyase in wheat reduces aphid resistance. Plant biotechnology journal 8, 1023–1032 (2010).
Skibbe, M., Qu, N., Galis, I. & Baldwin, I. T. Induced plant defenses in the natural environment: Nicotiana attenuata WRKY3 and WRKY6 coordinate responses to herbivory. The Plant Cell 20, 1984–2000 (2008).
Zheng, Z., Qamar, S. A., Chen, Z. & Mengiste, T. Arabidopsis WRKY33 transcription factor is required for resistance to necrotrophic fungal pathogens. The Plant Journal 48, 592–605 (2006).
Kim, K.-C., Lai, Z., Fan, B. & Chen, Z. Arabidopsis WRKY38 and WRKY62 transcription factors interact with histone deacetylase 19 in basal defense. The Plant Cell 20, 2357–2371 (2008).
Xing, D.-H. et al. Stress-and pathogen-induced Arabidopsis WRKY48 is a transcriptional activator that represses plant basal defense. Molecular plant 1, 459–470 (2008).
Liu, D., Leib, K., Zhao, P., Kogel, K.-H. & Langen, G. Phylogenetic analysis of barley WRKY proteins and characterization of HvWRKY1 and-2 as repressors of the pathogen-inducible gene HvGER4c. Molecular genetics and genomics 289, 1331–1345 (2014).
Liu, X. et al. GhWRKY25, a group I WRKY gene from cotton, confers differential tolerance to abiotic and biotic stresses in transgenic Nicotiana benthamiana. Protoplasma, 1–17 (2015).
Yan, Y. et al. Overexpression of GhWRKY27a reduces tolerance to drought stress and resistance to Rhizoctonia solani infection in transgenic Nicotiana benthamiana. Frontiers in physiology 6 (2015).
Gao, Q.-M., Venugopal, S., Navarre, D. & Kachroo, A. Low oleic acid-derived repression of jasmonic acid-inducible defense responses requires the WRKY50 and WRKY51 proteins. Plant Physiology 155, 464–476 (2011).
Qiu, D. et al. Rice gene network inferred from expression profiling of plants overexpressing OsWRKY13, a positive regulator of disease resistance. Molecular Plant 1, 538–551 (2008).
Satapathy, L. et al. Transcriptome-wide analysis of WRKY transcription factors in wheat and their leaf rust responsive expression profiling. Molecular genetics and genomics 289, 1289–1306 (2014).
McCartney, C. et al. The evaluation of FHB resistance QTLs introgressed into elite Canadian spring wheat germplasm. Molecular Breeding 20, 209–221 (2007).
Somers, D. J. et al. Assembling complex genotypes to resist Fusarium in wheat (Triticum aestivum L.). Theoretical and Applied Genetics 111, 1623–1631 (2005).
Kushalappa, A. C. & Gunnaiah, R. Metabolo-proteomics to discover plant biotic stress resistance genes. Trends in plant science 18, 522–531 (2013).
Vinaixa, M. et al. A guideline to univariate statistical analysis for LC/MS-based untargeted metabolomics-derived data. Metabolites 2, 775–795 (2012).
Kumar, A. et al. Real-time quantitative PCR based method for the quantification of fungal biomass to discriminate quantitative resistance in barley and wheat genotypes to fusarium head blight. Journal of Cereal Science 64, 16–22 (2015).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. Journal of molecular biology 215, 403–410 (1990).
Livak, K. J. & Schmittgen, T. D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. methods 25, 402–408 (2001).
Chen, S., Songkumarn, P., Liu, J. & Wang, G.-L. A versatile zero background T-vector system for gene cloning and functional genomics. Plant physiology 150, 1111–1121 (2009).
Yoo, S.-D., Cho, Y.-H. & Sheen, J. Arabidopsis mesophyll protoplasts: a versatile cell system for transient gene expression analysis. Nature protocols 2, 1565–1572 (2007).
Wang, X., Fan, C., Zhang, X., Zhu, J. & Fu, Y.-F. BioVector, a flexible system for gene specific-expression in plants. BMC plant biology 13, 1 (2013).
Cakir, C., Gillespie, M. E. & Scofield, S. R. Rapid determination of gene function by virus-induced gene silencing in wheat and barley. Crop science 50, S-77–S-84 (2010).
Scofield, S. R., Huang, L., Brandt, A. S. & Gill, B. S. Development of a virus-induced gene-silencing system for hexaploid wheat and its use in functional analysis of the Lr21-mediated leaf rust resistance pathway. Plant Physiology 138, 2165–2173 (2005).
Ma, M., Yan, Y., Huang, L., Chen, M. & Zhao, H. Virus-induced gene-silencing in wheat spikes and grains and its application in functional analysis of HMW-GS-encoding genes. BMC plant biology 12, 141 (2012).
Burhenne, K., Kristensen, B. K. & Rasmussen, S. K. A New Class of N-Hydroxycinnamoyltransferases Purification, Cloning, and Expression of a Barley Agmatine Coumaroyltransferase (EC 2.3. 1.64). Journal of Biological Chemistry 278, 13919–13927 (2003).
Muroi, A. et al. Accumulation of hydroxycinnamic acid amides induced by pathogen infection and identification of agmatine coumaroyltransferase in Arabidopsis thaliana. Planta 230, 517–527 (2009).
Cai, J., Abramovici, H., Gee, S. H. & Topham, M. K. Diacylglycerol kinases as sources of phosphatidic acid. Biochimica et Biophysica Acta (BBA)-Molecular and Cell Biology of Lipids 1791, 942–948 (2009).
Chanda, B. et al. Glycerol-3-phosphate levels are associated with basal resistance to the hemibiotrophic fungus Colletotrichum higginsianum in Arabidopsis. Plant physiology 147, 2017–2029 (2008).
Gómez-Merino, F. C. et al. AtDGK2, a novel diacylglycerol kinase from Arabidopsis thaliana, phosphorylates 1-stearoyl-2-arachidonoyl-sn-glycerol and 1, 2-dioleoyl-sn-glycerol and exhibits cold-inducible gene expression. Journal of Biological Chemistry 279, 8230–8241 (2004).
Testerink, C. & Munnik, T. Phosphatidic acid: a multifunctional stress signaling lipid in plants. Trends in plant science 10, 368–375 (2005).
Loke, J. C. et al. Compilation of mRNA polyadenylation signals in Arabidopsis revealed a new signal element and potential secondary structures. Plant Physiology 138, 1457–1468 (2005).
Kalde, M., Barth, M., Somssich, I. E. & Lippok, B. Members of the Arabidopsis WRKY group III transcription factors are part of different plant defense signaling pathways. Molecular Plant-Microbe Interactions 16, 295–305 (2003).
Wang, D., Amornsiripanitch, N. & Dong, X. A genomic approach to identify regulatory nodes in the transcriptional network of systemic acquired resistance in plants. PLoS Pathog 2, e123 (2006).
Shimono, M. et al. Rice WRKY45 plays a crucial role in benzothiadiazole-inducible blast resistance. The Plant Cell 19, 2064–2076 (2007).
Kushalappa, A. C., Yogendra, K. N., Sarkar, K., Kage, U. & Karre, S. Gene discovery and genome editing to develop cisgenic crops with improved resistance against pathogen infection. Canadian Journal of Plant Pathology (2016b).
Okazaki, Y. & Saito, K. Roles of lipids as signaling molecules and mitigators during stress response in plants. The Plant Journal 79, 584–596 (2014).
Arisz, S. A., Testerink, C. & Munnik, T. Plant PA signaling via diacylglycerol kinase. Biochimica et Biophysica Acta (BBA)-Molecular and Cell Biology of Lipids 1791, 869–875 (2009).
Khajuria, C. et al. Mobilization of lipids and fortification of cell wall and cuticle are important in host defense against Hessian fly. BMC genomics 14, 423 (2013).
Zhang, W. et al. The oleate-stimulated phospholipase D, PLDδ, and phosphatidic acid decrease H2O2-induced cell death in Arabidopsis. The Plant Cell 15, 2285–2295 (2003).
Ishihara, A. et al. The tryptophan pathway is involved in the defense responses of rice against pathogenic infection via serotonin production. The Plant Journal 54, 481–495 (2008).
Muroi, A. et al. Acquired immunity of transgenic torenia plants overexpressing agmatine coumaroyltransferase to pathogens and herbivore pests. Scientific reports 2 (2012).
De Jong, C. F. et al. Phosphatidic acid accumulation is an early response in the Cf‐4/Avr4 interaction. The Plant Journal 39, 1–12 (2004).
Zhang, W., Chen, J., Zhang, H. & Song, F. Overexpression of a rice diacylglycerol kinase gene OsBIDK1 enhances disease resistance in transgenic tobacco. Molecules and cells 26, 258–264 (2008).
Mandal, M. K. et al. Glycerol-3-phosphate and systemic immunity. Plant signaling & behavior 6, 1871–1874 (2011).
Yang, Y. et al. Glycerol-3-phosphate metabolism in wheat contributes to systemic acquired resistance against Puccinia striiformis f. sp. tritici (2013).
Rushton, P. J. et al. Interaction of elicitor-induced DNA-binding proteins with elicitor response elements in the promoters of parsley PR1 genes. The EMBO Journal 15, 5690 (1996).
Ramegowda, V., Mysore, K. S. & Senthil-Kumar, M. Virus-induced gene silencing is a versatile tool for unraveling the functional relevance of multiple abiotic-stress-responsive genes in crop plants. Frontiers in plant science 5 (2014).
Hu, J. et al. Genome-wide identification of transcription factors and transcription-factor binding sites in oleaginous microalgae Nannochloropsis. Scientific reports 4 (2014).
Gong, X. et al. FcWRKY70, a WRKY protein of Fortunella crassifolia, functions in drought tolerance and modulates putrescine synthesis by regulating arginine decarboxylase gene. Plant, cell & environment (2015).
Bahrini, I., Ogawa, T., Kobayashi, F., Kawahigashi, H. & Handa, H. Overexpression of the pathogen-inducible wheat TaWRKY45 gene confers disease resistance to multiple fungi in transgenic wheat plants. Breeding science 61, 319–326 (2011).
Hahlbrock, K. et al. Non-self recognition, transcriptional reprogramming, and secondary metabolite accumulation during plant/pathogen interactions. Proceedings of the National Academy of Sciences 100, 14569–14576 (2003).
Acknowledgements
This project was funded by the Ministere de l’Agriculture, des Pecherieset de l’Alimentation du Quebec (MAPAQ), Quebec, Canada. Kage is thankful to the Indian Council of Agricultural Research (ICAR), New Delhi and Government of Karnataka, India for ICAR-International Fellowship and National Overseas Scholarship, respectively.
Author information
Authors and Affiliations
Contributions
U.K. and A.K. are designed the experiments. U.K. conducted all the lab and greenhouse experiments, analyzed data and wrote the manuscript. Y.K. helped in VIGS and luciferase experiments.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Kage, U., Yogendra, K. & Kushalappa, A. TaWRKY70 transcription factor in wheat QTL-2DL regulates downstream metabolite biosynthetic genes to resist Fusarium graminearum infection spread within spike. Sci Rep 7, 42596 (2017). https://doi.org/10.1038/srep42596
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep42596
- Springer Nature Limited
This article is cited by
-
Integrative systems biology of wheat susceptibility to Fusarium graminearum uncovers a conserved gene regulatory network and identifies master regulators targeted by fungal core effectors
BMC Biology (2024)
-
Unraveling the involvement of WRKY TFs in regulating plant disease defense signaling
Planta (2024)
-
Metabolome dynamics during wheat domestication
Scientific Reports (2022)
-
Metabolic pathway genes for editing to enhance multiple disease resistance in plants
Journal of Plant Research (2022)
-
Identification of a novel major QTL from Chinese wheat cultivar Ji5265 for Fusarium head blight resistance in greenhouse
Theoretical and Applied Genetics (2022)