
Abstract
Single nucleotide variations (SNVs) can result in loss or gain of protein functional sites. We analyzed the effects of SNVs on enzyme active sites, ligand binding sites, and various types of post translational modification (PTM) sites. We found that, for most types of protein functional sites, the SNV pattern differs between germline and somatic mutations as well as between synonymous and non-synonymous mutations. From a total of 51,138 protein functional site affecting SNVs (pfsSNVs), a pan-cancer analysis revealed 142 somatic pfsSNVs in five or more cancer types. By leveraging patient information for somatic pfsSNVs, we identified 17 loss of functional site SNVs and 60 gain of functional site SNVs which are significantly enriched in patients with specific cancer types. Of the key pfsSNVs identified in our analysis above, we highlight 132 key pfsSNVs within 17 genes that are found in well-established cancer associated gene lists. For illustrating how key pfsSNVs can be prioritized further, we provide a use case where we performed survival analysis showing that a loss of phosphorylation site pfsSNV at position 105 in MEF2A is significantly associated with decreased pancreatic cancer patient survival rate. These 132 pfsSNVs can be used in developing genetic testing pipelines.
Similar content being viewed by others


Introduction
With the advancement of high-throughput sequencing (HTS) technology, the cost of sequencing the human genome has dropped significantly1,2. However, while many biologists expected that genome sequencing could solve human health issues in a short period of time, complex diseases, such as cancer, still remain difficult to tackle3. In the field of cancer genomics, several international collaborations, such as The Cancer Genome Atlas (TCGA) (http://cancergenome.nih.gov/), International Cancer Genome Consortium (ICGC)4, have provided useful HTS based genomics data by sequencing a large number of tumor samples across cancer types5,6,7. The availability of large number of samples across different types of cancer enables pan-cancer analysis which explores via comparative analysis various cancer genomes originating from different tumor types8,9. By investigating the similarities and differences of cancer genomes and cellular characteristics across cancer types, tumor heterogeneity has been better understood10,11 and a number of cancer associated pathways and genes have been identified7,12,13,14. Furthermore, such analysis can reveal how mutations affect protein function. Our previous study8 shows the landscape of protein functional site affecting non-synonymous single-nucleotide variations (nsSNVs) across cancer types. In the current study we extensively investigate the abundance or depletion of SNV (both synonymous and non-synonymous) occurrence in different protein functional site type and the immediate region of the protein functional site. We also perform a comparative study on the SNV occurrence between germline and somatic mutations impacting different functional sites. Previous studies show that synonymous mutations are not always silent and they are able to cause changes in protein expression, conformation and function15,16,17,18,19. Therefore, we also compare the frequencies of synonymous and non-synonymous mutations on protein functional sites.
Since proteins are the foundational and functional blocks of living organisms, how genomic alterations of protein coding genes affect protein functionality is an important question. While many previous publications have focused on genes through pan-cancer analysis, our efforts extend the utility of a pan-cancer analysis by examining the effect of genomic alterations on protein functional sites. To this end, we have retrieved a comprehensive collection of SNVs and protein functional sites, including post-translational modification (PTM), ligand binding site, and enzyme active site, from a variety of data sources. Somatic mutations were retrieved from COSMIC20, UniProtKB21, TCGA (http://cancergenome.nih.gov/), and ICGC4. Germline mutations were retrieved from dbSNP22. All SNVs were unified and mapped to amino acid positions. To facilitate the pan-cancer analysis, the original annotated cancer types retrieved from source databases were mapped to Disease Ontology (DO) slim terms23. Protein functional sites were retrieved from UniProKB sequence feature (FT) line21, NCBI Conserved Domain Database (CDD)24, and dbPTM25. By integrating SNVs and protein functional sites, we can identify functional site affecting SNVs (pfsSNVs) for downstream analysis.
In this study, we first obtained a global perspective on how germline and somatic mutations are distributed at the proteome level, especially on various protein functional sites through integrating 3,342,377 SNVs (1,501,666 germline mutations and 1,840,711 somatic mutations) and 268,478 known and curated PTM sites, binding sites and enzyme active sites. Then we created a framework to facilitate this SNV prioritization process using observed frequency in patients and cancer type information.
Materials and Methods
SNV dataset
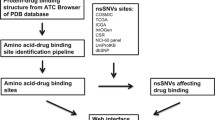
As the flowchart in Fig. 1 shows, somatic coding mutations were extracted from ICGC (version v0.10a), TCGA (release January 27, 2015), COSMIC (version v73), IntOGen (release 2014.12), and ClinVar (release 20150205). All somatic mutations were unified and then annotated using ANNOVAR26. Cancer types were mapped to DO Cancer Slim terms23 for cancer term unification. Frequency of a certain mutation was either calculated based on patient ID or was directly extracted from the downloaded files. All integrated information is stored and can be downloaded from the BioMuta database8. SNVs annotated as the same variation but from different sources/patients were collapsed into a single entry, but all relevant source information was maintained.

Flowchart of the distribution bias analysis of protein functional site affecting single nucleotide variations (pfsSNVs).
Germline coding mutations were collected from dbSNP (build 142) database. Minor Allele Frequency (MAF) and “Common/Rare SNP” tags were directly extracted from dbSNP. All SNVs were translated and mapped to the UniProtKB complete human proteome set (downloaded in January 2015) through a pairwise-alignment based pipeline for unification and downstream protein functional site analysis.
Protein functional site dataset
Protein post-translational modification (PTM), binding, and enzyme active site annotation were extracted from three different sources: dbPTM 3.025, UniProtKB/Swiss-Prot feature (FT) line (January 2015), and CDD features (January 2015). Only experimentally verified data were retrieved from dbPTM 3.0 and UniProtKB. Duplicates and conflicted accessions were removed. Variants with the same annotation from different sources were collapsed into a single data point while maintaining source information. Modification data was extracted using PTMlist, a controlled vocabulary provided by UniProtKB/Swiss-Prot. The NCBI CDD-based annotation of functional sites was retrieved using BATCH CD-Search against CDART database27. Entries such as domains, repeats, and motifs with longer than five consecutive amino acids were not considered. Filtered sites were categorized manually into various types of PTM sites, active sites, and binding sites with original annotations maintained in a separate column. Other PTM records were adopted based on dbPTM 3.0 which collects PTM data from more than 10 different sources25.
All entries were unified based on the UniProtKB complete human proteome set downloaded from UniProtKB on January 2015, which is identical to the proteome used for SNVs dataset unification.
Mapping SNVs to protein functional sites and the neighboring positions
The general process of mapping SNVs to protein functional sites includes loading the SNV file into matrix of “UniProt accession with UniProt Position” and match it to the protein functional site matrix. Once the protein accession and position are matched, additional steps were used to evaluate if this SNV caused a substitution at the functional site or not. If the SNV is a substitution, we also consider the known amino acid tolerance for corresponding PTM type, if the substitution replaces the original residue with a residue which cannot be modified as a PTM or function as an active site. The output file provides a tab-delimited file containing all SNVs and affected protein functional site information. A SNV ratio based on SNV numbers divided by proteome length was calculated for expected SNV number as well as the statistical significance using methods described earlier8. The SNV occurrence between protein functional site and all other amino acid located within +/− 20 amino acids was compared and the significance was evaluated through one sample t-test.
SNV-caused gain of protein phosphorylation and glycosylation site prediction
NetNGlyc (v1.0) and NetPhosK (v1.0) were used to predict SNV-caused gain of protein phosphorylation and N-glycosylation site28,29. 21 mer and 5 mer were set as the effective segment length of input sequences for phosphorylation and glycosylation site prediction respectively. For parameters, ESS filter and threshold 0.6 were applied for NetPhosK, while a score 0.6 is required for NetNGlyc prediction result. Both protein reference sequence and mutated sequence were used as input to the NetNGyc and NetPhos in order to minimize false positives by subtracting background predicted sites.
Statistical significance of amino acid based pfsSNV occurrence
To investigate whether the distinct frequency of SNV on protein functional sites is caused by different amino acid mutation rate, we conducted amino acid based binomial test on pfsSNV occurrence.
First, for each type of amino acid (denote as A), we first calculate the probability of A to be a F type of protein functional site, calculated as following:

where LA denotes total number of amino acid A on human proteome, nA(F) denotes the total number of positions for a specific functional site with amino acid A. Thus, amino acid based protein functional site rate pA(F) can be derived from our protein functional site dataset.
Then, we calculated the expected number of pfsSNVs nA(E) for each type of amino acid:

where NA is total number of variations with amino acid type A. nA(E) is then used to derive if the given type of pfsSNV occurrence on the given amino acid type A is enriched or depleted.
Next, after obtaining from our SNV dataset the value of observed pfsSNV nA(O) for a specific A and F, the binomial test was performed according to Mi et al.30, and the p-value was calculated as the total probabilities to observe nA the same as or more extreme (larger if nA(O) is larger than expected and smaller otherwise) than nA(O), which measures the deviance degree between an expected ratio (nA(E)/NA or pA(F)) and an observed ratio (nA(O)/NA):

Comparing to our previous study where the same expected SNV rate applying to all protein functional site8, advantage of this background SNV rate is that this allows each type of protein functional site having different expected SNV rates given different components of amino acid as their donor site.
Pan-cancer clustering of pfsSNV profiles
In order to investigate the somatic pfsSNV occurrence pattern in each cancer type, a pan-cancer analysis was performed. The observed and expected somatic mutation occurrence among each cancer type among different protein functional site type was calculated following same rule described under ‘Mapping SNVs to protein functional sites and the neighboring positions’. Basically the observed value is the mutation occurrence on a type of protein functional site while expected value is the average of neighboring mutation occurrence. And the fold change was used as a metric to perform hierarchical clustering (HC). The heat map was generated via the R package ggplot version 2.17.031.
pfsSNVs prioritization criteria
Two distinct criteria were used to prioritize pfsSNV: a) pfsSNVs that exist across 5 or more cancer types, b) pfsSNVs that are enriched in patients with certain cancers. To do this we leveraged TCGA patient counts mapped to our mutation dataset to identify key pfsSNVs. We combined pfsSNVs that can cause either a loss or gain of functional site. The Binomial test described above (section “Statistical significance of amino acid based pfsSNV occurrence”) was applied to identify pfsSNVs that is significantly associated with a certain cancer type based on enrichment in patients with that cancer. In this calculation, we calculated the expected probability of any type of pfsSNV occurring in a patient in a cancer type C:

where NC is the total number of patient in cancer type C, and nC(M) is the number of patient harboring a specific pfsSNV M in cancer type C. nC(E) is the expected number of patient in cancer type C for a given pfsSNV M for any functional types. Then the p-value was calculated as the sum of probabilities of observing number of patients the same as or more extreme (larger if the observed number of patients is larger than expected number, E(nC(M)), and lower if the observed number is smaller than E(nC(M))) than the observed number of patients nC(O) in the sample with the same cancer, NC.

This approach takes into consideration the differences in cancer’s mutational rate and rank the pfsSNVs enriched within cancers despite the sparseness of somatic mutation among patients.
After the log transformation, p-values are visualized in Manhattan plot where horizontal axis represent chromosome from 1 to 23. The cutoff line was calculated as 2E-6 using Bonferroni approach. Lastly, we compared our prioritized pfsSNVs with a well-known cancer gene list: significantly mutated gene (SMG)32 and cancer gene census (CGC)33 to further annotate the key pfsSNVs list.
Survival analysis
Identified key pfsSNVs were further investigated to see if any of them significantly affect patient survival. Patient clinical information was retrieved for TCGA samples from their FTP site (https://tcga-data.nci.nih.gov/tcgafiles/ftp_auth/distro_ftpusers/anonymous/tumor/). For each key pfsSNV in a specific cancer type that we identified through the prioritization process, based on the presence of the given pfsSNV, patients were divided into two groups with clinical factors that may affect patient survival. A log-rank test was applied to test the death time distributions between two groups. Then the Cox model was used to adjust factors like age at initial diagnosis, pathological stage and gender. SAS 9.3 was used to perform this analysis.
Results and Discussion
Impact of SNVs on protein functional sites
In this study, we expanded the scope of our previous study8 for better evaluation of mutational profile among various protein PTMs, active and binding sites. Tables 1 and 2 summarizes our data collection for both the current study and our previous study8. Table 1 shows total number of germline mutation, somatic mutation, and protein functional sites collected in both previous and current datasets. In Table 2, somatic and germline pfsSNV from Table 1 are split into major protein functional site types and summarized. The number of somatic mutations increased from 994,339 to 1,840,711 (1,272,878 non-synonymous, 476,087 synonymous and 91,746 stop codon). The number of germline mutations increased from 710,946 to 1,501,666 (937,634 non-synonymous, 541,029 synonymous and 23,003 stop codons). The number of protein functional sites increased from 259,216 to 268,478. After mapping both somatic and germline variations to protein functional site dataset, the number of pfsSNVs increases from 38,549 to 51,138 (31,999 somatic and 19,139 germline). We divided our pfsSNVs into four groups: non-synonymous germline mutation (non-SG), non-synonymous somatic mutation (non-SS), synonymous germline mutation (SG) and synonymous somatic mutation (SS) because each one of these mutation type has its own biological meaning, and therefore should be analyzed separately. Additionally, we enlarged the testable SNV dataset by incorporating predicted gain of N-linked glycosylation and phosphorylation site. It is common that SNV caused gain of PTM sites to be ignored in many HTS based proteome-wide analysis until recently34,35,36. We found a total number of 344,239 SNVs that cause gain of phosphorylation sites across 18,259 proteins and 17,921 SNVs that cause gain of N-linked glycosylation sites across 8,354 proteins.
In Fig. 2, for each protein functional site type, we calculated the percentage of its site impacted by somatic and germline SNVs (See Supplementary Table 1). In the scatter plot, X-axis and Y-axis indicate somatic and germline mutation percentages respectively while the dot and triangle represents non-synonymous and synonymous variation percentages respectively. Linear reference lines in the matrix show the global expected percentages. We can see from Fig. 2, for germline mutations, synonymous (lower reference line on Y axis) and non-synonymous (upper reference line on Y axis) SNVs cluster near the average reference lines. For somatic variations, synonymous and non-synonymous mutations also cluster near the averages (left reference line on X axis for synonymous; right reference line on X axis for non-synonymous). We can see that pfsSNV occurrence is around the global average percentages except for crotonylation sites, for which there are much more germline and somatic SNVs than the average. Outliers on the plot could be caused due to small sample size, for instance, crotonylation sites has higher synonymous and non-synonymous germline mutation occurrence than reference line but this is calculated based on just 79 data points.

The values on each axis show, for each PTM type, the percentage of its site occupied by SNVs. X-axis shows the somatic mutation percentage and Y-axis shows germline mutation percentage. Dot and triangle markings represent non-synonymous and synonymous mutations respectively. Each protein functional site type was shown in different color as per the legend. Linear lines in the figure show global ratio for each mutation type.
Instead of just focusing on the exact protein functional sites (such as PTM and active/binding sites) we also evaluated the preponderance of SNVs upstream and downstream of the functional site. Figure 3, plots all the SNV occurrence of residues with +/−20 amino acids around the functional site (see Supplementary Table 3a,b,c and d for plots of all 25 types). In most of the PTM sites, non-synonymous germline mutation (non-SG) shows either relatively low occurrence or similar rates when compared to neighboring regions. This result is consistent with the high evolutionary conservation of functional sites15,37. On the other hand, synonymous germline mutation shows mixed occurrence across different PTMs types with lower than expected occurrences for in some of the sites. It is interesting to note that several studies have shown that synonymous mutation can affect protein function16,38,39.

Occurrence ratio of synonymous somatic (SS), synonymous germline (SG), non-synonymous somatic (non-SS) and non-synonymous germline (non-SG) mutations +/−20 amino acid from protein functional sites. Y-axis shows fold of change of SNV occurrence on corresponding amino acid position. Different SNV types are represented as different colors. Value 0 on X-axis indicates the PTM site. T and P represent one sample t-test of the PTM site comparing with its neighboring. P represents the p-value of corresponding one sample t-test.
Out of 8,357 experimental confirmed acetylation sites in the human proteome, 691 lose acetylation site due to somatic mutation and 432 lose acetylation site due to germline mutations. In 22,524 ubiquitination sites in the human proteome, 1,562 ubiquitination sites are lost due to somatic mutations and 1052 ubiquitination sites are lost due to germline mutations. In comparison with our previous paper, the number of loss of acetylation sites and ubiquitination sites increased by 48 and 559 respectively. Dysregulation of both acetylation and ubiquitination processes may cause cancer initiation and it has been observed by others that there are frequent mutations in acetylation and ubiquitination sites which potentially can drive cancer40,41,42. For acetylation, different modified sites have distinct regulatory effects, even in the same protein (e.g. malate dehydrogenase 2)41. In another study, researchers found that both acetylation and deacetylation of p53 on different amino acids could either promote or block tumorigenesis43. Its complexity leads to the disunity of acetylation function in cancers. Our analysis shows low non-synonymous somatic (non-SS) mutation occurrence on acetylation sites suggesting that in cancer these sites are still less prone to mutations. In terms of ubiquitination, it can be seen that ubiquitination sites are less tolerant to SNVs (relatively conserved) compared with its neighboring region.
In our current dataset, we identified 7,373 somatic mutations and 5,282 germline mutations that cause loss of phosphorylation sites. Previous studies found high enrichment of mutations causing gain or loss of phosphorylation sites and they may be considered as key features in cancer occurrence34. High activity of kinases is essential to maintain the tumor malignant phenotype (oncogene addiction)44. It is consistent with our result that non-synonymous mutations (non-SS) show low occurrence at phosphorylation sites. It is also possible that the low occurrence on phosphorylation site may be caused by the relatively small number of cancer related genes45.
We identified 2,084 somatic mutations and 1,040 germline mutations that can cause loss of enzyme active site. In Fig. 3, the non-synonymous somatic mutation occurrence at active site is relatively higher than that at its surrounding regions. However, when the enzyme active site is considered, its role in cancer is also dependent on the feature of the protein (oncogene or tumor suppressor gene). For example, in breast cancer, overexpression of BCRP (breast cancer resistance protein) with its intact active site could cause drug resistance, while mutation in the active site of α-fetoprotein (AFP) could reduce breast cancer risk46. These mutations can impact enzymes to metabolize different substrates47, leading to pathological processes. In Fig. 3, the non-synonymous somatic mutation occurrence at active site is relatively higher than at its surrounding regions. Synonymous somatic mutation, on the other hand, has a low occurrence rate at active sites. This bias may be caused by the highly structure-dependent catalytic activity (stable structure is crucial for function)48. At ligand binding sites, 16,630 somatic mutations and 25,074 germline mutations were identified. For binding sites, studies have found their relationship with disease occurrence in terms of mutations49,50. Binding site analysis shows little SNV occurrence difference compared to its neighboring regions for SG, SS and non-SS, but overall low mutation occurrence in the entire functional region for non-SG. However, we would like to mention that binding sites can contain multiple sites which are not sequentially placed in the sequence. Our analysis focuses on short regions (see materials and methods), and counting each residue as one binding sites and the immediate region around it thus providing a practical and comparable evaluation of binding sites and other protein functional sites.
For methylation sites, we identified 208 somatic mutations and 74 germline mutations. It is interesting to note that the overall occurrence of SG, SS and non-SS is as high as two fold compared to the background occurrence. In particular, the non-SS mutation occurrence at the methylation sites is relatively higher than other mutation type and also their surrounding regions. Methylation regulates transcription factor binding affinity, and therefore, controls the expression level of the downstream target genes51. In consideration of cancer development, previous study suggests lysine-to-methionine substitution at methylation sites could cause loss of methylation and function in a variety of pathologies. And in our results, the relatively high non-SS mutation occurrence of methylation may suggest its primary role in either promoting oncogenes or suppressing tumor suppressor genes.
3,217 somatic mutations and 2,630 germline mutations were identified on N-linked glycosylation sites. Figure 3 also shows that the SNV occurrence at the N-linked glycosylation site and its surrounding amino acids (−1, +1 and +2) are much higher than others. Non-synonymous somatic mutation shows a deep dip at the N-linked glycosylation (0.67). In our previous study52, we found slightly lower frequency of all kinds of missense mutations in N (position 0) than the non-glycosylated motifs. This is also consistent with the higher conservation of glycoslylated asparagines as compared with the non-glycosylated ones53. Such a low mutation occurrence in the cancer genome implies its contribution and its role in cancer. In addition, somatic synonymous mutations (0.89) also show a similar trend at N-linked glycosylation sites. This also suggests that it is important to maintain N-linked glycosylation site undisrupted. Although, it is quite possible the overall functional impact is maintained through the heterogeneity of the glycans at the sites in normal vs. cancer tissues54.
The NX(S/T) amino acid sequon (asparagine for N, any amino acid except proline for X, and either serine or threonine for S/T) is considered as a requirement for N-glycosylation52. This could explain the low occurrence of the two types of synonymous mutations (germline and somatic) at the amino acid of position +1 (X) but higher rates for non-synonymous mutation, and high rate (SS: 1.64, SG: 1.75) at position +2 (alternation of serine and threonine, S/T). Additionally, we found that the amino acid at ‘−1’ position also has lower synonymous germline mutation occurrence, which suggests possible effects of “silent” mutations at this site.
In terms of O-linked glycosylation, 126 somatic mutations and 115 germline mutations were identified impacting the PTM site. O-linked glycosylation is known to be important in bearing tumor associated antigens and also involved in several physiological and pathological processes55,56,57. One interesting finding is that O-linked glycosylation sites is the only functional site type showing overall low occurrences across the entire functional site region in terms of all mutation types (non-SG: 0.60, SG: 0.64, SS: 0.57, non-SS: 0.59).
Pan-cancer view of somatic mutation occurrence on protein functional sites
For pan-cancer analysis, cancer Disease Ontology (DO) slim23 was used to unify the cancer types. The observed and expected somatic mutation occurrence on each functional type was then calculated. Figure 4 shows the pan-cancer heatmap of somatic mutation occurrences across functional sites (Fig. 4A: non-synonymous, Fig. 4B: synonymous). The mutation occurrence is indicated by ratio of change compared to the cancer type specific global ratio. Color in the figure indicates either the over-representation (red) or under-representation (blue) of pfsSNVs while white indicates no SNV occurrence difference between functional sites and neighboring sites. The grey color indicates the absence of pfsSNVs for the corresponding cancer type. Our assumption is that, since functional sites are generally conserved, the high/low ratio of somatic pfsSNVs occurrence on these sites implies the loss/gain of function for them and their possible roles in tumorigenesis.

Pan-caner hierarchical clustering of non-synonymous (A) and synonymous (B) somatic mutation occurrence on protein functional site region. Figure shows cancer SNV occurrence at PTM site vs somatic SNV occurrence at a neighboring region for different cancer types. Color indicates fold of change of somatic SNV occurrence. Red color indicates overrepresentation while blue indicates under-representation. Grey color means that there is no detected somatic SNV on corresponding PTM type for corresponding cancer.
The pan-cancer view of observed/expected SNVs shown in Fig. 4A displays unique patterns of nsSNV occurrence on functional sites (compared to neighboring site) in different cancer types. The variation occurrence at ubiquitination and acetylation sites is lower (blue color) at these PTM sites across almost all cancer types. On the other hand, the methylation site shows higher nsSNV occurrence (red color) in PTM site for majority of the cancer types. Active sites, binding sites, phosphorylation sites, and N-linked glycosylation sites show insignificant fold-change between PTM sites and neighboring sites. Similarly, for synonymous mutations (Fig. 4B), ubiquitination and acetylation site show an overall low somatic synonymous mutation occurrence at PTM sites across almost all the cancer types. However, unlike in non-synonymous mutation, methylation sites show mixed mutation occurrence across cancer types. Phosphorylation sites and N-linked glycosylation shows an increased synonymous mutation occurrence in multiple cancer types.
Identification of key pfsSNVs across multiple cancer types
Out of the 31,999 germline pfsSNVs and 19.139 somatic pfsSNVs, we found that 142 pfsSNVs exist across more than five cancer types, which we considered as key pfsSNVs (see Supplementary Table 6 for pfsSNVs in more than 3 cancer types). Table 3 displays the top 20 pfsSNVs with respect to number of associated cancer types. In addition, Fig. 5 shows their SNV-functional site relationship in the Circos plot58. From both Table 3 and Fig. 5 we can see that TP53, one of the most well-known oncogenes, with 79 out of 142 key pfsSNVs on that protein. We also want to emphasize pfsSNVs that exist on genes other than TP53. Since TP53 is a well-known oncogene, we emphasize top 20 pfsSNVs associated with multiple cancer types with TP53 excluded in Table 4: NRAS, CTNNB1, NRAS, GNAS, KRAS, HRAS and PTEN. It is clear that some genes harbor more key pfsSNVs than others as shown in Fig. 5. 14 out of 142 key pfsSNVs, including two of the top 20 pfsSNVs are found within CTNNB1 which is an important component of the canonical Wnt signaling pathway. It is interesting to note that all these key pfsSNVs are affecting protein phosphorylation sites between position 29 to 45. This finding confirms previous studies’59 claims that SNVs and overexpression of CTNNB1 are associated with many cancers: a large number of SNVs cluster on the N-terminal segment of CTNNB1, the β-TrCP binding motif.

Bands are colored by genes, and connect between gene and various types of protein functional sites. Note that, in 142 key pfsSNVs, all key pfsSNVs on CTNNB1 occur on phosphorylation site and all key pfsSNVs on RAS subfamily occur on binding site.
Other than TP53 and CTNNB1, many key members of Ras subfamily, such as NRAS, GNAS, KRAS and HRAS harbor SNVs across multiple cancer types. Figure 5 shows that virtually all the pfsSNVs on Ras subfamily are located on binding site. However, multiple alignment of NRAS, GNAS, KRAS and HRAS shows that most of the key pfsSNVs within these four genes occurs at the same position (RASN_HUMAN Q61), a well-known position responsible GAP-mediated GTP hydrolysis. SNVs on this residue disturb Ras signaling control and eventually trigger tumorigenesis by activate genes involved in cell growth, differentiation and survival60.
Identification of key pfsSNVs that are enriched in patients with specific cancer types
To ensure we do not miss any pfsSNVs that occur repetitively among patients within a specific cancer type, we performed Binomial test using a dataset combining known and predicted gaining/losing pfsSNV sites. This dataset includes 19,337 loss of functional site causing pfsSNVs, 10,991 gain of N-glycosylation sites, and 208,507 gain of phosphorylation sites. Log p-values for each pfsSNVs were used for visualization in Fig. 6 (See Supplementary Table 5 for all pfsSNVs with p-value). Based on our threshold (p-value = 2E-6 using the Bonferroni adjustment), a total number of 77 pfsSNVs (57 gain of phosphorylation site pfsSNVs, 3 gain of glycosylation site pfsSNVs, 12 loss of binding site pfsSNVs, 3 loss of phosphorylation site and 2 loss of active sites) were identified to be significant in specific cancer types. Table 5 shows the top 20 pfsSNVs with significant p-value associated with specific cancer types. [L] and [G] indicate loss of functional site and gain of functional site, respectively. Supplementary Table 5 shows p-values for all 24,668 pfsSNVs associated with specific cancer type. For example, the gain of phosphorylation site pfsSNV PIK3CA-545-E-K is significantly associated with as many as six cancer types (63 patients in breast cancer, 28 patients in head and neck cancer, 33 patients in cervical cancer, 19 patients in colon cancer, 14 patients in uterine cancer, 11 patients in stomach cancer).

X-axis indicates chromosome from 1 to 23 and X, Y in different colors. Each dot in the figure represents a pfsSNV with –log10 (p-value) calculated from a binomial test. Cutoff was set as -log10 (5e-8). A total number of 77 pfsSNVs are statistically significant in specific cancer type. [L] and [G] indicate loss of PTM/active/binding site and gain of PTM/active/binding site respectively. As marked in the figure, [L]NRAS-61-Binding Site and [G]PIK3CA-545/542-Phosphorylation significantly associate with multiple cancer type.
Pan-cancer analysis mentioned above identified a total number of 210 key pfsSNVs, among which 142 exist across more than five cancer types and 77 pfsSNVs are significantly enriched in patients with specific cancer type. All these 210 key pfsSNVs belong to 60 genes. For the purpose of comparison with key cancer genes found in other studies, we retrieved the significantly mutated gene (SMG) set found by MutSig suite32 and cancer gene census (CGC) from COSMIC33. By mapping SMG (260 genes), CGC (573 genes) and key pfsSNVs (60 genes), we found our key pfsSNVs map to 18 and 20 genes from SMG and CGC respectively. Moreover, we found 17 of them exist in all three datasets. Table 6 shows the list of these 17 genes with 132 pfsSNVs within them. These 17 genes and their key pfsSNVs which are 1) present in the list of 260 SMG set, 2) present in the list of 573 CGC gene set, 3) have key pfsSNVs which either exist across multiple cancer types or are significantly associated with specific cancer type.
Existing knowledge on 132 key pfsSNVs
Many of these 132 pfsSNVs and their genes have been described in the previous studies. One study showed BRAF is commonly activated by somatic point mutation in human cancer, and may suggest therapeutic potentials particularly in malignant melanoma61. The BRAF L597R missense mutation, which falls in the protein’s kinase domain, has been reported in primary ovarian cancer (OV)61 and lung adenocarcinoma (LUAD), and may become a chemotherapy target for a subset of LUAD patients. CTNNB1 mutation are found in the GSK3-beta phosphorylation sites, such as S37, T41, and such mutations have been implicated in ovarian tumorigenesis62. It has been also suggested that CTNNB1 has a higher rate of phosphorylation-related mutations in skin cancer and performs a critical role in hair matrix cell cancer development63. The EGFR T790M alteration is called the “gatekeeper” mutation, which is frequently described in lung cancers; it mediates resistance to maximally tolerated dosing of HKI-272 as well as EGFR kinase inhibitors (gefitinib and erlotinib) in about half of cases64,65,66. One study also indicated that this drug resistance mutation may also be linked with lung cancer genetic susceptibility67. HRAS shows high incidence of activating mutations at Q61 in drug-induced skin cancer68,69,70. As for KRAS mutations at Q61 in lung cancer, Q61R is observed in a great portion of urethane-induced tumors from wild-type mice, however, Q61L appears in the majority of tumors from KRAS heterozygous mice. KRAS Q61 mutations may also play an important role in melanoma photocarcinogenesis71. NRAS Q61 is predominant in malignant melanoma, being a potential therapeutic target in this cancer72. Mutated NRAS at Q61 also shares similarities in signaling among various cancer types, and inhibition of both the MAPK and PI3K/AKT/mTOR pathways reduces cell viability in all cancers harboring this mutation73. IDH1 mutation at R132 is demonstrated to be tissue-specific, and may play a special role in high-grade gliomas with prognostic value for survival74,75. IDH1 and IDH2 mutations are common in AML (acute myeloid leukemia), and are associated with the accumulation of metabolite 2-hydroxyglutarate, which is affected by neomorphic enzyme activity76,77,78.
Identification of key pfsSNVs that affect patient survival
Although there are many studies that attempt to connect mutations to patient survival there are very few attempts to connect loss or gain of protein function to mortality. Identifying SNVs that directly lead to gain or loss of functional sites can help biologists focus on specific biochemical processes that might be impacted due to the variation. The amount of clinical data available from TCGA is limited because of the length of the study and the type of information that has been collected so far. Nonetheless, it is possible to showcase how one can filter SNVs for further evaluation which can assist in translating genomics efforts to actionable therapeutics and diagnostics. Below is an example of how such analysis can be performed.
We started from the pool of above identified 77 key pfsSNVs that are significantly enriched in a specific cancer type after adjusting multiple testing. Then we grouped patients based on ‘having’ or ‘not having’ a pfsSNV. From Cox regressions adjusting for age at initial pathological diagnosis, gender and clinical stage 27 out of the 77 mutations increase the risk of cancer (Hazard Ratio >1). Most of the candidate pfsSNVs occur in very small number of patients, and sometimes no death case has been observed in the small number of patients with the candidate pftSNVs, in which cases data cannot provide a valid hazard ratio estimate. We identified 3 pfsSNVs causing significant higher or lower mortality risks (See Fig. 7, Supplementary Figures 1 and 2). One example found in pancreatic cancer patients with and without the MEF2A-Y105C-Phosphorylation site was found to have statistically significant different mortality risk (p-value = 0.0012) from a log-rank test. Even after adjusting for age at initial diagnosis, pathological stage and gender using a Cox model, the mortality risks in the presence of MEF2A-Y105C is 2.348 times higher than those without MEF2A -Y105C (adjusted p-value = 0.0255). Figure 7 shows that the Kaplan-Meier estimates in the survival probabilities over days since diagnosis of the two groups are well separated. MEF2A is a transcriptional factor which binds to MEF2 element and activates numerous growth factors. It plays diverse roles in the control of cell growth, survival and apoptosis79,80,81. Although many studies have been performed on MEF2’s role in muscle and neuron, its role in pancreatic cancer remains unclear.

X-axis indicate days of survival and Y-axis indicates survival probability. Red and blue lines indicate survival time of pancreatic cancer patients with and without such mutation respectively. Log-rank test shows that, comparing with patients with MEF2A-105-Y-C-Phosphorylation, patients without this pfsSNV survive significantly longer with adjusted p-value of 0.0255. The hazard ratio is 2.348.
Although we found few pfsSNVs which appear to be associated with survival, we would like to point out the limitation of the survival analysis as our sample size is quite small among ‘having mutation’ group and ‘no mutation’ groups. When no death has been observed in the small group of participants with a specific mutation, the impact of the pftSNV on mortality are not estimable. Because no studies can enroll an infinite sample size or follow participants indefinitely, this analysis has no intention to overcome this inherent limitation of data, while just as we stated in the beginning of this section, it serves a showcase of potential impact of pfsSNVs to evoke more interest for follow-up studies.
Conclusion
We have comprehensively investigated the interplay between protein functional sites and SNVs. Each type of protein functional site shows a distinct SNV frequency in synonymous somatic mutations, synonymous germline mutations, non-synonymous somatic mutations, and non-synonymous germline mutations. Our experiments show that, at least for the majority of protein functional site types, non-synonymous germline mutations occur less frequently. We believe that these sites are, as expected, more evolutionarily conserved because of their functionality. Except for acetylation and ubiquitination sites, other protein functional site types show diverse variation frequencies between synonymous germline SNVs, synonymous somatic SNVs and non-synonymous somatic SNVs. Investigation of whether the protein functional sites of tumor tissue tends to accumulate or reject SNVs at functional sites provides insights on the effect of SNVs impacting each type of protein functional sites. For synonymous variations, although previous studies show that such variations can affect protein function by changing expression level, current understanding of the effects of synonymous mutations is still limited. However, some protein functional site types show significant synonymous variation frequency changes, for example, O-linked glycosylation site contains significantly high frequency of both germline (t statistic = −19.35, p-value = 6.5E-22) and somatic mutation (t statistic = −16.54, p-value = 1.80E-19).
Although a number of studies have been conducted to discover significant mutated genes in cancer82,83,84, our study takes steps forward by targeting the impact of key SNVs on amino acids which can be further evaluated through wet-laboratory experimentations. The top pfsSNVs exist among well-known oncogenes, such as DNA binding sites and zinc binding sites within TP53, and GEF interaction sites within NRAS. This study identified several highly mutated regions such as position 29–45 in CTNNB1. To make the key pfsSNVs comprehensive, we conducted a binomial test using both loss and gain of functional site causing pfsSNVs. This approach identified 77 pfsSNVs enriched in patients with specific cancer types which are good candidates for further investigation in terms of their biological function and effect in tumor growth. The identification of key pfsSNVs has its value not only in facilitating the investigation of tumorigenesis mechanism, but also in evaluating the risk of developing cancer. Identified pfsSNVs can be further evaluated using resources such as MutationAligner85,86, and other mutation analysis services87,88,89.
Additional Information
How to cite this article: Pan, Y. et al. Distribution bias analysis of germline and somatic single-nucleotide variations that impact protein functional site and neighboring amino acids. Sci. Rep. 7, 42169; doi: 10.1038/srep42169 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Mardis, E. R. Anticipating the 1,000 dollar genome. Genome biology 7, 112, doi: 10.1186/gb-2006-7-7-112 (2006).
Service, R. F. Gene sequencing. The race for the $1000 genome. Science 311, 1544–1546, doi: 10.1126/science.311.5767.1544 (2006).
Kilpinen, H. & Barrett, J. C. How next-generation sequencing is transforming complex disease genetics. Trends in genetics: TIG 29, 23–30, doi: 10.1016/j.tig.2012.10.001 (2013).
Hudson, T. J. et al. International network of cancer genome projects. Nature 464, 993–998, doi: 10.1038/nature08987 (2010).
The Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 517, 576–582, doi: 10.1038/nature14129 (2015).
Agrawal, Nishant et al. Integrated genomic characterization of papillary thyroid carcinoma. Cell 159, 676–690, doi: 10.1016/j.cell.2014.09.050 (2014).
Hoadley, K. A. et al. Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell 158, 929–944, doi: 10.1016/j.cell.2014.06.049 (2014).
Wu, T. J. et al. A framework for organizing cancer-related variations from existing databases, publications and NGS data using a High-performance Integrated Virtual Environment (HIVE). Database: the journal of biological databases and curation 2014, bau022, doi: 10.1093/database/bau022 (2014).
Wan, Q. et al. BioXpress: an integrated RNA-seq-derived gene expression database for pan-cancer analysis. Database: the journal of biological databases and curation 2015, doi: 10.1093/database/bav019 (2015).
Morris, L. G. et al. Pan-cancer analysis of intratumor heterogeneity as a prognostic determinant of survival. Oncotarget, doi: 10.18632/oncotarget.7067 (2016).
Andor, N. et al. Pan-cancer analysis of the extent and consequences of intratumor heterogeneity. Nature medicine 22, 105–113, doi: 10.1038/nm.3984 (2016).
Neapolitan, R., Horvath, C. M. & Jiang, X. Pan-cancer analysis of TCGA data reveals notable signaling pathways. BMC cancer 15, 516, doi: 10.1186/s12885-015-1484-6 (2015).
Leiserson, M. D. et al. Pan-cancer network analysis identifies combinations of rare somatic mutations across pathways and protein complexes. Nature genetics 47, 106–114, doi: 10.1038/ng.3168 (2015).
Fredriksson, N. J., Ny, L., Nilsson, J. A. & Larsson, E. Systematic analysis of noncoding somatic mutations and gene expression alterations across 14 tumor types. Nature genetics 46, 1258–1263, doi: 10.1038/ng.3141 (2014).
Supek, F., Minana, B., Valcarcel, J., Gabaldon, T. & Lehner, B. Synonymous mutations frequently act as driver mutations in human cancers. Cell 156, 1324–1335, doi: 10.1016/j.cell.2014.01.051 (2014).
Kimchi-Sarfaty, C. et al. A “silent” polymorphism in the MDR1 gene changes substrate specificity. Science 315, 525–528, doi: 10.1126/science.1135308 (2007).
Zhang, G., Hubalewska, M. & Ignatova, Z. Transient ribosomal attenuation coordinates protein synthesis and co-translational folding. Nature structural & molecular biology 16, 274–280, doi: 10.1038/nsmb.1554 (2009).
Zhou, M. et al. Non-optimal codon usage affects expression, structure and function of clock protein FRQ. Nature 495, 111–115, doi: 10.1038/nature11833 (2013).
Sauna, Z. E. & Kimchi-Sarfaty, C. Understanding the contribution of synonymous mutations to human disease. Nature reviews. Genetics 12, 683–691, doi: 10.1038/nrg3051 (2011).
Forbes, S. A. et al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic acids research 39, D945–950, doi: 10.1093/nar/gkq929 (2011).
Update on activities at the Universal Protein Resource (UniProt) in 2013. Nucleic acids research 41, D43-47, doi: 10.1093/nar/gks1068 (2013).
Sherry, S. T. et al. dbSNP: the NCBI database of genetic variation. Nucleic acids research 29, 308–311 (2001).
Wu, T. J. et al. Generating a focused view of disease ontology cancer terms for pan-cancer data integration and analysis. Database: the journal of biological databases and curation 2015, bav032, doi: 10.1093/database/bav032 (2015).
Marchler-Bauer, A. et al. CDD: a Conserved Domain Database for the functional annotation of proteins. Nucleic acids research 39, D225–229, doi: 10.1093/nar/gkq1189 (2011).
Lu, C. T. et al. DbPTM 3.0: an informative resource for investigating substrate site specificity and functional association of protein post-translational modifications. Nucleic acids research 41, D295–305, doi: 10.1093/nar/gks1229 (2013).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic acids research 38, e164, doi: 10.1093/nar/gkq603 (2010).
Geer, L. Y., Domrachev, M., Lipman, D. J. & Bryant, S. H. CDART: protein homology by domain architecture. Genome research 12, 1619–1623, doi: 10.1101/gr.278202 (2002).
Blom, N., Gammeltoft, S. & Brunak, S. Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. Journal of molecular biology 294, 1351–1362, doi: 10.1006/jmbi.1999.3310 (1999).
Blom, N., Sicheritz-Ponten, T., Gupta, R., Gammeltoft, S. & Brunak, S. Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics 4, 1633–1649, doi: 10.1002/pmic.200300771 (2004).
Mi, H. & Thomas, P. PANTHER pathway: an ontology-based pathway database coupled with data analysis tools. Methods Mol Biol 563, 123–140, doi: 10.1007/978-1-60761-175-2_7 (2009).
Team, R. C. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. URL http://www.R-project.org/, 2016).
Lawrence, M. S. et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 505, 495–501, doi: 10.1038/nature12912 (2014).
Forbes, S. A. et al. COSMIC: exploring the world’s knowledge of somatic mutations in human cancer. Nucleic acids research 43, D805–811, doi: 10.1093/nar/gku1075 (2015).
Radivojac, P. et al. Gain and loss of phosphorylation sites in human cancer. Bioinformatics 24, i241–247, doi: 10.1093/bioinformatics/btn267 (2008).
Vogt, G. et al. Gains of glycosylation comprise an unexpectedly large group of pathogenic mutations. Nature genetics 37, 692–700, doi: 10.1038/ng1581 (2005).
Nicolaou, N. et al. Gain of glycosylation in integrin alpha3 causes lung disease and nephrotic syndrome. The Journal of clinical investigation 122, 4375–4387, doi: 10.1172/JCI64100 (2012).
Chuma, S., Hosokawa, M., Tanaka, T. & Nakatsuji, N. Ultrastructural characterization of spermatogenesis and its evolutionary conservation in the germline: germinal granules in mammals. Molecular and cellular endocrinology 306, 17–23, doi: 10.1016/j.mce.2008.11.009 (2009).
Chamary, J. V., Parmley, J. L. & Hurst, L. D. Hearing silence: non-neutral evolution at synonymous sites in mammals. Nature reviews. Genetics 7, 98–108, doi: 10.1038/nrg1770 (2006).
Zhou, T. et al. Non-silent story on synonymous sites in voltage-gated ion channel genes. PloS one 7, e48541, doi: 10.1371/journal.pone.0048541 (2012).
Zhou, M. J., Chen, F. Z. & Chen, H. C. Ubiquitination involved enzymes and cancer. Med Oncol 31, 93, doi: 10.1007/s12032-014-0093-6 (2014).
Xu, W., Li, Y., Liu, C. & Zhao, S. Protein lysine acetylation guards metabolic homeostasis to fight against cancer. Oncogene 33, 2279–2285, doi: 10.1038/onc.2013.163 (2014).
Narayan, S., Bader, G. D. & Reimand, J. Frequent mutations in acetylation and ubiquitination sites suggest novel driver mechanisms of cancer. Genome medicine 8, 55, doi: 10.1186/s13073-016-0311-2 (2016).
Zhang, J., Shen, L. & Sun, L. Q. The regulation of radiosensitivity by p53 and its acetylation. Cancer letters 363, 108–118, doi: 10.1016/j.canlet.2015.04.015 (2015).
Ruprecht, B. & Lemeer, S. Proteomic analysis of phosphorylation in cancer. Expert review of proteomics 11, 259–267, doi: 10.1586/14789450.2014.901156 (2014).
Reimand, J., Wagih, O. & Bader, G. D. The mutational landscape of phosphorylation signaling in cancer. Scientific reports 3, 2651, doi: 10.1038/srep02651 (2013).
Jacobson, H. I., Andersen, T. T. & Bennett, J. A. Development of an active site peptide analog of alpha-fetoprotein that prevents breast cancer. Cancer Prev Res (Phila) 7, 565–573, doi: 10.1158/1940-6207.CAPR-13-0405 (2014).
Hartmann, T. et al. The impact of single nucleotide polymorphisms on human aldehyde oxidase. Drug metabolism and disposition: the biological fate of chemicals 40, 856–864, doi: 10.1124/dmd.111.043828 (2012).
Parker, J. A. & Mattos, C. The Ras-Membrane Interface: Isoform-specific Differences in The Catalytic Domain. Molecular cancer research: MCR 13, 595–603, doi: 10.1158/1541-7786.MCR-14-0535 (2015).
Begovich, A. B. et al. A missense single-nucleotide polymorphism in a gene encoding a protein tyrosine phosphatase (PTPN22) is associated with rheumatoid arthritis. American journal of human genetics 75, 330–337, doi: 10.1086/422827 (2004).
Di Lullo, G. A., Sweeney, S. M., Korkko, J., Ala-Kokko, L. & San Antonio, J. D. Mapping the ligand-binding sites and disease-associated mutations on the most abundant protein in the human, type I collagen. The Journal of biological chemistry 277, 4223–4231, doi: 10.1074/jbc.M110709200 (2002).
Hamamoto, R., Saloura, V. & Nakamura, Y. Critical roles of non-histone protein lysine methylation in human tumorigenesis. Nature reviews. Cancer 15, 110–124, doi: 10.1038/nrc3884 (2015).
Mazumder, R., Morampudi, K. S., Motwani, M., Vasudevan, S. & Goldman, R. Proteome-wide analysis of single-nucleotide variations in the N-glycosylation sequon of human genes. PloS one 7, e36212, doi: 10.1371/journal.pone.0036212 (2012).
Park, C. & Zhang, J. Genome-wide evolutionary conservation of N-glycosylation sites. Molecular biology and evolution 28, 2351–2357, doi: 10.1093/molbev/msr055 (2011).
Kang, J. G., Ko, J. H. & Kim, Y. S. Application of cancer-associated glycoforms and glycan-binding probes to an in vitro diagnostic multivariate index assay for precise diagnoses of cancer. Proteomics, doi: 10.1002/pmic.201500553 (2016).
Hounsell, E. F., Davies, M. J. & Renouf, D. V. O-linked protein glycosylation structure and function. Glycoconjugate journal 13, 19–26 (1996).
Van den Steen, P., Rudd, P. M., Dwek, R. A. & Opdenakker, G. Concepts and principles of O-linked glycosylation. Critical reviews in biochemistry and molecular biology 33, 151–208, doi: 10.1080/10409239891204198 (1998).
Slawson, C. & Hart, G. W. O-GlcNAc signalling: implications for cancer cell biology. Nature reviews. Cancer 11, 678–684, doi: 10.1038/nrc3114 (2011).
Krzywinski, M. et al. Circos: an information aesthetic for comparative genomics. Genome research 19, 1639–1645, doi: 10.1101/gr.092759.109 (2009).
Morin, P. J. Beta-catenin signaling and cancer. BioEssays: news and reviews in molecular, cellular and developmental biology 21, 1021–1030, doi: 10.1002/(SICI)1521-1878(199912)22:1<1021::AID-BIES6>3.0.CO;2-P (1999).
Lu, S., Jang, H., Nussinov, R. & Zhang, J. The Structural Basis of Oncogenic Mutations G12, G13 and Q61 in Small GTPase K-Ras4B. Scientific reports 6, 21949, doi: 10.1038/srep21949 (2016).
Davies, H. et al. Mutations of the BRAF gene in human cancer. Nature 417, 949–954, doi: 10.1038/nature00766 (2002).
Sagae, S. et al. Mutational analysis of beta-catenin gene in Japanese ovarian carcinomas: frequent mutations in endometrioid carcinomas. Japanese journal of cancer research: Gann 90, 510–515 (1999).
Chan, E. F., Gat, U., McNiff, J. M. & Fuchs, E. A common human skin tumour is caused by activating mutations in beta-catenin. Nature genetics 21, 410–413, doi: 10.1038/7747 (1999).
Godin-Heymann, N. et al. The T790M “gatekeeper” mutation in EGFR mediates resistance to low concentrations of an irreversible EGFR inhibitor. Molecular cancer therapeutics 7, 874–879, doi: 10.1158/1535-7163.MCT-07-2387 (2008).
Bean, J. et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proceedings of the National Academy of Sciences of the United States of America 104, 20932–20937, doi: 10.1073/pnas.0710370104 (2007).
Suda, K., Onozato, R., Yatabe, Y. & Mitsudomi, T. EGFR T790M mutation: a double role in lung cancer cell survival? Journal of thoracic oncology: official publication of the International Association for the Study of Lung Cancer 4, 1–4, doi: 10.1097/JTO.0b013e3181913c9f (2009).
Bell, D. W. et al. Inherited susceptibility to lung cancer may be associated with the T790M drug resistance mutation in EGFR. Nature genetics 37, 1315–1316, doi: 10.1038/ng1671 (2005).
Cox, A. D. & Der, C. J. The RAF inhibitor paradox revisited. Cancer cell 21, 147–149, doi: 10.1016/j.ccr.2012.01.017 (2012).
Balmain, A., Ramsden, M., Bowden, G. T. & Smith, J. Activation of the mouse cellular Harvey-ras gene in chemically induced benign skin papillomas. Nature 307, 658–660 (1984).
Leon, J., Kamino, H., Steinberg, J. J. & Pellicer, A. H-ras activation in benign and self-regressing skin tumors (keratoacanthomas) in both humans and an animal model system. Molecular and cellular biology 8, 786–793 (1988).
Yang, G., Curley, D., Bosenberg, M. W. & Tsao, H. Loss of xeroderma pigmentosum C (Xpc) enhances melanoma photocarcinogenesis in Ink4a-Arf-deficient mice. Cancer research 67, 5649–5657, doi: 10.1158/0008-5472.CAN-06-3806 (2007).
Uguen, A. et al. NRAS (Q61R), BRAF (V600E) immunohistochemistry: a concomitant tool for mutation screening in melanomas. Diagnostic pathology 10, 121, doi: 10.1186/s13000-015-0359-0 (2015).
Vujic, I. et al. Mutant NRASQ61 shares signaling similarities across various cancer types–potential implications for future therapies. Oncotarget 5, 7936–7944, doi: 10.18632/oncotarget.2326 (2014).
Bleeker, F. E. et al. IDH1 mutations at residue p.R132 (IDH1(R132)) occur frequently in high-grade gliomas but not in other solid tumors. Human mutation 30, 7–11, doi: 10.1002/humu.20937 (2009).
Bleeker, F. E. et al. The prognostic IDH1(R132) mutation is associated with reduced NADP+ -dependent IDH activity in glioblastoma. Acta neuropathologica 119, 487–494, doi: 10.1007/s00401-010-0645-6 (2010).
Abbas, S. et al. Acquired mutations in the genes encoding IDH1 and IDH2 both are recurrent aberrations in acute myeloid leukemia: prevalence and prognostic value. Blood 116, 2122–2126, doi: 10.1182/blood-2009-11-250878 (2010).
Ward, P. S. et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer cell 17, 225–234, doi: 10.1016/j.ccr.2010.01.020 (2010).
Gross, S. et al. Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. The Journal of experimental medicine 207, 339–344, doi: 10.1084/jem.20092506 (2010).
Zhao, M. et al. Regulation of the MEF2 family of transcription factors by p38. Molecular and cellular biology 19, 21–30 (1999).
Okamoto, S. et al. Dominant-interfering forms of MEF2 generated by caspase cleavage contribute to NMDA-induced neuronal apoptosis. Proceedings of the National Academy of Sciences of the United States of America 99, 3974–3979, doi: 10.1073/pnas.022036399 (2002).
Shalizi, A. et al. A calcium-regulated MEF2 sumoylation switch controls postsynaptic differentiation. Science 311, 1012–1017, doi: 10.1126/science.1122513 (2006).
Watson, I. R., Takahashi, K., Futreal, P. A. & Chin, L. Emerging patterns of somatic mutations in cancer. Nature reviews. Genetics 14, 703–718, doi: 10.1038/nrg3539 (2013).
Ding, L., Wendl, M. C., McMichael, J. F. & Raphael, B. J. Expanding the computational toolbox for mining cancer genomes. Nature reviews. Genetics 15, 556–570, doi: 10.1038/nrg3767 (2014).
Dees, N. D. et al. MuSiC: identifying mutational significance in cancer genomes. Genome research 22, 1589–1598, doi: 10.1101/gr.134635.111 (2012).
Miller, M. L. et al. Pan-Cancer Analysis of Mutation Hotspots in Protein Domains. Cell systems 1, 197–209, doi: 10.1016/j.cels.2015.08.014 (2015).
Gauthier, N. P. et al. MutationAligner: a resource of recurrent mutation hotspots in protein domains in cancer. Nucleic acids research 44, D986–991, doi: 10.1093/nar/gkv1132 (2016).
Ng, S. et al. Paradigm-Shift predicts the function of mutations in multiple cancers using pathway impact analysis. Bioinformatics 28, i640–i646, doi: 10.1093/bioinformatics/bts402 (2012).
Adzhubei, I., Jordan, D. M. & Sunyaev, S. R. Predicting functional effect of human missense mutations using PolyPhen-2. Current protocols in human genetics/editorial board, Jonathan L. Haines … [et al.] Chapter 7, Unit7 20, doi: 10.1002/0471142905.hg0720s76 (2013).
Ng, P. C. & Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic acids research 31, 3812–3814 (2003).
Acknowledgements
We want to thank Hayley Dingerdissen for useful comments. This project was funded in part by The McCormick Genomic & Proteomic Center funds and GW funds to RM.
Author information
Authors and Affiliations
Contributions
Y.P., C.Y. and R.M. conceived, designed and coordinated the study. Y.P. and C.Y. developed the specific algorithms and were responsible for statistical and functional analysis, and wrote the manuscript. Q.W., J.T.-R. and Y.F. participated in data integration. Q.P. directed the statistical analysis and Y.H. developed the BioMuta web interface to browse the data. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Pan, Y., Yan, C., Hu, Y. et al. Distribution bias analysis of germline and somatic single-nucleotide variations that impact protein functional site and neighboring amino acids. Sci Rep 7, 42169 (2017). https://doi.org/10.1038/srep42169
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep42169
- Springer Nature Limited