
Abstract
It is still unclear whether there exist functional constraints on the evolution of protein ubiquitination sites, because most previous studies regarded all protein ubiquitination sites as a whole or only focused on limited structural properties. We tried to clarify the relation between functional constraints and ubiquitination sites evolution. We investigated the evolutionary conservation of human ubiquitination sites in a broad evolutionary scale from G. gorilla to S. pombe, and we found that in organisms originated after the divergence of vertebrate, ubiquitination sites are more conserved than their flanking regions, while the opposite tendency is observed before this divergence time. By grouping the ubiquitination proteins into different functional categories, we confirm that many functional constraints like certain molecular functions, protein tissue expression specificity and protein connectivity in protein-protein interaction network enhance the evolutionary conservation of ubiquitination sites. Furthermore, by analyzing the gains of ubiquitination sites at different divergence time and their functional characters, we validate that the emergences of ubiquitination sites at different evolutionary time were also affected by the uncovered functional constraints. The above results suggest that functional constraints on the adaptive evolution of ubiquitination sites increase the opportunity for ubiquitination to synthetically regulate various cellular and developmental processes during evolution.
Similar content being viewed by others

Introduction
Proteins ubiquitination at lysine residues mediates 80~85% of the protein degradation in eukaryotic cells, and this ATP-dependent process is efficient and highly specific1,2. Ubiquitination is involved not only in protein degradation, but also in a broad spectrum of cellular processes including cell cycle progression3, apoptosis4, transcriptional regulation5, DNA damage repair6 and immune response7. Disorder of ubiquitination is found to be related with various human diseases, such as cancer8,9,10 and neuronal disorders11,12,13. The specificity of ubiquitination sites extends the functions of ubiquitination, making it possible to regulate various biological processes. Exploring the evolution of ubiquitination sites and functional constraints would be helpful to understand the underlying mechanism and function of ubiquitination.
Taking 753 ubiquitination sites of 626 proteins from public database as a whole, Hagai et al.14 analyzed ubiquitination sites evolution and found that ubiquitination sites are weakly more conserved than unmodified lysine residues, and ubiquitination sites tend to evolve faster in disordered regions than in ordered domains. By analyzing 281 ubiquitination sites of 252 proteins that first appeared along the human lineage, Kim et al.15 found that these novel ubiquitination sites could be involved in the evolution of protein degradation and other regulatory networks. However, as ubiquitination can be involved in a wide range of biological functions, the functional constraints might also influence the evolutionary conservation of the ubiquitination sites. As previous studies regarded all ubiquitination sites as a whole in a limited evolutionary period or only focused on their structural propensity, it remains to be explored whether there exists a relation between functional constraints and ubiquitination sites adaptive evolution.
To acquire more accurate and comprehensive understanding of the relation between functional constraints and ubiquitination sites evolution events, we first investigated the evolutionary conservation of the human ubiquitination sites in a broad evolutionary scale by aligning them with their orthologs from G. gorilla to S. pombe (Fig. 1) and we found that in organisms originated after the divergence of vertebrate, ubiquitination sites are more conserved than their flanking regions, while the opposite tendency is observed before this divergence time; then, by grouping the ubiquitination proteins into different functional categories, we also found that many functional constraints like certain functions (such as enzyme binding, cellular macromolecule metabolic process and developmental process), protein tissue expression specificity and protein connectivity in protein-protein network enhance the evolutionary conservation of ubiquitination sites; finally, by analyzing the gains of ubiquitination sites at different divergence time and their functional characters, we validates that the emergence of ubiquitination sites at different evolutionary time were affected by the uncovered functional constraints. Above results suggest that functional constraints on the adaptive evolution of ubiquitination sites increase the opportunity for ubiquitination to synthetically regulate various cellular and developmental processes during evolution.

The phylogeny of these organisms was obtained by the tool of Taxonomy Common Tree from NCBI59, and the phylogenetic tree was plotted using FigTree (http://tree.bio.ed.ac.uk/software/figtree/).
Results and Discussion
Conservation of ubiquitination sites versus their flanking regions over a broad evolutionary scale
Previous studies pointed out that ubiquitination sites are more conserved compared with their flanking regions14, however, it is still unclear how the evolutionary rate of ubiquitination sites changes at different evolutionary periods. To answer this question, we investigated the evolutionary conservation of human ubiquitination sites (Supplementary Table S1) in a broad evolutionary scale by aligning them with their orthologs in 14 common model organisms: G. gorilla, M. mulatta, R. norvegicus, M. musculus, G. gallus, X. tropicalis, D. rerio, D. melanogaster, A. gambiae, C. elegans, K. lactis, S. cerevisiae, N. crassa and S. pombe (Fig. 1). Then we introduced Poisson distance16 to estimate the evolutionary conservation of ubiquitination sites. The Poisson distance can correct multiple substitutions at a site and has the linear relationship with time16, and this strategy has been successfully used to measure the evolutionary conservation of phosphorylation sites17. Following Wang et al.17, we selected the ten flanking residues around a ubiquitination site other than other lysine residues as the background to correct the potential biases17,18 (see Materials and Methods).
Then we performed z-score test to examine whether there exists significant difference between the Poisson distance of ubiquitination sites and that of flanking regions in each individual reference organisms. In addition to obtaining the P-value for each individual reference organisms through z-score test, we employed chi-square test to obtain an overall P-value to assess the significance of the difference between the Poisson distance of ubiquitination sites and that of flanking regions across multiple reference organisms (see Materials and Methods).
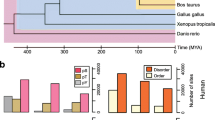
To obtain novel finding for the evolutionary tendency of ubiquitination sites, we adopted more ubiquitination datasets and reference organisms in a broad evolutionary scale. We observed that using individual reference organism before the divergence of vertebrate, ubiquitination sites evolved faster than flanking regions, while using individual reference organism after the divergence of vertebrate, ubiquitination sites are more conserved than flanking regions. Moreover, we calculated overall P-value for multiple reference organisms, and we also observed significant differences. On the overall level, ubiquitination sites evolve faster before the vertebrate divergence time, while after that, ubiquitination sites are more conserved (Fig. 2), which is consistent with analysis in individual reference organism. This finding suggests that there might be some functional constraints increasing the opportunity for fine regulation of the ubiquitination-mediated cellular and developmental processes during evolution19, which leads to the promotion in evolutionary conservation of ubiquitination sites.

14 reference organisms were arranged along the X axis in the order of their evolutionary distance to H. sapiens. The dashed line indicates the divergence time of vertebrate. In organisms originated after the divergence of vertebrate (on the left side of the dashed line), ubiquitination sites are more conserved than their flanking regions, while the opposite tendency is observed before this divergence time (on the right side of the dashed line). U: ubiquitination sites; F: flanking regions.
Constraints of various biological factors on ubiquitination sites evolution
Constraints shape phenotypic evolution together with adaption20. Although the basis of the mechanism for phenotypic evolution proposed by Darwin (1859) was adaptation21, it is recognized that the response to natural selection is subject to various constraints that place qualitative and quantitative limits on the course or outcome of adaptive evolution22,23,24,25,26,27. These constraints can be biomechanical-chemical, developmental, genetic and functional. As for proteins, systematic surveys indicate that protein evolution is not determined exclusively by selection on biological processes, but also affected by their expression patterns and their position in biological networks. In this study, we tried to explore what functional constraints influence ubiquitination sites conservation by classifying proteins into various categories.
KEGG pathway categories
We mapped 8245 non-redundant human ubiquitination proteins with 35197 ubiquitination sites to 283 KEGG pathways28. These pathways were further grouped into six large functional categories: metabolism, genetic information processing, environmental information processing, cellular processes, organismal systems and human diseases. Using Poisson distance, we pair-wisely compared the evolutionary conservation of ubiquitination sites between six KEGG pathway categories. As a result, different KEGG pathway categories have different evolutionary conservations across multiple reference organisms (Fig. S1). In particular, ubiquitination sites involved in the metabolism pathway evolved at a higher rate than those in other pathways (Fig. 3A). However, we infer that this may be due to that proteins in metabolism pathway are more conserved during evolution regardless of ubiquitination. Therefore, we applied the same analysis to flanking regions, and found that flanking regions in metabolism pathway also evolve at a higher rate than those in other pathways (Fig. 3B). This finding confirms that protein conservation difference introduced by different protein categories cannot be ignored. To eliminate this influence, we calculated relative Poisson distance, which is the ratio of Poisson distance of the ubiquitination sites to that of their flanking regions in each group (see Materials and Methods). Using this relative Poisson distance, we found that ubiquitination sites in metabolism pathway show lower conservation compared with those in other pathways in multiple reference organisms (Fig. 3C). Furthermore, the small overall P-value across all the reference organisms demonstrates that the ubiquitination sites in the metabolism pathway (especially amino acid metabolism, metabolism of cofactors and vitamins, carbohydrate metabolism and lipid metabolism pathways. Fig. S2) evolved significantly more quickly than those in other pathways, which indicates that KEGG pathways like genetic information processing, environmental information processing, cellular processes, organismal systems and human diseases are likely to be constraints on the evolution of ubiquitination, while metabolism pathway is not much involved (it should be pointed out that ubiquitin mediated proteolysis is not included in KEGG metabolism pathway).

(A,B) Using Poisson distance to evaluate the conservation of ubiquitination sites/flanking regions in metabolism pathway and other pathways. (C) Using relative Poisson distance (see Materials and Methods) to compare the evolutionary conservations of ubiquitination sites between metabolism pathway and other pathways.
Gene Ontology terms
We also classified ubiquitination proteins according to Gene Ontology terms29 and used relative Poisson distance to measure the evolutionary rate of ubiquitination sites. According to the result, ubiquitination sites in molecular function terms like poly (a) RNA binding (Fig. 4A), enzyme binding (Fig. 4B) and transcription factor binding (Fig. S3A) evolve with lower rates than those in other functional terms, while ubiquitination sites in term like oxidoreductase activity evolve with higher rate (Fig. S3B). These results suggest that ubiquitination plays more important roles in certain molecular functions. As for cellular component, we found that ubiquitination sites are more conserved in components of nucleus (Fig. 4C) and ribonucleoprotein complex (Fig. S3C) than in others, which suggests ubiquitination is important to these cellular components, this finding is in accordance with previous study30. In addition, we found that ubiquitination sites of intracellular organelle (Fig. 4D) have a lower evolutionary rate than those of other proteins, while ubiquitination sites of extracellular matrix have a higher evolutionary rate (Fig. S3D), which indicates ubiquitination is more important for intracellular proteins. For the ontology of biological process, result shows that ubiquitination sites in groups like developmental process (Fig. 4E) and cellular macromolecule metabolic process (Fig. 4F) are more conserved compared with those in other biological processes; on the contrary, ubiquitination sites in lipid metabolic process (Fig. S3D) and small molecule metabolic process (Fig. S3E) are less conserved than those in other biological processes. These findings are consistent with our previous hypothesis: ubiquitination sites are likely to participate in the fine regulation of cellular and developmental processes, and this improves the evolutionary conservation of ubiquitination sites during evolution.

Relative Poisson distance of the ubiquitination sites for the Gene Ontology terms of poly (a) RNA binding (A), enzyme binding (B), nucleus (C), intracellular organelle (D), developmental process (E) and cellular macromolecule metabolic process (F).
Protein tissue expression specificity
An obvious correlate of pleiotropy in multicellular organisms is breadth of expression: proteins that are expressed in many tissues have to operate under diverse cellular conditions and might interact with diverse proteins. It has been proved that in mammals, insects and plants, broadly expressed proteins evolve more slowly than tissue-specific proteins31. Therefore, we anticipate that ubiquitination sites evolution might be affected by their protein tissue expression specificity.
For each protein, we calculated its tissue-specificity score TSPS32 to measure the extent to which its expression distribution departs from the null distribution of uniform expression across all tissues. According to the definition of TSPS, a minimal TSPS = 0 means proteins uniform expression across all tissues, while a maximal TSPS ≈ 5 means only in a single tissue. Following Ravasi et al.32, we define proteins with widespread expression (TSPS < 1) as “facilitators”, and those with high tissue specificity (TSPS ≥ 1) as “specifiers”. Using relative Poisson distance, we found that ubiquitination sites in “facilitators” are more conserved compared with those in “specifiers” (Fig. 5A). This preference suggests that protein expression pattern can act as a constraint on ubiquitination site evolution and ubiquitination plays more important roles in broadly expressed “facilitators”.

(A) Constraint of protein tissue expression specificity. The protein tissue expression specificity is measured by TSPS. Following Ravasi et al.32, we define proteins with widespread expression (TSPS < 1) as “facilitators”, and those with high tissue specificity (TSPS ≥ 1) as “specifiers”. (B) Constraint of protein connectivity in protein-protein interaction network. The protein connectivity is measured by protein degree (number of interaction partners in the human protein interaction network from HPRD Release 9). We define proteins with degree larger than 20 as hub proteins.
Protein connectivity in protein-protein interaction network
Protein interaction networks are principal components of a system-level description of the cell33,34,35,36, and many previous studies have explored global aspects of network topology, clearly linking it to protein function, expression dynamics and other genomic features37,38,39,40. Here, we adopted protein degree (number of direct interaction partners) to measure protein connectivity in human protein-protein interaction network (HPRD41 Release 9), and we define proteins with degree larger than 20 as hub proteins. Using relative Poisson distance to evaluate ubiquitination sites conservation, we found that ubiquitination sites on hub proteins are more conserved than those on other proteins (Fig. 5B), which suggests that hub proteins exert stronger constraints on ubiquitination sites evolution. As hub proteins play essential roles in biological processes42, this constraint and subsequent adaptive evolution of ubiquitination sites increase the opportunity for ubiquitination to function widely.
In conclusion, by classifying ubiquitination sites into various categories instead of taking all ubiquitination sites as a whole, we found that various cellular and developmental processes, protein tissue expression specificity and protein connectivity in protein-protein interaction network can act as functional constraints on the evolution of ubiquitination sites. All these functional constraints increase the opportunity for the complex regulation of the ubiquitination-mediated cellular and developmental processes during evolution, leading to the promotion in evolutionary conservation of ubiquitination sites. Considering that functionally more important molecules, or portions of molecules, evolve slower than less important ones43, we think that ubiquitination play more important roles in these functional categories. These findings provide a novel view for the research of ubiquitination evolutionary events.
Gains and functional distribution of novel ubiquitination sites during evolution
The above analysis suggests that certain biological functions impose constraints on ubiquitination sites evolutionary conservation, and we wonder whether the emergence of ubiquitination sites at different evolutionary time were affected by these functional constraints. To answer this question, we performed multiple sequence alignment to identify ubiquitination sites emerged at different evolutionary time (Fig. 6A), and functional enrichment analysis to explore the functional characters of these corresponding ubiquitination proteins (Fig. 6B). We have listed all the cases of newly gained ubiquitination sites at different evolutionary stages in Supplementary Table S1.

(A) Detection and timing of gains of ubiquitination sites during evolution. The numbers of newly gained ubiquitination sites were plotted at different evolutionary time, and 14 reference organisms were arranged along the X axis in the order of their evolutionary distance to H. sapiens. Five key points of evolutionary divergence time (eukaryote, animal, vertebrate, mammalian and primate) with great promotion of ubiquitination sites were marked by the dashed line. (B) Functional distribution of newly gained ubiquitination lysines. GOfact54 was used to calculate the category enrichment for the newly gained ubiquitination proteins. Significant enrichment/depletion was defined as P-value < 0.05 (hyper geometry distribution test). The number in each table cell is the number of observed proteins in certain functional category, and the number in brackets is the random expectation. (C–F) Four examples of gains of ubiquitination sites. Central ubiquitination lysines and the surrounding regions for proteins RRAGA, PCNA, PEX5 and RAB11A are shown based on multiple sequence alignment. The gained ubiquitination sites are painted in black. The residues that are the same as those in human sequence are represented as dots (.). Dash (−) is alignment gap.
As shown in Fig. 6A, at the divergence time of eukaryote, ubiquitination sites begin to emerge with large amount, indicating that ubiquitination starts to play important roles in eukaryotic biological processes. There follows four notable incensements of ubiquitination sites at the divergence time of animal, vertebrate, mammalian and primate respectively, which are all critical evolutionary points. This phenomenon is supposed to promote the complex regulation of ubiquitination-mediated cellular and developmental processes.
From Fig. 6B, we found that at the divergence time of eukaryote, there gains a significantly improved portion of ubiquitination sites for the process of protein modification. This finding is consistent with the phenomenon that ubiquitination mediates 80~85% of the protein degradation in eukaryotic cells (although ubiquitin like protein Pup exist in prokaryote, it doesn’t play as important role of protein degradation as in eukaryotic cells44). We also found that proteins of newly gained ubiquitination sites at divergence time of eukaryote are enriched in function categories like cell cycle, cell division, cell death, protein localization, intracellular signal transduction and DNA repair. For example, K244 ubiquitination site of protein RRAGA emerged at divergence time of eukaryote (Fig. 6C). The K244 was found to be attached by a K63-linked ubiquitin chain mediated by RNF152 and this event can regulate mTORC1 signal transduction45. Another example (Fig. 6D) is mono-ubiquitination of PCNA on K164 (gained at eukaryote), which is essential for DNA repair by translation DNA synthesis46.
At the divergence time of vertebrate, there are great proportions of newly gained ubiquitination sites enriched in functions like regulation of cellular process, cell differentiation, anatomical structure morphogenesis, cell motility, receptor metabolic process and pexophagy, which are typical cellular and developmental processes. For example, PEX5 gained a ubiquitination site K209 at vertebrate (Fig. 6E), this ubiquitination site is recognized by the autophagy adaptor protein p62, directing the autophagosome to peroxisomes to induce pexophagy47. Another example is that co-expression of ADRB2 (Beta-2 adrenergic receptor) with HACE1 induces the ubiquitination and activation of Rab11a at K145 (gained at vertebrate) (Fig. 6F), which in turn regulates ADRB2 receptor metabolic process48.
We found that during evolution, the proteins containing newly gained ubiquitination sites are significantly enriched in the functions of protein localization, protein modification, cell cycle, cell differentiation and anatomical structure morphogenesis, which are all fundamental cellular and developmental processes for the survival of the cell. This finding validates our hypothesis that the emergences of ubiquitination sites at different evolutionary time were also affected by multiple functional constraints. This suggests that ubiquitination events play more and more important roles in the fine-tuning mechanism of cell survival in the whole process of evolution.
As Hagai et al. pointed out, there exist three levels of ubiquitination sites evolutionary conservation: conserved, compensated and uncompensated, while the latter two belong to the situation of non-conserved ubiquitination sites14. In this paper, we only focus on the strict definition of conservation, trying to answer the question that whether the ubiquitination sites are conserved along the evolution and what the functional constrains are. Therefore, we didn’t perform ubiquitination sites compensation analysis in this study, although the compensation mechanism is also very important for ubiquitination sites evolution14. In addition, it is impossible for us to implement the statistical evolutionary analysis for this compensation mechanism now, because there is not enough functional annotation information available to determine whether a nearby lysine is functionally compensated or not.
Materials and Methods
Preprocessing Data
Datasets of human ubiquitination proteins and sites were retrieved from PhosphoSitePlus49. Protein sequences were downloaded from UniProtKB50. The orthologs of the ubiquitination proteins in G. gorilla, M. mulatta, R. norvegicus, M. musculus, G. gallus, X. tropicalis, D. rerio, D. melanogaster, A. gambiae, C. elegans, K. lactis, S. cerevisiae, N. crassa and S. pombe were downloaded from Inparanoid V8.051. Clustal Omega52 was used for sequence alignment, and the orthologous sites of the human ubiquitination sites were extracted from the alignments. Local protein structures were predicted by the VSL2 software53. Pathway assignment was performed based on the KEGG database28 (Release 53.0). The enrichment of specific GO terms was tested using a hyper geometry distribution test, followed by the Bonferroni multiple testing correction to control for the false discovery rate54. Protein-protein interaction data were retrieved from HPRD41 (Release 9). Protein tissue expression data were downloaded from ProteomicsDB55 (20150420).
Calculating Poisson distance
For each ubiquitination protein in human, we retrieved its orthologous protein from the reference organisms. Then we used Clustal Omega to align the sequence of human ubiquitination protein and that of its ortholog. The orthologous site of human protein ubiquitination site was extracted from the alignments to estimate whether the ubiquitination site is remained or changed in reference organism.
For a protein group i in human, we calculated the proportion of different residues between the ubiquitination sites of human protein and their orthologous sites in organism o as pi,o, which is used for comparing the extent of homologous ubiquitination sites divergence:

where  denotes the number of different residues at the ubiquitination site and ni,o denotes the number of all residues. Here,
denotes the number of different residues at the ubiquitination site and ni,o denotes the number of all residues. Here,  follows binomial distribution, therefore, the variance of pi,o is given by
follows binomial distribution, therefore, the variance of pi,o is given by

as the reference Nei and Kumar16, we took the Poisson correction to transform variance proportion pi,o value into Poisson distance di,o to measure the degree of conservation, which can correct multiple substitutions at a site and has the linear relationship with time.

then the variance of  is given by
is given by

and then, Poisson distance di,o will be applied to measure the evolutionary distance of ubiquitination sites between human and reference organism. The larger the Poisson distance, the higher the evolutionary rate and the lower the evolutionary conservation.
Contrasting ubiquitination sites with their flanking regions
Referring to phosphorylation sites evolution analysis17,18, we selected the ten flanking residues around a ubiquitination site rather than other lysine sites or random sample of sites as the background based on following speculations. PTM (post-translational modification) sites can be conserved for many other reasons rather than ubiquitination alone, such as protein dispensability, expression abundance, and others31. Besides, ubiquitination sites locate preferentially in disordered regions, the local structure will impose constraints on the conservation of ubiquitination sites56,57. Therefore comparing ubiquitination sites with a random sample of sites or other lysine residues can be misleading. Whether ubiquitination sites are more conserverd than non-ubiquitination sites can only be estimated when local protein structures are taken into account17,18,58.
The flanking regions were defined as the ten residues centered on a ubiquitination site. All flanking regions in a protein category i were extracted and combined together, and the Poisson distance of the flanking regions was estimated with the same method as we did for the ubiquitination sites. The z-score was used to assess whether the ubiquitination sites and the flanking regions evolved at the same rate17, which is calculated as:

where di,o and di′,o are the Poisson distance for the ubiquitination sites and the flanking regions, respectively. Under the null hypothesis, the z-score follows the standard normal distribution approximately. The obtained z-score Zii′,o can be used to contrast ubiquitination sites with flanking regions in a single organism o. To compare the evolutionary conservation of the ubiquitination sites with that of flanking regions in the protein category i across multiple organisms, we further calculate:

which follows a chi-square distribution with the degree of freedom equal to n (n denotes the number of organisms considered).
Calculating relative Poisson distance
We introduced relative Poisson distance to eliminate difference of evolutionary conservations between protein categories caused by protein intrinsic properties. For reference organism o, the relative Poisson distance of ubiquitination sites in protein category i is calculated as:

where di,o and di′,o denote the Poisson distance of ubiquitination sites and flanking regions in protein category i, respectively. The estimate variance will be:

where var(di,o) and var(di′,o) represent estimated variance for the Poisson distance of ubiquitination sites and flanking regions in protein category i, respectively. The larger the relative Poisson distance, the higher the evolutionary rate and the lower the evolutionary conservation.
Comparing ubiquitination sites between categories
The z-score was also calculated to assess the difference of relative Poisson distance between two categories i and j:

where ri,o and rj,o denote the relative Poisson distance of ubiquitination sites in protein categories i and j respectively, var(ri,o) and var(rj,o) represent estimated variance for the relative Poisson distance of ubiquitination sites in protein categories i and j respectively. The z-score follows the standard normal distribution approximately. The obtained z-score Zij,o reflects the difference of ubiquitination sites evolutionary conservation between the categories i and j in a single organism o. To assess the difference of ubiquitination sites conservation between the categories i and jacross multiple organisms, we further calculate:

which follows a chi-square distribution with the degree of freedom equal to n(n denotes the number of organisms considered) under the null hypothesis.
Calculating protein tissue expression specificity score
Using relative entropy, protein tissue expression specificity score TSPSi32 is calculated as:

where  (the fractional expression level of the protein i expression level in tissue j) is computed as the ratio of the protein iexpression level in tissue j to its sum total expression level across all tissues, and qi was the fractional expression of protein i under a null model assuming uniform expression across tissues.
(the fractional expression level of the protein i expression level in tissue j) is computed as the ratio of the protein iexpression level in tissue j to its sum total expression level across all tissues, and qi was the fractional expression of protein i under a null model assuming uniform expression across tissues.
Additional Information
How to cite this article: Lu, L. et al. Functional constraints on adaptive evolution of protein ubiquitination sites. Sci. Rep. 7, 39949; doi: 10.1038/srep39949 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Hershko, A. & Ciechanover, A. The ubiquitin system for protein degradation. Annu. Rev. Biochem. 61, 761–807 (1992).
Hochstrasser, M. Ubiquitin-dependent protein degradation. Annu. Rev. Genet. 30, 405–439 (1996).
Nakayama, K. I. & Nakayama, K. Ubiquitin ligases: cell-cycle control and cancer. Nat. Rev. Cancer 6, 369–381 (2006).
Yang, Y., Fang, S., Jensen, J. P., Weissman, A. M. & Ashwell, J. D. Ubiquitin protein ligase activity of IAPs and their degradation in proteasomes in response to apoptotic stimuli. Science 288, 874–877 (2000).
Muratani, M. & Tansey, W. P. How the ubiquitin-proteasome system controls transcription. Nat. Rev. Mol. Cell Biol. 4, 192–201 (2003).
Hoege, C., Pfander, B., Moldovan, G.-L., Pyrowolakis, G. & Jentsch, S. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature 419, 135–141 (2002).
Liu, Y.-C. Ubiquitin ligases and the immune response. Annu. Rev. Immunol. 22, 81–127 (2004).
Semenza, G. L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 3, 721–732 (2003).
Burger, A. M. & Seth, A. K. The ubiquitin-mediated protein degradation pathway in cancer: therapeutic implications. Eur. J. Cancer Oxf. Engl. 1990 40, 2217–2229 (2004).
Bertrand, M. J. M. et al. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol. Cell 30, 689–700 (2008).
Shimura, H. et al. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat. Genet. 25, 302–305 (2000).
Shimura, H. et al. Ubiquitination of a new form of alpha-synuclein by parkin from human brain: implications for Parkinson’s disease. Science 293, 263–269 (2001).
Ciechanover, A. & Brundin, P. The ubiquitin proteasome system in neurodegenerative diseases: sometimes the chicken, sometimes the egg. Neuron 40, 427–446 (2003).
Hagai, T., Tóth-Petróczy, Á., Azia, A. & Levy, Y. The origins and evolution of ubiquitination sites. Mol. Biosyst. 8, 1865–1877 (2012).
Kim, D. S. & Hahn, Y. Gains of ubiquitylation sites in highly conserved proteins in the human lineage. BMC Bioinformatics 13, 306 (2012).
Nei, M. & Kumar, S. Molecular Evolution and Phylogenetics (Oxford University Press, 2000).
Wang, Z. et al. Evolution of protein phosphorylation for distinct functional modules in vertebrate genomes. Mol. Biol. Evol. 28, 1131–1140 (2011).
Nguyen, B. A. N. & Moses, A. M. Evolution of characterized phosphorylation sites in budding yeast. Mol Biol Evol. 27, 2027–2037 (2010).
Semple, C. A. M., RIKEN GER Group & GSL Members. The comparative proteomics of ubiquitination in mouse. Genome Res. 13, 1389–1394 (2003).
Ghalambor, C. K., Reznick, D. N. & Walker, J. A. Constraints on adaptive evolution: the functional trade-off between reproduction and fast-start swimming performance in the Trinidadian guppy (Poecilia reticulata). Am. Nat. 164, 38–50 (2004).
Rose, M. R. & Lauder, G. V. Adaptation (Academic Press, 1996).
Dawson, W. R., Bartholomew, G. A. & Bennett, A. F. A Reappraisal of the Aquatic Specializations of the Galapagos Marine Iguana (Amblyrhynchus cristatus). Evolution 31, 891–897 (1977).
Gould, S. J. & Lewontin, R. C. The Spandrels of San Marco and the Panglossian Paradigm: A Critique of the Adaptationist Programme. Proc. R. Soc. Lond. Ser. B 205, 581–598 (1979).
Smith, J. M. et al. Developmental Constraints and Evolution: A Perspective from the Mountain Lake Conference on Development and Evolution. Q. Rev. Biol. 60, 265–287 (1985).
Arnold, S. J. Constraints on phenotypic evolution. Am. Nat. 140, S85–S107 (1992).
Perrin, N. & Travis, J. On the use of constraints in evolutionary biology and some allergic reactions to them. Funct. Ecol. 6, 361–363 (1992).
Lande, R. Quantitative Genetic Analysis of Multivariate Evolution, Applied to Brain: Body Size Allometry. Evolution 33, 402–416 (1979).
Kanehisa, M. & Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 28, 27–30 (2000).
Ashburner, M. et al. Gene Ontology: tool for the unification of biology. Nat. Genet. 25, 25–29 (2000).
Creixell, P. & Linding, R. Cells, shared memory and breaking the PTM code. Mol. Syst. Biol. 8, 598 (2012).
Pál, C., Papp, B. & Lercher, M. J. An integrated view of protein evolution. Nat. Rev. Genetics. 7, 337–348 (2006).
Ravasi, T. et al. An atlas of combinatorial transcriptional regulation in mouse and man. Cell 140, 744–752 (2010).
Barabási, A.-L. & Oltvai, Z. N. Network biology: understanding the cell’s functional organization. Nat. Rev. Genet. 5, 101–113 (2004).
Gavin, A.-C. et al. Proteome survey reveals modularity of the yeast cell machinery. Nature 440, 631–636 (2006).
Jeong, H., Mason, S. P., Barabási, A.-L. & Oltvai, Z. N. Lethality and centrality in protein networks. Nature 411, 41–42 (2001).
Jansen, R. et al. A Bayesian Networks Approach for Predicting Protein-Protein Interactions from Genomic Data. Science 302, 449–453 (2003).
de Lichtenberg, U., Jensen, L. J., Brunak, S. & Bork, P. Dynamic complex formation during the yeast cell cycle. Science 307, 724–727 (2005).
Kelley, R. & Ideker, T. Systematic interpretation of genetic interactions using protein networks. Nat. Biotechnol. 23, 561–566 (2005).
Han, J.-D. J. et al. Evidence for dynamically organized modularity in the yeast protein–protein interaction network. Nature 430, 88–93 (2004).
Lee, I., Date, S. V., Adai, A. T. & Marcotte, E. M. A Probabilistic Functional Network of Yeast Genes. Science 306, 1555–1558 (2004).
Keshava Prasad, T. S. et al. Human Protein Reference Database—2009 update. Nucleic Acids Res. 37, D767–D772 (2009).
Kim, P. M., Lu, L. J., Xia, Y. & Gerstein, M. B. Relating three-dimensional structures to protein networks provides evolutionary insights. Science 314, 1938–1941 (2006).
Kimura, M. The Neutral Theory of Molecular Evolution (Cambridge University Press, 1984).
Festa, R. A. et al. Prokaryotic ubiquitin-like protein (Pup) proteome of Mycobacterium tuberculosis [corrected]. PloS One 5, e8589 (2010).
Deng, L. et al. The Ubiquitination of RagA GTPase by RNF152 Negatively Regulates mTORC1 Activation. Mol. Cell 58, 804–818 (2015).
Terai, K., Abbas, T., Jazaeri, A. A. & Dutta, A. CRL4Cdt2 E3 Ubiquitin Ligase Monoubiquitinates PCNA to Promote Translesion DNA Synthesis. Mol. Cell 37, 143–149 (2010).
Zhang, J. et al. ATM functions at the peroxisome to induce pexophagy in response to ROS. Nat. Cell Biol. 17, 1259–1269 (2015).
Lachance, V. et al. Ubiquitylation and activation of a Rab GTPase is promoted by a β2AR-HACE1 complex. J. Cell Sci. 127, 111–123 (2014).
Hornbeck, P. V. et al. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 43, D512–D520 (2015).
Consortium, T. U. UniProt: a hub for protein information. Nucleic Acids Res. gku989, doi: 10.1093/nar/gku989 (2014).
Sonnhammer, E. L. L. & Östlund, G. InParanoid 8: orthology analysis between 273 proteomes, mostly eukaryotic. Nucleic Acids Res. gku1203, doi: 10.1093/nar/gku1203 (2014).
Sievers, F. et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, n/a–n/a (2011).
Peng, K., Radivojac, P., Vucetic, S., Dunker, A. K. & Obradovic, Z. Length-dependent prediction of protein intrinsic disorder. BMC Bioinformatics 7, 1–17 (2006).
Li, D. et al. An integrated strategy for functional analysis in large-scale proteomic research by gene ontology. Prog. Biochem. Biophys. 32, 1026–1029 (2005).
Wilhelm, M. et al. Mass-spectrometry-based draft of the human proteome. Nature 509, 582–587 (2014).
Edwards, Y. J., Lobley, A. E., Pentony, M. M. & Jones, D. T. Insights into the regulation of intrinsically disordered proteins in the human proteome by analyzing sequence and gene expression data. Genome Biol. 10, 100–105 (2009).
Radivojac, P. et al. Identification, analysis, and prediction of protein ubiquitination sites. Proteins. 78, 365–380 (2010).
Gnad, F. et al. PHOSIDA (phosphorylation site database): management, structural and evolutionary investigation, and prediction of phosphosites. Genome Biol. 8, 561–570 (2007).
Federhen, S. The NCBI Taxonomy database. Nucleic Acids Res. 40, D136–D143 (2012).
Acknowledgements
We would like to thank Jun Qin, Xianming Wu, Jian Wang and Dong Yang for fruitful discussion. This work was supported by the Chinese Program of International S&T Cooperation [2014DFB30020], Chinese National Key Program of Basic Research [2012CB910300], the Program of Precision Medicine [2016YFC0901905], Innovation Project [16CXZ027], the grant of National Instrumentation Program [2013YQ19046707] and grant of Shenzhen Science & Technology Program [JCYJ20151029154245758].
Author information
Authors and Affiliations
Contributions
F.H., D.L. and J.X. provided guidance and revised the manuscript. L.L., Y.L. and D.L. designed the study. L.L. wrote the manuscript. Z.L., F.L., F.G., D.W., S.Y. and Y.H. participated in the study. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Lu, L., Li, Y., Liu, Z. et al. Functional constraints on adaptive evolution of protein ubiquitination sites. Sci Rep 7, 39949 (2017). https://doi.org/10.1038/srep39949
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep39949
- Springer Nature Limited