
Abstract
The white-backed planthopper, Sogatella furcifera (Horváth) (Hemiptera: Delphacidae), is a serious pest of rice in Asia. However, little is known regarding the migration of this pest insect from the Greater Mekong Subregion (GMS) including Cambodia, Laos, Myanmar (Burma), Thailand, and Vietnam, into China’s Yunnan Province. To determine the migration patterns of S. furcifera in the GMS and putative secondary immigration inside China’s Yunnan Province, we investigated the population genetic diversity, genetic structure, and gene flow of 42 S. furcifera populations across the six countries in the GMS by intensive sampling using mitochondrial genes. Our study revealed the potential emigration of S. furcifera from the GMS consists primarily of three major sources: 1) the S. furcifera from Laos and Vietnam migrate into south and southeast Yunnan, where they proceed to further migrate into northeast and central Yunnan; 2) the S. furcifera from Myanmar migrate into west Yunnan, and/or central Yunnan, and/or northeast Yunnan; 3) the S. furcifera from Cambodia migrate into southwest Yunnan, where the populations can migrate further into central Yunnan. The new data will not only be helpful in predicting population dynamics of the planthopper, but will also aid in regional control programs for this economically important pest insect.
Similar content being viewed by others
Introduction
The white-backed planthopper, Sogatella furcifera (Horváth) (Hemiptera: Delphacidae), is a serious pest of rice in Asia1,2. S. furcifera can damage rice directly by feeding on the rice or indirectly by transmitting plant viruses such as southern rice black-streaked dwarf virus (SRBSDV)3,4,5. The planthopper can also be responsible for causing sudden and unexpected devastation to local rice crops due to their ability to annually migrate long distances6. Therefore, knowledge of the migration pattern and routes taken by the pest is important both for the control of the virus vector and the vectored virus7. Many previous studies, based on trajectory analyses and migration simulations, indicated that the East Asian populations of S. furcifera overwinter in Vietnam and southern Hainan Province, and in spring migrate to eastern China, Japan, and Korea, then migrate back to their overwintering areas in autumn8,9. Very little is known about the migration of this pest insect, however, in the Greater Mekong Subregion (GMS) which includes Cambodia, Laos, Myanmar (Burma), Thailand, Vietnam, and China’s Yunnan Province7.
Sogatella furcifera is one the most destructive pests of rice production in the GMS. The migration information of S. furcifera in the region, especially the immigration source into China’s Yunnan Province, has mainly been derived from trajectory analyses and migration simulations. Previous studies based on weather during the rice growth period and using the HYSPLIT method showed that although a small number of S. furcifera do successfully overwinter in China’s Yunnan Province, the immigrants from the spring migration are the major source of this pest. The source of the April to early May migration into Yunnan were estimated to have originated mainly in Myanmar, while immigrations occurring in mid-May were thought to have come from northern Vietnam10,11,12. Until now, there haven’t been any molecular markers available for this planthopper to help determine the migration source and the migration routes7.
It is difficult to determine potential migration source of the insect using conventional approaches such as atmospheric current analysis, fluorescent marker dyes, and radar monitoring primarily due to the small size of the insects coupled with their relatively short lifespan13,14. Approaches involving population genetics such as the use of mitochondrial and nuclear microsatellite markers can provide essential tools for overcoming these problems15,16,17,18,19,20. Previous study has demonstrated the genetic differentiation in S. furcifera from five disjunct localities in Korea, the Philippines, China (only two populations), Malaysia, and Vietnam using mitochondrial sequences. Genetic diversity of S. furcifera from Yunnan Province and three Southeastern Asia countries (Vietnam, Laos, and Myanmar) was also demonstrated using the inter-simple sequence repeat (ISSR) technique6. Although very useful from a genetics standpoint, these studies have not, however, substantially contributed to clarifying global migration patterns of the pest in the GMS.
In the present study, we investigated the genetic structure of 42 S. furcifera populations from across the six countries in the GMS by intensive sampling using mitochondrial genes. The objective of this study was to reveal the migration pattern of S. furcifera in the GMS and putative secondary immigration in China’s Yunnan Province. These results will provide insight into migrations of S. furcifera in the GMS countries, and should also form a basis for sustainable management of this economically important pest insect in this region.
Results
Mitochondrial COI haplotype network and distribution
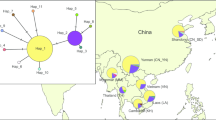
The haplotype network tree of mtCOI formed two major groups (Fig. 1), which obviously displayed a star-like pattern with the most common haplotypes in the star’s center. Each of the dominant haplotypes was present in all of the GMS countries. A total of 73 mtCOI haplotypes (abbreviated as H1-H73, respectively) (Table S1) were identified in this study, among which 37 unique haplotypes were found in China’s Yunnan Province, and 23 in the other GSM countries. Thirteen haplotypes were shared by different populations.

The haplotype network of the mitochondrial genes of COI.
*The haplotype network of mtCOI genes were inferred using the median-joining algorithm 31 and the software, Network v.4.6.1.0 (Fluxus Technology Ltd, England). The Star contraction method and “Frequency.1” criterion were used for the calculations. After the calculation, the MP calculation was used to identify and remove unnecessary median vectors and links41. The network’s results were drawn and prepared using the software, Network Publisher v.2.0.0.1 (Fluxus Technology Ltd, England). Colors within the nodes: red, China’s Yunnan Province; orange, Laos; yellow, Thailand; green, Cambodia; blue, Vietnam; indigo, Myanmar.
Among the 13 shared haplotypes, seven haplotypes were shared by different populations from China’s Yunnan and other GSM countries as follows: 1) Haplotypes H1 and H3 were the dominant shared haplotypes in every population, occupying 75–100% in each population; 2) H6 was shared by the populations from Laos (L2 and L5 populations), Vietnam (V3 population), and China’s Yunnan (XP, YUJ, JP, and FN populations); 3) H12 was shared by the populations from Vietnam (V3 population) and China’s Yunnan (JP population); 4) H57 was shared by the populations from Cambodia (C2 population), and China’s Yunnan (GM and BS populations); 5) H34 was shared by the populations from Myanmar (M1 population), and China’s Yunnan (MS, MD, and SJ populations); 6) H61 was shared by the populations from Myanmar (M4 population) and China’s Yunnan (GM population).
Among the other six shared haplotypes, (except H73 which was only found in the M1 population from Myanmar), the remaining five haplotypes were shared only by populations within China’s Yunnan Province: 1) H30 was shared by Yunnan’s MS and SM populations; 2) H20 was shared by the province’s SJ and XP populations; 3) H5 was shared by the YUJ and YS populations; 4) H53 was shared by the JP and FN populations; and 5) H28 was shared by the ZY and KY populations.
Mitochondrial COI diversity and genetic differentiation
Genetic diversity analysis based on mtCOI varied among different populations (Table 1). For example, the Hd of the populations from China’s Yunnan Province ranged from 0.297 to 0.725 while those from the other countries ranged from 0.173 to 0.830. The average Hd value from China’s Yunnan (0.5043) had no significant difference with those from the neighboring countries (0.4609) (P > 0.05). Thirty-one of the 38 populations had negative Fu’s F and Tajima’s D indices, suggesting a recent post-bottleneck population expansion21,22.
The pairwise Fst difference based on mitochondrial genes showed significant differentiation in 74 of the 861 population pairs (Table 2). Several populations, such as YS, FN, MD, MH, C2, C3, and M1, had more Fst values that were significantly different than other populations. The Mantel test results produced an r value of −0.0238 for mitochondrial genes (P = 0.3470) (Fig. 2), indicating that no correlation was found between genetic distance and geographical distance among the populations of S. furcifera in the GMS countries.

Relationship between genetic distance and log of geographical distance for pairwise population comparisons.
*The line represents the regression line and circles represent the logarithm transformation of distance.
Gene flow based on mitochondrial data
Analysis between each pair of the 42 populations showed the presence of high gene flow among different populations. Unidirectional estimates of M ranged from 28.6 (from M2 to T1) to 950.9 (from L4 to CX) (Table S2). When the M values were translated into effective migrants per generation (Nem) (Table 3), a high numbers of total migrants (Nem > 1000) in the GSM (excluding China’s Yunnan Province) were found in several populations, including L5 of Laos, C1-C3 of Cambodia, and V2 in Vietnam. The numbers of total migrants in Myanmar and other GSM localities, however, were relatively low.
In China’s Yunnan Province, a relatively high numbers of migrants (Nem > 1300) were found in a number of populations, including those from south Yunnan (e.g., JP), southwest Yunnan (e.g., MH), southeast Yunnan (e.g., YS), and central Yunnan (e.g., CX and YUJ), although, the numbers in some of the populations from these areas in Yunnan were very low, including KY in south Yunnan (Nem = 0.4), FN in southeast Yunnan (Nem = 0.7), GM in southeast Yunnan (Nem = 0.7), and SM in central Yunnan (Nem = 1.3). A number of the populations from western and northeastern Yunnan were also relatively low. This was especially true in the majority of those from western Yunnan where the numbers were quite low (Nem < 1.8) except in the MS population (Nem = 320.3). Interestingly, the average value for number of migrants into south Yunnan was the highest of all areas while that of west Yunnan was the lowest among all of the Yunnan populations.
Discussion
Evidence for intensive gene flow of S. furcifera in the GMS
The mtCOI haplotype network showed that the haplotypes H1 and H3 were widely distributed throughout the GMS, indicating the occurrence of extensive gene flow. The low ratio of population pairs having significant differentiation reinforces the postulation. In addition, the negative Fu’s F and Tajima’s D indices of most populations also demonstrated the occurrence of an extensive population expansion. These results are consistent with previous studies using trajectory analyses and migration simulations that the S. furcifera from the GMS countries can immigrate into China’s Yunnan Province7,8,9. Furthermore, the Mantel test results confirming that none of the populations of S. furcifera in the GMS are geographically isolated were also highly consistent with the postulated intensive gene flow.
However, the genetic diversity among different populations was fairly distinct and a number of migrants were also obviously different (Tables 1 and 3), suggesting that the amount of gene flow between different population varied. For example, the number of total migrants in the L5 population of Laos was higher than in the other populations from Laos.
Potential migration sources of S. furcifera in the GMS
Based on shared mtCOI haplotypes, a close association was found to exist between: 1) the S. furcifera populations from southeast Yunnan, south Yunnan, Laos, and Vietnam; 2) the S. furcifera populations from Myanmar, west Yunnan, central Yunnan, and northeast Yunnan; 3) and the S. furcifera populations from Cambodia and southwest Yunnan. According to the seasonal weather data, we can infer that potential immigrations into Yunnan, China may occur in at least three main potential sources: 1) a source from Laos and Vietnam into adjacent southeastern and southern Yunnan; 2) a source from Myanmar into the adjacent western Yunnan, with subsequent migrations into central and northeastern Yunnan; 3) a source from Cambodia into southwestern Yunnan. For the potential source populations from Myanmar or Cambodia, they may be introduced into China’s Yunnan indirectly because of the long geographical distance, which should be further explored in the future researches.
In Yunnan Province, the haplotype composition of the populations in central Yunnan is closely linked with that from southeast Yunnan, west Yunnan, and southwest Yunnan since the H20, H30, and H5 haplotypes discovered in central Yunnan are also found in the latter three regions. When combined with the seasonal weather patterns, the populations in central Yunnan may have several sources such as from the neighboring regions. The haplotypes of the populations in south Yunnan are also closely associated with that found in northeast Yunnan, suggesting a potential immigration source from south Yunnan into northeast Yunnan.
Based on the combined data mentioned above, the potential emigration of S. furcifera from the GMS consists primarily of three major sources: 1) the S. furcifera from Laos and Vietnam migrate into south and southeast Yunnan, where they proceed to further migrate into northeast and central Yunnan; 2) the S. furcifera from Myanmar migrate into west Yunnan, and/or central Yunnan, and/or northeast Yunnan; 3) the S. furcifera from Cambodia migrate into southwest Yunnan, where the populations can migrate further into central Yunnan.
These postulated multiple sources of the S. furcifera populations in Yunnan, China are consistent with previous studies. Based on their analysis using the HYSPLIT model, Shen et al.9 concluded that the main source areas of the early migration of S. furcifera into Yunnan, China in 2009 were located in Myanmar, and that secondary source areas were in Laos and Vietnam, with a few coming from the Imphal area in Manipur, India. The frequent westerly winds coupled with the tremendous increase in rice production in the Myanmar source areas were considered to be the principal reasons causing the mass migration of S. furcifera into Yunnan Province9. Jiang et al.10 found similar results confirming that the main source areas of the early migration of S. furcifera in May were located in the middle of Myanmar, and secondarily in northern Thailand and Vietnam. The westerly and southwesterly wind carried massive numbers of S. furcifera into Funing (FN), a site in southeastern Yunnan. Wind shear combined with the occurrence of threshold flying temperatures associated with rainfall, caused a mass descent of S. furcifera. An analysis by Zheng et al.12 also pointed out that the source areas of an early April migration into Shizong (SZ), a site in northeastern Yunnan were mainly located in northeastern Myanmar and that secondary source areas were in northern Vietnam and the Golden Triangle area of GMS.
Our study not only reveals the potential migratory sources from Myanmar, Vietnam, and Laos, but also adds Cambodia as another source of S. furcifera migrants. This data will be helpful in predicting the population dynamics of this economically important planthopper, and will aid in regional control of this major pest insect.
Future research on the S. furcifera in the GMS
The genetic diversity of species is closely associated with their ecological adaptations, which have been explored in many species especially in invasive alien species19,23,24. In this study, unique mtCOI haplotypes have been identified in many populations from the GMS, which indicates that the adaptation of these haplotypes are a response to the local environment including the unique climate, host plants, and agricultural activities experienced by the population from that particular region. The widespread mtCOI haplotypes may have robust adaptive abilities to the diverse ecological factors. On the other hand, the genetic diversity based on mitochondrial DNA may be inconsistent with that based on nuclear DNA. For example, a relatively high nuclear genetic diversity was revealed in introduced populations of the invasive species, Bemisia tabaci biotype Q in Shandong, China, while the mitochondrial genetic diversity was considerably lower23, suggesting that mitochondrial DNA may not be indicative of the level of diversity in the nuclear DNA. Therefore, more attention should be focused on the genetic diversity in nuclear DNA, which will help us further understand the relationship between genetic variation and ecological adaptation.
Although, based on mtCOI data, it is apparent that migrations from and within the GMS consist of many sources, detailed migration patterns may be more complex than expected due to variations of yearly ecological factors occurring in this region. In addition, recent studies have revealed the differences resulting from using mitochondrial and nuclear markers18,23,25,26. The combining molecular markers of distinct modes of inheritance, such as the combination of the mitochondrial and nuclear markers, can provide extra, complementary information on gene flow27,28. Further studies, including application of the nuclear markers, may lead to more effective research into migration pattern and population dynamics in various geographic regions, which will be essential in developing sustainable pest management strategies.
Conclusions
The genetic diversity and structures found in the S. furcifera populations analyzed in this study enabled us to infer the planthopper’s migration sources. Based on our results, we can speculate that the potential migration of S. furcifera from the GMS consisted primarily of three major sources: 1) the S. furcifera from Laos and Vietnam migrate into southern and southeastern Yunnan. These populations can later migrate into northeast and central Yunnan; 2) the S. furcifera originating from Myanmar migrate into western Yunnan, and/or central Yunnan, and/or northeastern Yunnan; 3) the S. furcifera from Cambodia migrates into southeastern Yunnan, where the populations can further migrate into central Yunnan. Our study not only reaffirmed the detailed potential migration source from Myanmar, Vietnam, and Laos, but also demonstrated the migration of S. furcifera from Cambodia. The added data will be helpful in predicting the population dynamics of this pest insect, which, in turn will be useful in regional control programs for the planthopper.
Materials and Methods
Field sampling and DNA extraction
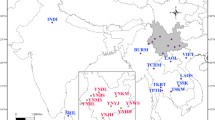
Adult S. furcifera samples were collected from 42 locations in the GMS during 2014–2015 (Fig. 3; Table 4). The samples included 20 populations from China’s Yunnan Province, four from Vietnam, eight from Laos, two from Thailand, four from Cambodia, and four from Myanmar. The specimens were fixed in 95% ethanol and stored at −20 °C until DNA was extracted. Genomic DNA was individually extracted from each adult planthopper using the DNAzol kit (Molecular Research Center, Inc., Cincinnati, OH) and stored at −20 °C.

Geographical distribution of Sogatella furcifera populations in the Greater Mekong Subregion (GMS).
*The map of Southeast Asia’s mainland was modified from the map generated using GeoMapApp (version 2) (http://www.geomapapp.org/).
Mitochondrial COI amplification and sequencing
The cytochrome coxidase subunit I gene of the mitochondrial DNA (mtCOI) was used to determine the genetically distinct populations of S. furcifera. All individual DNA samples were first amplified using the mtCOI primers CI-J-2183 (5′-CAACATTTATTTTGATTTTTTGG-3′) and L2-N-3014 (5′-TCCAATGCACTAATC TGCCATATTA-3′) and then sequenced29. The PCR reactions were performed in 20 μL buffer containing 2 μL 10× buffer, 1.5 mM MgCl2, 0.2 μM dNTPs, 1 unit Taq DNA polymerase, 2 μL template DNA, and 0.2 μM of each primer. PCR amplification was carried out as follows: initial denaturation at 94 °C for 5 min, followed by 35 cycles each of 30 s at 94 °C, 30 s at 50 °C, and 60 s at 72 °C, and a final elongation step at 72 °C for 30 min.
Mitochondrial COI haplotype analysis
These sequences of mtCOI were aligned with Clustal W30 and were then checked for indels and numts. The haplotype network of mtCOI genes was inferred using the median-joining algorithm31. All calculations were conducted using the software program Network v.4.6.1.0 (Fluxus Technology Ltd., England).
The genetic diversity indices of each population which were analyzed based on mtCOI using DnaSP v.5.032 included the number of polymorphic (segregating) sites (S), the total number of mutations (η)33, the average number of nucleotide differences (K)34, the number of haplotypes (H), the haplotype diversity (Hd)35, the nucleotide diversity (π)35, the nucleotide diversity with Jukes and Cantor correction (π (JC))36, and the number of net nucleotide substitutions per site between populations with Jukes and Cantor correction, Da (JC)35. Tajima’s D (D)21 and Fu ‘s F test22 were performed to detect deviation from neutrality.
The Weir and Cockerham’s fixation index Fst37, the traditional population differentiation approach, was calculated using ARLEQUIN v.3.5 software38. The correlation between genetic differentiation and geographic distance was examined using the Mantel test with IBDWS v.3.15 software39.
Gene flow analysis based on mitochondrial COI data
The dispersal of different S. furcifera populations in the GMS, was determined by calculating the effective numbers of migrants per generation Nem using mitochondrial COI data. Nem is ϴM (ϴ = Neμ, where μ is the mutation rate per site per generation; M = m/μ, where m is the migration rate) calculated using Bayesian search strategies in MIGRATE v. 3.2.16 software40.
Additional Information
How to cite this article: Li, X.-y. et al. Possible Source Populations of the White-backed Planthopper in the Greater Mekong Subregion Revealed by Mitochondrial DNA Analysis. Sci. Rep. 6, 39167; doi: 10.1038/srep39167 (2016).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Khan, Z. R. & Saxena, R. C. Electronically recorded waveforms associated with the feeding behavior of Sogatella furcifera (Homoptera: Delphacidae) on susceptible and resistant rice varieties. J. Econ. Entomol. 77(6), 1479–1482 (1984).
Xiao, T. & Tang, J. Effects of the susceptibility of rice varieties to Sogatella furcifera on nymphal development and reproduction of Microvelia Horváthi through a food chain. Insect. Sci. 14(4), 317–321 (2007).
Zhou, G. H. et al. Southern rice black-streaked dwarf virus: a new proposed Fijivirus species in the family Reoviridae. Chin. Sci. Bull. 53, 3677–3685 (2008).
Zhou, G. H. et al. Occurrence and damage analysis of a new rice dwarf disease caused by Southern rice black-streaked dwarf virus. Plant Prot. 36, 144–146 (2010).
Zhou, G. H. et al. Southern rice black-streaked dwarf virus: a white-backed planthopper transmitted fijivirus threatening rice production in Asia. Front. Microbiol. 4, 270 (2013).
Liu, J. N., Gui, F. R. & Li, Z. Y. Genetic diversity of the planthopper, Sogatella furcifera in the Greater Mekong Subregion detected by inter-simple sequence repeats (ISSR) markers. J. Insect. Sci. 10(52), 239–287 (2010).
Otuka, A. Migration of rice planthoppers and their vectored re-emerging and novel rice viruses in East Asia. Frontiers in Microbiology. 4(309), 1–11 (2013).
Shen, H. M. & Chen Analysis on the early immigration of rice planthoppers in southern Guangxi. Chin. J. Appl. Entomol. 48, 1268–1277 (in Chinese) (2011a).
Shen, H. M. et al. Analysis of the source areas of the early immigration of the white-backed planthopper, Sogatella furcifera (Horváth) (Homoptera: Delphacidae) in Fujian Province, China. Acta Entomologica Sinica (in Chinese) (2011b).
Shen, H. et al. Source areas and landing mechanism of early immigration of white-backed planthoppers Sogatella furcifera (Horváth) in Yunnan 2009. Acta Ecologica Sinica (in Chinese) (2011c).
Jiang, C. X. et al. Study on source area and landing mechanism of early immigration of white-backed planthopper (Sogatella furcifera (Horváth)) in Funning, Yunnan. J. Sichuan Agric. Uni. 30(2), 232–237 (in Chinese) (2012).
Zheng, D. B. et al. Source areas and landing mechanisms of early immigrant population of white-backed planthoppers Sogatella furcifera (Horváth) in Shizong, Yunnan Province. Acta Ecol.Sin. 34(15), 4262–4271 (in Chinese) (2014).
Chapman, J. W., Drake, V. A. & Reynolds, D. R. Recent insights from radar studies of insect flight. Ann. Rev. Entomol. 56(1), 337–356 (2011).
Wei, S. J. et al. Genetic structure and demographic history reveal migration of the diamondback moth Plutella xylostella (Lepidoptera: Plutellidae) from the southern to northern regions of China. PLoS ONE 8(4), e59654- e59654 (2013).
Porretta, D. et al. Improving insect pest management through population genetic data: a case study of the mosquito Ochlerotatus caspius (Pallas). J. Appl. Ecol. 44(3), 682–691 (2007).
Chuan, M. A. et al. Mitochondrial genomes reveal the global phylogeography and dispersal routes of the migratory locust. Mol. Ecol. 21(17), 4344–4358 (2012).
Duan, H. S. et al. Sudden widespread distribution of western flower thrips in Shandong Province, China. Fla. Entomol. 96(3), 933–940 (2013a).
Duan, H. S. et al. Genetic diversity and inferences on potential source areas for the introduced western flower thrips, Frankliniella occidentalis in Shandong, China based on mitochondrial and microsatellite markers. Fla. Entomol. 96(3), 963–972 (2013b).
Chu, D. et al. Evidence for rapid spatiotemporal changes in genetic structure of an alien whitefly during initial invasion. Sci. Rep. 4(11), 4396 (2013).
Zhou, H. X. et al. Invasion genetics of woolly apple aphid (Hemiptera: Aphididae) in China. J. Econ. Entomol. 108(3), 1040–1046 (2015).
Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genet. 123(3), 585–595 (1989).
Fu, Y. X. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genet. 147(2), 915–925 (1997).
Chu, D. et al. Investigation of the genetic diversity of an invasive whitefly in China using both mitochondrial and nuclear DNA markers. Bull. Entomol. Res. 101(4), 467–475 (2011).
Wang, J. et al. Peach fruit moth, Carposina sasakii (Lepidoptera: Carposinidae) in its native range consists of two sympatric cryptic lineages as revealed by mitochondrial COI gene sequences. J. Insect. Sci. 15(1) (2015).
Zhang, R. M. et al. Two putative bridgehead populations of Aphelinus mali (Hymenoptera: Aphelinidae) introduced in China. Fla. Entomol. 97(2), 401–405 (2014).
Zhou, H. X. et al. Analysis of genetic diversity and structure of two clades of Aphelinus mali (Hymenoptera: Aphelinidae) in China. Fla. Entomol. 97(2), 699–706 (2014).
Torres-Leguizamon, M., Mathieu, J., Decaens, T. & Dupont, L. Genetic structure of earthworm populations at a regional scale: inferences from mitochondrial and microsatellite molecular markers in Aporrectodea icterica (Savigny 1826). PLoS ONE 9(9), e101597–e101597 (2014).
Wei, S. J. et al. Population genetic structure and approximate Bayesian computation analyses reveal the southern origin and northward dispersal of the oriental fruit moth Grapholita molesta (Lepidoptera: Tortricidae) in its native range. Mol. Ecol. 24(16), 4094–4111 (2015).
Simon, C. et al. Evolution, weighting, and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers. Ann. Entomol. Soc. Amer. 87(6), 651–701 (1994).
Thompson, J. D., Higgins, D. G. & Gibson, T. J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position specific gap penalties, and weight matrix choice. Nucleic. Acids. Res. 22 (1994).
Bandelt, H. J., Forster, P. & Röhl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 16(1), 37–48 (1999).
Rozas, J., Sanchez-Delbarrio, J. C., Peypoch, X. M. & Rozas, R. Dnasp, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 19(18), 2496–2497 (2003).
Watterson, G. A. On the number of segregating sites in genetical models without recombination. Int. J. Publ. Opin. Res. 7(2), 256–276 (1975).
Tajima, F. Evolutionary relationship of DNA sequences in finite populations. Genet. 105(2), 437–460 (1983).
Nei, M. Molecular Evolutionary Genetics. (Columbia Univ. Press, New York, 1987).
Lynch, M. & Crease, T. J. The analysis of population survey data on DNA sequence variation. Mol. Biol. Evol. 7, 377–394 (1990).
Weir, B. S. & Cockerham, C. C. Estimating F-statistics for the analysis of population structure. Evolution 38(6), 1358–1370 (1984).
Excoffier, L. & Lischer, H. E. L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Res. 10(3), 564–567 (2010).
Jensen, J. L., Bohonak, A. J. & Kelley, S. T. Isolation by distance web service. BMC. Genet. 6, doi: 10.1186/1471-2156-6-13 (2005).
Beerli, P. & Felsenstein, J. Maximum likelihood estimation of a migration matrix and effective population sizes in n subpopulations by using a coalescent approach. Proc. Natl. Acad. Sci. USA 98(8), 4563–4568 (2001).
Polzin, T. & Daneshmand, S. V. On Steiner trees and minimum spanning trees in hypergraphs. Oper. Res. Lett. 31(1), 12–20 (2010).
Acknowledgements
We are very grateful to the people for kind helps during the sample population: Prof. Tao Dayun (International Cooperation Division of Yunnan Academy of Agricultural Sciences, China), Prof. Li Lu (Institute of Food Crops of Yunnan Academy of Agricultural Sciences, China), Prof. Yang Qinzhong and Prof. Zhang Fudou (Agriculture Environment and Resources Institute of Yunnan Academy of Agricultural Sciences, China). This research was supported by the Yunnan Joint Funds of the National Natural Science Foundation of China (No. U1202266), the Yunnan Province Science and Technology Plan (No. 2014IA009), and the Yunnan Rice Industry Technology System to A. Chen, and the Shandong Modern Agricultural Technology & Industry System (SDAIT-17-07) to D. Chu. The funders had no role in study design, data population and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: A.C., D.C. Performed the experiments: X.L., Y.Y. X.Z., A.C., D.C. Analyzed the data: A.C., D.C. Contributed reagents/materials/analysis tools: A.C. Wrote the paper: D.C., A.C. Contributed sample collection: S.K., D.B., K.M., K.M., V.N., C.N.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Li, Xy., Chu, D., Yin, Yq. et al. Possible Source Populations of the White-backed Planthopper in the Greater Mekong Subregion Revealed by Mitochondrial DNA Analysis. Sci Rep 6, 39167 (2016). https://doi.org/10.1038/srep39167
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep39167
- Springer Nature Limited
This article is cited by
-
Migration of Sogatella furcifera between the Greater Mekong Subregion and northern China revealed by mtDNA and SNP
BMC Evolutionary Biology (2020)
-
Extensive gene flow of white-backed planthopper in the Greater Mekong Subregion as revealed by microsatellite markers
Scientific Reports (2017)