
Abstract
As highly prized, popular mushrooms, morels are widely distributed in the northern hemisphere, with China as a modern centre of speciation and diversity. Overharvesting of morels has caused concern over how to effectively preserve their biological and genetic diversity. However, little is known about their population biology and life cycle. In this study, we selected two sympatric phylogenetic species, Mel-13 (124 collections from 11 geographical locations) and Morchella eohespera (156 collections from 14 geographical locations), using fragments of 4 DNA sequences, to analyse their genetic structure. Our results indicated significant differentiation among geographic locations in both species, whereas no obvious correlation between genetic and geographic distance was identified in either species. M. eohespera exhibited a predominantly clonal population structure with limited recombination detected in only 1 of the 14 geographic locations. In contrast, relatively frequent recombination was identified in 6 of the 11 geographic locations of Mel-13. Our analysis indicated that the sympatric species Mel-13 and M. eohespera might have divergent evolutionary patterns, with the former showing signatures of recent population expansion and the latter being relatively stable. Interestingly, we found no heterozygosity but strong evidence for genealogical incongruence, indicating a high level of inbreeding and hybridisation among morel species.
Similar content being viewed by others

Introduction
Morels are among the world’s most prized edible fungi, and are collected by both fungal enthusiasts and food specialists due to their unique culinary flavour and rarity. Although cultivation of Morchella rufobrunnea1 and M. importuna2 has been achieved separately in the USA and China, morels collected from the wild still dominate the markets. To meet the demand created by their growing popularity, wild morels are heavily harvested and traded extensively in China, India, Turkey, Mexico and the USA3. As with many other wild gourmet mushrooms, the loss and fragmentation of habitat and exploitative over-harvesting have created significant challenges for the management of natural populations3,4. A better understanding of their population biology is needed to facilitate the development of conservation strategies to maintain wild populations of morels.
Fungal mating systems play important roles in shaping the genetic structure of fungal populations5,6,7,8,9,10, yet little is known about the life cycle and reproductive systems of morels. Such knowledge is critical to understanding their population dynamics and the effects of habitat fragmentation and harvesting practices on their genetic structure. Determining the patterns of genetic variation using molecular markers will provide information on the population genetics of morels, which can also be used to infer their life cycle and mode of reproduction. Several studies have indicated that species in the Elata and Esculenta clades of Morchella might be heterothallic and could outcross in nature11,12,13,14,15. However, the reproductive modes of Morchella species are still not clearly known, and it is unknown whether inbreeding or clonality exists in the life cycle of morels. Dalgleish and Jacobson11 and Pagliaccia et al.14 emphasised the importance of further research to resolve important aspects of the morel life cycle regarding heterokaryosis and inbreeding potential. To address these issues, more extensive sampling and the use of highly polymorphic co-dominant molecular markers are needed to detect genetic variation within and between populations and to infer critical issues regarding the mating system of morels.
China is known for its complex geological and ecological diversity. Our recent study identified China as the modern species diversity centre of Morchella16. Interestingly, several closely related species are sympatric (unpublished data). The high species diversity and sympatric species distribution pattern make China an ideal region in which to analyse and compare the genetic diversity, patterns of genetic variation and modes of reproduction of morels. In this study, we analysed and compared the genetic structure of two sympatric morel species (Mel-13 Morchella sp. proved as the valid phylogenetic species16 and M. eohespera) from China using multi-locus sequence data. We aimed to address the following questions. (1) What is the spatial genetic structure, the level of genetic diversity within and among populations and the degree of population differentiation within each of the two sympatric species? (2) Are their evolutionary histories similar to or divergent from each other? (3) What are their potential modes of reproduction? Are their reproductive modes dominated by clonality, recombination or a mixture of both? Our findings will help in understanding the reproductive modes and the distributional patterns of genetic variability in natural populations of morels, which is essential for developing effective strategies for the management and conservation of these two morel species.
Results
Sequence variation within four sequenced fragments of Mel-13 and M. eohespera
We successfully obtained all of the sequences from four DNA fragments for 280 samples (124 samples of Mel-13 and 156 samples of M. eohespera). No heterozygosity was observed for any site in the four genetic makers of both species.
For Mel-13, the observed polymorphic sites (percentage)/total aligned nucleotide sites for B1, F1, F2 and ITS rDNA were 1 (0.16%)/638, 6 (0.85%)/706, 4 (0.59%)/673 and 11 (0.17%)/666, respectively, separating the 124 collections into 2, 8, 4 and 7 sequence types or haplotypes, respectively. For M. eohespera, the observed polymorphic sites (percentage)/total aligned nucleotide sites for B1, F1, F2 and ITS were 5 (0.78%)/638, 8 (1.24%)/644, 10 (1.5%)/670 and 16 (2.3%)/690, respectively, separating the 124 collections into 5, 6, 8 and 15 sequence types or haplotypes, respectively.
Population Structure
Mel-13
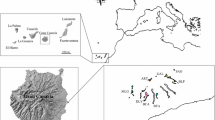
Twenty-two variable sites were found among the 2682 aligned nucleotide sites for the combined four-gene dataset, the variable site proportion of which was 0.82%. Of the 29 haplotypes detected in this species (Table 1), 23 (79.3%) were private haplotypes, each found in only one population. Most of these private haplotypes were represented by one sample each. The remaining six haplotypes were shared by collections from two or more geographic locations. Haplotypes H7, H14, H22 and H23 were widely distributed. The SXFS population had the highest haploid diversity and nucleotide diversity. Interestingly, none of the four common haplotypes (H7, H14, H22 and H23) were found in this population. The median joining network showed that the remaining 25 haplotypes were linked to these 4 main haplotypes in a ‘star-like’ network. Except for the QHZM population, which had only one haplotype (H23), each population had at least two haplotypes (Fig. 1).

(a) Geographic location of the 14 populations of Mel-13 analyzed in the present study and the distribution of the 29 haplotypes detected (see Table 1 for population codes). Pie chart size corresponds to the sample size of each population. (b) Median joining phylogenetic networks among haplotypes of Mel-13. Maps were generated using ArcView GIS 3.2 (ESRI, Redlands, CA, USA) to estimate the distance between genets and locations.
The molecular genetic diversity indices, Hd and π, for each population are summarised in Table 1. The highest haplotype diversity (Hd = 0.889) was detected in the XJZS and SXFS populations, with nucleotide diversity (π) of 0.00083 and 0.0015, respectively. The QHZM population had the lowest Hd (0), with only one haplotype (H23).
Nei’s genetic distance between pairwise members of the population ranged from 0.009 (between QHBB and QHZG) to 0.663 (between SCJZ and SCMEK) (Table S1). The estimated mean gene flow, Nm, among populations was 0.53. Based on a MultiLocus analysis, the 11 geographic locations showed significant genetic differentiation with a value of 0.4877, significantly larger than the randomised datasets (p < 0.001).
The SAMOVA analysis showed that the ΦCT value was highest when K = 9. However, as it made little difference when the 11 populations were defined as 9 groups, we do not define the groups in Mel-13 here.
The AMOVA revealed that 49% of genetic variation was within populations and 51% between populations (Table 2), and the results were statistically significant. Mantel tests (Figure S1) were conducted to determine whether the observed genetic differentiation was related to geographical and altitudinal differences. A positive correlation was found for geographical distance (p = 0.2) and a negative correlation for altitude (p = 0.19), although the correlations were not statistically significant.
The unimodal graphs illustrating the mismatch distribution analyses (Figure S3) for Mel-13 indicated a sudden population expansion, inferred by the near-perfect fit between the observed and the expected mismatch distribution, and supported by non-significant SSD and HRI statistics (Table 3). The star-like pattern exhibited by the neighbouring joining network (Fig. 1) was also characteristic of recent population expansion. However, the neutrality tests produced insignificant positive values for Fu’s FS and Tajima’s D statistics, as expected for stationary populations (Table 3).
M. eohespera
Among the 2642 aligned nucleotide sites in the combined four-gene dataset, 39 variable sites were found, thus the proportion of variable sites was 1.48%. Of the 33 haplotypes detected in this species, 17 (51.5%) were private haplotypes, each found in only one population (Table 1). H22 was the most abundant haplotype, distributed in 12 of the 14 geographic locations. XZRW harboured the highest number of haplotypes among all of the populations, and also had the richest and most widely distributed haplotype, H22. All populations had two or more haplotypes each. The median-joining haplotype network of these 33 haplotypes is shown in Fig. 2.

(a) Geographic location of the 14 populations of M. eohespera analyzed in the present study and the distribution of the 33 haplotypes detected (see Table 1 for population codes). Pie chart size corresponds to the sample size of each population. (b) Median joining phylogenetic networks among haplotypes of M. eohespera. Maps were generated using ArcView GIS 3.2 (ESRI, Redlands, CA, USA) to estimate the distance between genets and locations.
The molecular genetic diversity indices, Hd and π, for each population are summarised in Table 1. The XZLL population had the highest haplotype diversity (Hd = 0.9) and XZRW had the highest number of haplotypes (n = 10, Hd = 0.867). The XJWLMQ and GSZQ populations had the highest nucleotide diversity (π, 0.00318) and the lowest Hd (0.417), respectively.
Nei’s genetic distance between pairwise members of the population ranged from 0.001 (between GSDB and GSZQ) to 0.254 (between QHBB and XZRW) (Table S2). The average Nei’s genetic distance among populations was 0.0552. The estimated mean gene flow, Nm, among populations was 0.27. A MultiLocus analysis of the 14 populations showed significant genetic differentiation with a value of 0.3675, significantly larger than the randomised datasets (p < 0.001).
In the SAMOVA analysis with K = 2, two groups were well defined and the ΦCT value was the highest, corresponding to the XZRW population as one group and the remaining populations as the other. Based on the groups defined by SAMOVA, the AMOVA analysis revealed that 35% of genetic variation was within populations, 7% between populations and 58% between groups (Table 2), all of which were statistically significant. Mantel tests were conducted to determine whether the observed genetic differentiation was related to their geographical and altitudinal differences. Negative correlations were found for both geographical distance (p = 0.25) and altitude (p = 0.52), but they were not statistically significant (Figure S2).
The multimodal graphs from the mismatch distribution analyses (Figure S4) for M. eohespera indicated stationary populations, and this was supported by the non-significant positive value of Fu’s FS and Tajima’s D generated in the neutrality tests. However, the SSD and HRI statistics were non-significant, which is unexpected for stationary populations (Table 3).
Evidence for clonality and recombination
We performed multilocus linkage disequilibrium statistical tests, including the IA and phylogenetic incompatibility tests, on our data using two methods. First, to detect whether recombination existed within each gene fragment, we conducted these tests respectively in B2, F1, F2 and ITS rDNA fragments for the total samples of Mel-13 and M. eohespera. Each variable nucleotide site was treated as a locus and different nucleotides at the same site were viewed as different alleles. The results (Table 4) showed no recombination in the F2 and ITS DNA fragments in Mel-13, except for the B2 gene fragment, which was excluded because only one polymorphic site was detected. However, the phylogenetic incompatibility test identified evidence of recombination within the F1 gene fragment, which was indicative of limited recombination. In M. eohespera, no recombination was detected in the B2, F1 and F2 gene fragments, but limited recombination was found in the ITS region according to the rBarD value, which was not significantly different from the null hypothesis.
Second, it is easy to bring the linkage disequilibrium within a gene fragment to that between DNA fragments when using nucleotide variation within and among genes. Thus, to examine the associations between the alleles of the four genes, we defined four DNA fragments as four loci and used the allelic data from each locus to analyse the relationships among the alleles from different loci. Based on this revised data, multilocus linkage disequilibrium statistical tests were conducted for two sample types: the total samples and samples in each population (Table 5).
For the total samples in both Mel-13 and M. eohespera, our analyses revealed that the rBarD values were significantly different from the null hypothesis of random recombination, but the phylogenetic incompatibility test identified limited evidence of recombination. These results indicate that limited recombination partly contributed to differentiation in both species.
Of the 11 populations within Mel-13, the rBarD and PrC values showed significant evidence of recombination in the SXFS population; five populations (XJZS, XJWLMQ, SXYA, SCJZ and YNLJS) were indicative of limited recombination by the PrC values; and the other five (QHZM, QHMH, QHBB, QHZG and SCMEK, accounting for 45.5%) showed no evidence of recombination. Of the fourteen populations in M. eohespera, the PrC value indicated limited recombination only in the XZRW population. Thus, Mel-13 showed more evidence of recombination than M. eohespera.
Hybridisation or horizontal gene transfer and sympatric distribution
During the preliminary screening of the 496 samples collected in China using sequences from the four gene fragments, three of the markers showed largely consistent gene genealogical patterns, but that of the F1 fragment was different. Among the 496 strains, 7 showed evidence of hybridisation or horizontal gene transfer: D129, D165, D390, D502, D518, D546 and D554 (Fig. 3). According to the individual phylogenetic analysis of ITS, B2 and F2, D129 and D165 were nested within M. importuna, D502 and D546 nested within Mel-13, D518 nested within M. eximioides, D390 and D554 nested within M. eohespera. However, the results based on the F1 gene fragment showed that D129 was nested within M. eximia, D390 and D554 were nested within Mel-13, D165 and D502 were nested within M. eohespera and D518 and D546 were nested within Mel-34 (Fig. 3; ITS and 43 collections were used as the representatives). To confirm that these collections did have the unusual sequences, we re-extracted their DNA and sequenced the four loci again. All sequences were found to be the same as those obtained in the initial sequencing. Our results suggest that the special alleles in F1 among these six species were probably due to recent hybridisation or horizontal gene transfer. Analyses of additional markers should help to reveal the extent of hybridisation among the lineages. Due to the conflicting findings for the F1 DNA fragments and the other potential hybridisation individuals, they were not included in the population studies.

The tree for the left and the right correspond to ITS and F1, respectively. These seven collections probably experienced hybridization or horizontal gene transfer, which were indicated by seven different symbols shown on the trees, respectively.
Sympatric distribution was also found to be common in Morchella. For example, the 19 samples collected from the SCMEK population (found on a small mountain) belonged to three species, Mel-13 (n = 10), Mel-14 (n = 7) and M. eohespera (n = 2). Similarly, in the HBES population, 11 samples collected less than 30 m apart were found to represent 3 species, M. eohespera (n = 1), Mel-21 (n = 9) and M. pulchella (n = 1). In the SCSJS population, 9 samples collected less than 50 m apart represented 2 species, Mel-14 (n = 1) and M. eohespera (n = 8). In the SCQC population, 10 samples collected from less than 100 m apart represented two species, Mel-21(n = 8) and M. pulchella (n = 2). Their sympatric distribution affords plenty of opportunities for hybridisation among the lineages.
Discussion
This is the first study conducted on the population biology of two closely related sympatric morels species. Both species showed rich and divergent genetic diversity. The ITS region and the B2 gene fragment, respectively, harboured the most polymorphic sites and the fewest polymorphic sites in both species and all four DNA fragments showed more variation in M. eohespera than in Mel-13. Some studies have suggested that increases in genetic polymorphism, mutation and recombination might ensure higher levels of genetic diversity, thus providing greater potential for genetic adaptation in changing environments17,18,19,20,21. No heterozygosity was observed in either species, even for the ITS region, which is known to be multi-copied within the nuclear ribosomal gene cluster. The lack of heterozygosity suggests that the fruiting bodies derived from haploid mycelia and that these four DNA fragments represent single copy gene markers. Yoon et al.22 indicated that species in the M. esculenta complex (Esculenta Clade) were haploid due to no heterozygosity found, as similar results were observed in Mel-13 and M. eohespera (Elata Clade). It is presumed that selfing might be very common in these morel species or they were homothallic and their fruiting bodies were developed from haploid mycelia (see the latter discussion).
Significant differentiation was observed among geographic locations within Mel-13 and M. eohespera. However, no correlation was detected between genetic distance, geographic distance and altitude (Figure S1 and Figure S2). Although geographic separation has been shown to be an important contributor to genetic differentiation in many species, environmental factors can also play a significant role23,24. For example, habitat fragmentation has been found to alter the structure, distribution and functioning of natural ecosystems25, and to increase population genetic divergence, inbreeding or selfing and reduce gene flow26,27,28. The rich private haplotypes observed in Mel-13 (79.3%) and M. eohespera (51.5%) indicated potential multiple habitat fragmentation events. Considering that the speciation time of Mel-13 and M. eohespera has been estimated at around 2.855 Mya and 0.443 Mya respectively16, these habitat fragmentation events were likely to have been influenced by the Quaternary Glaciation. Moreover, mating patterns tended to shift towards an increased rate of selfings buffering the genetic effects of habitat fragmentation29. Consistent with the prediction, we found clear evidence of clonality/selfing in Mel-13 and M. eohespera.
Although Mel-13 and M. eohespera have sympatric distributions in southern China, our results indicated that they probably had different evolutionary histories. The mismatch distribution analysis and the median joining network indicated that Mel-13 probably experienced a sudden population bottleneck followed by expansion, while M. eohespera did not. The speciation time for Mel-13 has been estimated at about 2.855 Mya. During its evolutionary history, the climatic oscillations of the Quaternary led to repeated drastic environmental changes (<2.0 Mya)30,31. These changes probably promoted the sudden expansion of Mel-13 and stimulated the emergence of very rich private haplotypes (79.3%) and more frequent recombination detected in 54.5% of the Mel-13 population, which may have helped it to survive the extensive environmental changes and habitat fragmentation. Different genotypes may display different fitness in different environments, so the highly localised genetic diversity could have accelerated adaptation to heterogeneous and changing biotic and abiotic environments32,33.
It has been estimated that M. eohespera emerged around 0.443 Mya, during the Pleistocene16. Four major glaciations have been proposed in the Qinghai-Tibet Plateau during the Quaternary, and they became progressively less extensive after the largest Naynayxungla Glaciation (c.0.72–0.5 Mya)34,35,36. It is thus unlikely that the last glaciation had a strong influence on M. eohespera, which partly explains why fewer private haplotypes (51.5%) were found in M. eohespera than in Mel-13, and why limited recombination was identified only in the XZRW population. Our results suggested that the XZRW population was the centre of diversity of M. eohespera in China because this population possessed the most haplotypes (n = 10), including the most broadly distributed haplotype, H22, and the SAMOVA analysis separated it from the other populations.
The reproduction mode of morels has been debated by various authors. Studies have indicated that collections identified as M. esculenta, M. deliciosa and M. snyderi are heterothallic and could be outcrossed in the laboratory12,14,37,38. However, Dalgleish and Jacobson11 and Pagliaccia14 raised the possibility that some populations from the US probably incorrectly reported as M. esculenta and some individuals of M. snyderi were clonal or had selfing as their dominant reproduction mode.
Our current results identified limited recombination within the four combined loci of the total samples for both species, and within the F1 gene fragment of Mel-13 and the ITS region of M. eohespera, however, no evidence of recombination was found in the other DNA fragments in either species (Table 4). In Mel-13, 45.5% of the populations (QHZM, QHMH, QHBB, QHZG and SCMEK) appeared to be asexual, and five populations (XJZS, XJWLMQ, SXYA, SCJZ and YNLJS) appeared to have limited recombination. Notably, only one population (SXFS) showed evidence of recombination according to both rBarD and PrC values, and this population had the highest haploid diversity and nucleotide diversity. Among the 14 populations in M. eohespera, 13 showed no evidence of recombination, consistent with asexual reproduction. Only the XZRW population showed limited recombination according to the PrC value. Similar to Mel-13, XZRW, the only population where recombination was detected in M. eohespera, also had high haploid and nucleotide diversity. Interestingly, two other populations, XJWLMQ and XZLL, in M. eohespera also had high haploid and nucleotide diversity, but no recombination was identified in them.
Taylor et al.39 proposed that clonality and recombination can be temporally or spatially separated in a single fungal species. Indeed, many fungal populations have been shown to contain signatures of both recombination and clonal expansion of a few genotypes40,41,42,43,44. Our analysis revealed a general picture of admixture. A predominantly clonal reproductive strategy with limited recombination was inferred for M. eohespera. The results were supported not only by the results of a multilocus linkage disequilibrium analysis, but also by the identification of a few over-represented genotypes (H3 and H22), consistent with clonal expansion. This strategy might facilitate the dispersal of individuals colonising a new habitat, as proposed by Dalgleish and Jacobson11 and documented in mycorrhizal basidiomycetes45,46,47. Clonal reproductive modes were also prevalent in Mel-13, but recombination events were much more frequent in Mel-13 than in M. eohespera, which probably stimulated the increase in genetic diversity that might have enhanced the adaptation of some populations to hostile environmental and climate changes. The different populations in Mel-13 may have had different reproductive strategies, whereas most populations in M. eohespera possesses clonal reproductive mode, and the levels of genotypic diversity were at least partly associated with the level of recombination.
It is not known how variation in reproductive mode across morel populations is controlled, or whether these distinct populations use sexual and/or asexual reproductive modes to regulate gene flow, select more rapidly adaptive genotypes and respond to heterogeneous environments. Extensive sampling and laboratory mating assays would help to gain a full understanding of the mating behaviour and determine the relative contribution of asexual versus sexual reproductive modes in morels. We cannot reject the possibility that both species are heterothallic, and speculate that the unknown mating system may also facilitate outcrossing considering the high level of genetic variation in the populations of both species.
If the life cycles of Mel-13 and M. eohespera were heterothallic, the lack of heterozygosity in any of the samples from any of the four DNA fragments would suggest that inbreeding must be highly prevalent. The presence of hybridisation, as suggested by the gene genealogical incongruence, indicates that mating must have occurred in nature. If this were true, the lack of heterozygosity would indicate that inbreeding must have been the predominant mode of sexual reproduction.
At least five species of Morchella were found to be sympatric in our study, but whether there is reproductive isolation between them and what’s the isolation degree is unknown. Some studies have proposed greater reproductive isolation is involved in sympatric species than allopatric ones48,49. However, Le Gac and Giraud50 indicated no significant difference in the degree of reproductive isolation between sympatric and allopatric pairs of ascomycota, and found that many pairs of closely related sympatric species in ascomycota were not isolated by strong premating barriers. This is supported by our observation that crossing experiments on these morel species under laboratory conditions showed little evidence of reproductive isolation (unpublished data). Le Gac and Giraud50 proposed two potential explanations for the maintenance of sibling species in the same geographic area without the evolution of strong premating barriers: (1) sexual reproduction may not have been frequent in nature, thus impeding strong selection for enhanced premating isolation and the fusion of insufficiently isolated populations, as we observed that the reproductive mode of some morels in nature was predominantly clonal; and (2) the sibling species may have shared only a restricted geographic contact zone (parapatry). Finally, we still have not identified the mating-type locus in morels, so the role that it plays in reproduction of different sympatric species is unknown. How these sympatric species remain reproductively isolated remains to be determined.
Conclusions
The population genetics of two sympatric closely related species of Morchella was conducted for the first time in this study. Rich genetic diversity and significant population differentiation were found in Mel-13 and M. eohespera, however, M. eohespera possessed more variation than Mel-13 at the four molecular markers. We conclude that the XZRW population was probably the diversity centre of M. eohespera in China. Prevalent clonality, limited local recombination and potential hybridisation or horizontal gene transfer were detected in the Morchella taxa, but the mating systems of both species remain to be determined. Taken together, our findings suggest that the two sympatric species have a potentially divergent evolutionary history.
Genomic studies have become a research hotspot. Sequencing morel genomes will provide unprecedented insights on fruiting-related genes, the mating system and genes essential for sexual reproduction of morels.
Methods
Sampling
Over 1000 morels were collected from different regions of China between 2003 and 2014, covering 26 collecting sites in 10 provinces. As it is difficult to distinguish closely related morels species by morphology, We first selected 496 samples from these 26 collecting to represent each collecting site, and then screened them using sequences of four gene fragments, ITS, B2, F1 and F2. The same fragments were also used in our subsequent population genetic analyses. The screening identified 11 species from the 496 samples: M. sexelata, M. eximia, M. exuberans, M. importuna, Mel-13, Mel-14, M. eximioides, M. eohespera, Mel-21, M. pulchella and Mel-34. Based on the results combined with the phylogenetic and biogeographic results in Du et al.16, and considering that the target species should be closely related and sympatric species, more widely distributed than other species in China and represent enough samples in order to detect and compare their evolutionary histories and potential reproductive modes, we chose Mel-13 and M. eohespera for conducting comparative population genetic studies. Additionally, Mel-13 and M. eohespera were also respectively distributed in India and Europe, we tried to obtain collections from both regions for this study but failed. More effort will be made in the future for a study on the continental disjunction distribution and evolutionary history of morels. Finally, 124 collections of 11 populations representing Mel-13 distributed in 6 provinces (Fig. 1) and 156 collections of 14 populations representing M. eohespera distributed in 6 provinces (Fig. 2), covering the main distribution of both species in China, were selected for the analysis. Here, each population represented each collecting site. Maps of these collecting sites were generated using ArcView GIS 3.2 (ESRI, Redlands, CA, USA). The sample size, geographical coordinates and altitudes for each population are presented in Table 1.
Selection of genetic markers
To develop genetic markers for our population genetic analyses, we first extracted genomic DNA from the fruiting body of isolate D139, which belonged to Mel-13, according to the modified CTAB method51. A random shotgun genomic library was constructed using genomic DNA from isolate D139 according to the methods described previously52.
The cloned fragments in the range 0.5–1.0 kb were amplified using vector primers and the PCR products were confirmed using agarose gel electrophoresis and then sequenced. We developed PCR primers from 15 sequences chosen from the cloned fragments using the online software Primer3 to directly amplify and sequence the DNA from four isolates: D139 (positive control), D47, D300 and D112 (the latter three strains were geographically different from D139), all of which belonged to Mel-13. Sequences obtained from the four strains were compared to identify single-nucleotide substitutions. Most of these primer pairs showed inconsistent PCR amplification of the samples and/or low DNA polymorphism. Finally, 3 of the 15 fragments respectively coded as B2, F1 and F2 by us, with different mutation rates and consistent PCR amplification success, never used before in morels study, were selected based on the comparison of sequences of the above four samples. The blasting results of B2, F1 and F2 fragments shown no similar results to them found in NCBI and their identity was unknown. These 3 markers together with the internal transcribed spacer (ITS) region with high nucleotide variation rates in morels, were chosen to genotype all 280 strains belonging to Mel-13 and M. eohespera from China.
PCR amplification and sequencing
Amplifications of ITS, B2, F1 and F2 were conducted using ITS4 and ITS553, B2F (5′ TGACGAGGATTGCCTTAACA 3′) and B2R (5′ AGGAGCATCATCTCCGGGTA 3′), F1F (5′ GGCTAAGATACGAGCTACGAGA 3′) and F1R (5′ ACATCAATGAGAGCCATTCG 3′) and F2F (5′ GAGCCATTCGTGCTCGTTAC 3′) and F2R (5′ ACCTGTTCGCCAGAGTTCAT 3′) for ITS, B2, F1 and F2, respectively. Each PCR reaction contained 1 μl of 20 ng/ul genomic DNA, 2.5 μl of 10 × PCR reaction buffer, 0.5 μl dNTP mix (10 mmol), 2 μl each of primer (5 umol), 1.5 μl bovine serum albumin (20 mg/ml) and 1.5 U of Taq DNA polymerase (Biomed, China). The final volume was adjusted to 25 ul with sterile distilled H2O. PCRs were conducted in an Applied Biosystems 2720 thermocycler (ABI, Foster City, CA), using the following cycling parameters: 94°C for 3 min, 35 cycles of 94 °C for 1 min, 50 °C (ITS and B2), 49 °C (F1) and 53 °C (F2) for 30 s, 72 °C for 1 min, followed by a final extension of 10 min at 72 °C. Amplicons were electrophoresed in 1.2% agarose in 1 × TAE, stained with GoldView™ (Guangzhou Geneshun Biotech Ltd., Guangdong, China), then photographed over an ultraviolet transilluminator. PCR products were purified using a Bioteke DNA Purification Kit (Bioteke Corporation, Beijing, China). They were used for bidirectional sequencing using the PCR primers with ABI BigDye v3.1 (Sangon Co., Ltd., Shanghai, China) and run on an ABI 3730 DNA analyser. All newly acquired sequences have been deposited as KR073664-KR073911 in GenBank.
Data analysis
The raw sequence data of each gene fragment was edited in SeqMan (DNAStar Package, Madison, WI) and they were aligned using MUSCLE v3.8.3154. Aligned sequences were visually inspected and manually adjusted using BioEdit v7.0.955 (http://www.mbio.ncsu.edu/bioedit/bioedit.html). As no sequence ambiguities or heterozygosity were detected, the genetic profile exhibited by each individual on the basis of four DNA fragments was treated as a haplotype on the assumption that they were haploid fungi.
Genetic diversity and population structure
To identify the overall population structure and compare potential differences in the structure, in the following analysis we combined the four DNA fragments into one dataset respectively comprising Mel-13 and M. eohespera. Each variable nucleotide site was treated as a locus and different nucleotides at the same site were viewed as different alleles.
Indices of nucleotide diversity (π) and haplotype diversity (Hd) were calculated for each population and each species using DNASP v5.1056. A Nei’s pairwise genetic distance matrix among populations was generated using GenAlEx v6.157. The same software was used to perform the analysis of molecular variance (AMOVA)58 and Mantel test to estimate the relative contributions of geographic separation to the overall genetic variation and detect the influence of their geographic distances and altitudinal differences on their population genetic structures. The genetic relationships between geographic locations were further estimated by theta59 using MultiLocus v1.360.
A median-joining haplotype network was constructed using NETWORK v4.5.1 (available at http://www.fluxus-engineering.com) with the MP criterion applied. The single ambiguity (closed loop) observed in the network was resolved using the frequency criterion and the geographical criterion based on coalescence theory61,62.
Neutrality tests and Pairwise mismatch distributions were implemented in ARLEQUIN v3.5.158 to test the historical population dynamics. Negative statistics in neutrality tests were taken to indicate recent population expansions, whereas positive statistics indicated stationary populations. In mismatch distributions analysis, multimodal mismatch distributions and unimodal distributions are respectively expected for stationary populations and populations with recent demographic expansions63,64. The validity of the expansion model can be evaluated with the sum of squared deviations (SSD) and the raggedeness index (HRI) of observed distributions.
The spatial genetic structuring of haplotypes was analysed using spatial analysis of molecular variance analysis software, SAMOVA v1.065, based on a simulated annealing approach to define groups of populations (K) that are geographically homogeneous and maximally differentiated from each other. An ΦCT index66 of genetic differentiation among K initial groups was computed and the largest ΦCT value was retained as the best population grouping.
Clonality and recombination
The following two tests were conducted to examine evidence of recombination in an individual population and the role of sexual recombination in natural populations. First, we calculated the index of association (IA) and rBarD. The null hypothesis for IA was that there were random associations (recombination, linkage equilibrium) between alleles at different loci. A p value of < 0.05 indicated that the null hypothesis should be rejected. We standardised the IA value by the number of loci using the rBarD algorithm, because the value of the traditional IA can be influenced by the number of loci analysed. In the second test, the proportion of pairwise loci that were phylogenetically incompatible (PI) was calculated. A lack of phylogenetic incompatibility implies asexual reproduction. The incompatibility ratio (IR), where IR = (number of incompatible pairs of sites in the test dataset)/(number of incompatible pairs of sites in a randomly shuffled dataset), can be used as a test of statistical significance60. The phylogenetic incompatibility and the rBarD tests were conducted using MultiLocus v1.360.
Additional Information
Accession codes: Nucleotide sequences associated with this article are available through GenBank (Accession nos: KR073664-KR073911).
How to cite this article: Du, X.-H. et al. High inbreeding, limited recombination and divergent evolutionary patterns between two sympatric morel species in China. Sci. Rep. 6, 22434; doi: 10.1038/srep22434 (2016).
References
Kuo, M. Morchella tomentosa, a new species from western North America, and notes on M. rufobrunnea . Mycotaxon 105, 441 (2008).
Zhao, Q., Xu, Z., Cheng, Y., Qi, S. & Hou, Z. Bionic cultivation of Morchella conica . Southwest China J Agr Sci 22, 1690–1693 (2009).
Pilz, D. Ecology and management of morels harvested from the forests of western North America, 161 (DIANE Publishing, General Technical Report PNW-GTR-710. Portland: US Department of Agriculture, Forest Service, Pacific Northwest Research Station, 2008).
Nantel, P., Gagnon, D. & Nault, A. Population viability analysis of American ginseng and wild leek harvested in stochastic environments. Conserv Biol 10, 608–621 (1996).
Douhan, G., Peever, T. & Murray, T. Multilocus population structure of Tapesia yallundae in Washington State. Mol Ecol 1, 2229–2239 (2002).
Hemmati, R., Javan-Nikkhah, M. & Linde, C. C. Population genetic structure of Sclerotinia sclerotiorum on canola in Iran. Eur J Plant Pathol 125, 617–628 (2009).
Henk, D. A. et al. Clonality despite sex: the evolution of host-associated sexual neighborhoods in the pathogenic fungus Penicillium marneffei . PLoS pathog 8, e1002851, doi: 10.1371/journal.ppat.1002851 (2012).
Milgroom, M. G. Recombination and the multilocus structure of fungal populations. Annu Rev Phytopathol 34, 457–477 (1996).
Peever, T., Ibanez, A., Akimitsu, K. & Timmer, L. Worldwide phylogeography of the citrus brown spot pathogen, Alternaria alternata. Phytopathology 92, 794–802 (2002).
Sun, S. & Heitman, J. Is sex necessary? BMC Biol 9, 56 (2011).
Dalgleish, H. & Jacobson, K. A first assessment of genetic variation among Morchella esculenta (Morel) populations. J Hered 96, 396–403 (2005).
Hervey, A., Bistis, G. & Leong, I. Cultural studies of single ascospore isolates of Morchella esculenta . Mycologia, 1269–1274 (1978).
King, H. & Jacobson, K. Genetic diversity within and between populations of morel mushrooms (Morchella esculenta). In: Proceedings of the Iowa Academy of Science Meeting, Des Moines, April 22. Cedar Falls: Iowa Academy of Science. (2000).
Pagliaccia, D. et al. Development of molecular markers and preliminary investigation of the population structure and mating system in one lineage of black morel (Morchella elata) in the Pacific Northwestern USA. Mycologia 103, 969–982 (2011).
Volk, T. J. & Leonard, T. J. Experimental studies on the morel. I. Heterokaryon formation between monoascosporous strains of Morchella. Mycologia, 523–531 (1989).
Du, X.-H., Zhao, Q., O’Donnell, K., Rooney, A. P. & Yang, Z. L. Multigene molecular phylogenetics reveals true morels (Morchella) are especially species-rich in China. Fungal Genet Biol 49, 455–469 (2012).
Cao, Y. et al. Structure, gene flow, and recombination among geographic populations of a Russula virescens Ally from southwestern China. PloS one 8, e73174, doi: 10.1371/journal.pone.0073174 (2013).
Kis-Papo, T., Kirzhner, V., Wasser, S. P. & Nevo, E. Evolution of genomic diversity and sex at extreme environments: fungal life under hypersaline Dead Sea stress. P Natl Acad Sci USA 100, 14970–14975 (2003).
Korol, A., Preygel, I. & Preygel, S. Recombination variability and evolution. Chapman Hall, London (1994).
Nevo, E. Evolution of genome–phenome diversity under environmental stress. P Natl Acad Sci USA 98, 6233–6240 (2001).
West, S. A., Lively, C. M. & Read, A. F. A pluralist approach to sex and recombination. J Evolution Biol 12, 1003–1012 (1999).
Yoon, C.-S., Gessner, R. V. & Romano, M. A. Population genetics and systematics of the Morchella esculenta complex. Mycologia, 227–235 (1990).
Liu, J., Zhu, Z., Liu, G., Qi, D. & Li, F. AFLP variation analysis on the germplasm recources of Leymus chinensis . Acta Bot Sin 44, 845–851 (2001).
Liu, H. et al. Genetic differentiation within and between Leymus chinensis populations from different zones of Mid-Eastern Inner Mongolia steppe. Acta Ecol Sin 24, 2157–2164 (2004).
Saunders, D. A., Hobbs, R. J. & Margules, C. R. Biological consequences of ecosystem fragmentation: a review. Conserv Biol 5, 18–32 (1991).
Lowe, A., Boshier, D., Ward, M., Bacles, C. & Navarro, C. Genetic resource impacts of habitat loss and degradation; reconciling empirical evidence and predicted theory for neotropical trees. Heredity 95, 255–273 (2005).
Sork, V. L., Nason, J., Campbell, D. R. & Fernandez, J. F. Landscape approaches to historical and contemporary gene flow in plants. Trends in Ecol Evol 14, 219–224 (1999).
Young, A., Boyle, T. & Brown, T. The population genetic consequences of habitat fragmentation for plants. Trends in Ecol Evol 11, 413–418 (1996).
Honnay, O. & Bossuyt, B. Prolonged clonal growth: escape route or route to extinction? Oikos 108, 427–432 (2005).
Hewitt, G. The genetic legacy of the Quaternary ice ages. Nature 405, 907–913 (2000).
Hewitt, G. Genetic consequences of climatic oscillations in the Quaternary. Philos T R Soc London, Ser. B 359, 183–195 (2004).
Smith, J. M. Evolution in sexual and asexual populations. Am Nat, 469–473 (1968).
Xu, J. The prevalence and evolution of sex in microorganisms. Genome 47, 775–780 (2004).
Owen, L. A. et al. Climatic and topographic controls on the style and timing of Late Quaternary glaciation throughout Tibet and the Himalaya defined by 10 Be cosmogenic radionuclide surface exposure dating. Quaternary Sci Rev 24, 1391–1411 (2005).
Shi, Y., Li, J. & Li, B. Uplift and Environmental Changes of Qinghai-Tibetan Plateau in the Late Cenozoic. Guangdong Science and Technology Press. Guangzhou, China. Guangzhou: Guangdong Science and Technology Press (1998).
Zheng, B., Xu, Q. & Shen, Y. The relationship between climate change and Quaternary glacial cycles on the Qinghai–Tibetan Plateau: review and speculation. Quatern Int 97, 93–101 (2002).
Gessner, R. V., Romano, M. A. & Schultz, R. W. Allelic variation and segregation in Morchella deliciosa and M. esculenta . Mycologia, 683–687 (1987).
Volk, T. J. & Leonard, T. J. Cytology of the life-cycle of Morchella . Mycol Res 94, 399–406 (1990).
Taylor, J. W., Turner, E., Townsend, J. P., Dettman, J. R. & Jacobson, D. Eukaryotic microbes, species recognition and the geographic limits of species: examples from the kingdom Fungi. Philos T R Soc London, Ser. B 361, 1947–1963 (2006).
Campbell, L. T. et al. Clonality and recombination in genetically differentiated subgroups of Cryptococcus gattii . Eukaryot Cell 4, 1403–1409 (2005).
Hiremath, S. S. et al. Long-distance dispersal and recombination in environmental populations of Cryptococcus neoformans var. grubii from India. Microbiol 154, 1513–1524 (2008).
Xu, J. Xu, J. (ed.) 2005. Evolutionary Genetics of Fungi. Norfolk, UK: Horizon Bioscience (2005).
Xu, J., Yan, Z. & Guo, H. Divergence, hybridization, and recombination in the mitochondrial genome of the human pathogenic yeast Cryptococcus gattii . Mol ecol 18, 2628–2642 (2009).
Zhang, Y., Yu, Z. F., Xu, J. & Zhang, K. Q. Divergence and dispersal of the nematode‐trapping fungus Arthrobotrys oligospora from China. Env Microbiology Rep 3, 763–773 (2011).
Jacobson, K. & Miller Jr, O. Postmeiotic mitosis in the basidia of Suillus granulatus: implications for population structure and dispersal biology. Mycologia, 511–516 (1994).
Tommerup, I., Bougher, N. & Malajczuk, N. Laccaria species ectomycorrhizal with Eucalypts: why does the ecology of bisporic species differ from quadrisporic species? In Proceedings of the 8th North American Conference on Mycorrhizae. Jackson, Wyoming, USA 284 (1990).
Tommerup, I. Significance of post-meiotic mitoses in basidiomes of ectomycorrhizal fungi. In Proceedings of the Eighth North American Conference on Mycorrhizae ( Allen M. & Williams S. L., eds). Laramie: University of Wyoming Vol. 282 (1990).
Coyne, J. A. & Orr, H. A. Patterns of speciation in Drosophila. Evol, 362–381 (1989).
Dettman, J. R., Jacobson, D. J., Turner, E., Pringle, A. & Taylor, J. W. Reproductive isolation and phylogenetic divergence in Neurospora: comparing methods of species recognition in a model eukaryote. Evol 57, 2721–2741 (2003).
Le Gac, M. & Giraud, T. Existence of a pattern of reproductive character displacement in Homobasidiomycota but not in Ascomycota . J Evolution Biol 21, 761–772 (2008).
Doyle, J. J. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem bull 19, 11–15 (1987).
Xu, J., Guo, H. & Yang, Z.-L. Single nucleotide polymorphisms in the ectomycorrhizal mushroom Tricholoma matsutake . Microbiol 153, 2002–2012 (2007).
White, T. J., Bruns, T., Lee, S. & Taylor, J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. PCR protocols: a guide to methods and applications (eds Innis, M. A. et al.) Academic Press, San Diego, CA 18, 315–322 (1990).
Edgar, R. C. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC bioinformatics 5, 113 (2004).
Hall, T. A. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. in Nucleic acids symposium series Vol. 41, 95–98 (1999).
Librado, P. & Rozas, J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452 (2009).
Peakall, R. & Smouse, P. E. GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol notes 6, 288–295 (2006).
Excoffier, L. & Lischer, H. E. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10, 564–567 (2010).
Weir, B. S. & Ott, J. Genetic data analysis II. Trends Genet 13, 379 (1997).
Agapow, P. M. & Burt, A. Indices of multilocus linkage disequilibrium. Mol Ecol Notes 1, 101–102 (2001).
Crandall, K. A. & Templeton, A. R. Empirical tests of some predictions from coalescent theory with applications to intraspecific phylogeny reconstruction. Genet 134, 959–969 (1993).
Templeton, A. R. & Sing, C. F. A cladistic analysis of phenotypic associations with haplotypes inferred from restriction endonuclease mapping. IV. Nested analyses with cladogram uncertainty and recombination. Genet 134, 659–669 (1993).
Harpending, H. Signature of ancient population growth in a low-resolution mitochondrial DNA mismatch distribution. Hum Biol, 91–600 (1994).
Rogers, A. R. & Harpending, H. Population growth makes waves in the distribution of pairwise genetic differences. Mol Biol Evol 9, 552–569 (1992).
Dupanloup, I., Schneider, S. & Excoffier, L. A simulated annealing approach to define the genetic structure of populations. Mol Ecol 11, 2571–2581 (2002).
Wright, S. & Wright S. Variability Within and Among Natural Populations, Evolution and the Genetics of Populations. University of Chicago Press, Chicago, 1st edn, Vol. 4, 580 (1978).
Acknowledgements
The authors thank Dr. Bang Feng (Kunming Institute of Botany, CAS), Mr. Yong-Song Zhang, Dr. Bo Xiao (Chongqing Institute of Medicinal Plant Cultivation), Dr. Rui Xing (Northwest Institute of Plateau Biology, CAS), and Dr. Yu-Guang Fan (Changbai Mountain Academy of Sciences) for their help during the authors’ field trip. Thanks also are due to the anonymous reviewers for constructive comments and suggestions. This work was supported by the National Basic Research Program of China (973 Program, No. 2014CB138305), the National Natural Science Foundation of China (No. 31300022), West Light Foundation of The Chinese Academy of Sciences and the CAS/SAFEA International Partnership Program for Creative Research Teams.
Author information
Authors and Affiliations
Contributions
Z.L.Y. and X.-H.D. designed the study. X.-H.D. and Q.Z. conducted the laboratory work. X.-H.D., J.X. and Q.Z. analyzed the data. X.-H.D. wrote the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Du, XH., Zhao, Q., Xu, J. et al. High inbreeding, limited recombination and divergent evolutionary patterns between two sympatric morel species in China. Sci Rep 6, 22434 (2016). https://doi.org/10.1038/srep22434
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep22434
- Springer Nature Limited
This article is cited by
-
Genome assembly of M. spongiola and comparative genomics of the genus Morchella provide initial insights into taxonomy and adaptive evolution
BMC Genomics (2024)
-
Ten decadal advances in fungal biology leading towards human well-being
Fungal Diversity (2022)
-
Multilocus phylogenies reveal three new truffle-like taxa and the traces of interspecific hybridization in Octaviania (Boletaceae, Boletales)
IMA Fungus (2021)
-
The saprotrophic Pleurotus ostreatus species complex: late Eocene origin in East Asia, multiple dispersal, and complex speciation
IMA Fungus (2020)
-
Heterothallism and potential hybridization events inferred for twenty-two yellow morel species
IMA Fungus (2020)