
Abstract
The green algal phylum Chlorophyta has six diverse classes, but the phylogenetic relationship of the classes within Chlorophyta remains uncertain. In order to better understand the ancient Chlorophyta evolution, we have applied a site pattern sorting method to study compositional heterogeneity and the model fit in the green algal chloroplast genomic data. We show that the fastest-evolving sites are significantly correlated with among-site compositional heterogeneity and these sites have a much poorer fit to the evolutionary model. Our phylogenomic analyses suggest that the class Chlorophyceae is a monophyletic group and the classes Ulvophyceae, Trebouxiophyceae and Prasinophyceae are non-monophyletic groups. Our proposed phylogenetic tree of Chlorophyta will offer new insights to investigate ancient green algae evolution and our analytical framework will provide a useful approach for evaluating and mitigating the potential errors of phylogenomic inferences.
Similar content being viewed by others


Introduction
Chloroplast phylogenomics has become a useful approach to elucidate the enigmatic evolutionary relationships of different taxonomic levels of plants1,2,3,4. However, resolving the ancient phylogenetic history remains difficult because of non-phylogenetic signal5, or use of simplistic substitution models6 in the large-scale molecular data. It is known mathematically that the phylogenetic signal fall off exponentially with time at the deepest divergences7, thus accurately reconstructing ancient divergence is a difficult task. It has long been suggested that non-phylogenetic signal exist in ancient divergences8 and these signal are frequently generated by fast-evolving or compositionally heterogeneous sites9,10,11,12. Simplistic substitution models have a poor fit to the data, resulting in un-reliable phylogenetic reconstruction4,13,14. Non-phylogenetic signal and simplistic models are possibly major causes of errors in the large-scale genomic data and it is critical to reduce the impact of these errors for deep-level phylogenetic reconstruction.
Green algae are estimated to have originated over 1.8 billion years ago15 and it splits early into two lineages: the Charophytes and the Chlorophyta16,17. Resolving the Charophytes phylogeny has received attention because of their close evolutionary relationship to the land plants12,18,19,20, but a reliable phylogeny of Chlorophyta is important to understanding ancient evolution and diversification of morphological and cytological characters of green algae.
Chlorophyta comprises six diverse morphological groups that live in both marine and freshwater environments and includes Prasinophyceae, Ulvophyceae, Trebouxiophyceae, Chlorophyceae, Chlorodendrophyceae and Pedinophyceae21,22. It has been previously suggested that Ulvophyceae, Trebouxiophyceae and Chlorophyceae are all monophyletic groups and the common term “UTC clade” defines the grouping of these three classes23,24,25. However, the monophyly of Ulvophyceae and Trebouxiophyceae is not strongly supported based on several molecular studies22,26,27 and the new term “core Chlorophyta” has been recently used to define UTC groups plus Chlorodendrophyceae and Pedinophyceae21,22. Thus the phylogenetic relationships within Chlorophyta remain controversial. The three phylogenetic topologies between Ulvophyceae, Trebouxiophyceae and Chlorophyceae have been hypothesized: (1) Ulvophyceae be placed as sister to Chlorophyceae28,29,30; (2) Ulvophyceae sister to Trebouxiophyceae31,32; (3) Chlorophyceae sister to Trebouxiophyceae33,34.
Here, we attempt to reconstruct a more reliable phylogenetic tree of Chlorophyta both by using a site-heterogeneous substitution model and by removing non-phylogenetic signal. By estimating the relative evolutionary rate and among-site compositional heterogeneity, we demonstrate that fastest-evolving sites show strong compositional heterogeneity and have a much poorer fit to the evolutionary model. By removing these fastest-evolving sites and using a site-heterogeneous model, we produced a congruent phylogenetic tree of Chlorophyta supporting the following: (1) The class Chlorophyceae is a monophyletic group. (2) The class Ulvophyceae is a polyphyletic group. (3) The class Trebouxiophyceae is a paraphyletic group. (4) The class Chlorophyceae is likely close to a group uniting Chlorodendrophyceae and one clade of Ulvophyceae.
Results and Discussion
The original data set included 30,240 nucleotide positions of 53 protein-coding genes from 53 green algae taxa (Supplementary Table S1) and the posterior predictive test indicated that the assumption of compositional homogeneity is strongly violated in the original data (global z-score = 14.52). To reduce the among-lineage compositional heterogeneity of the data set, we excluded the taxa with the most strongly deviating nucleotide composition. The test statistic for individual taxa indicated that the nucleotide composition of 10 taxa is significantly deviating (z-score ranges from 16.10 to 34.68; Supplementary Table S1). When these 10 taxa were excluded from the original data set, the among-lineage compositional heterogeneity is strongly reduced (global z-score = 4.08). Furthermore, two extremely long-branch Trentepohliales (Ulvophyceae) taxa (Trentepohlia annulata and Cephaleuros parasiticus, see Supplementary Fig. S1) were removed to minimize the potential long-branch attraction artifact35 and further prevent the violation of the assumption of compositional homogeneity (global z-score = 3.68). The final data matrix consisted of 53 protein-coding genes from 41 taxa.
It has been demonstrated that fastest-evolving sites tend to mask genuine phylogenetic signal and support an erroneous topology4,36,37. First, we investigated the among-site compositional heterogeneity of the final data matrix. The final 30,240 sites of 41 taxa were sorted from most variable to least variable by using the TIGER method and the compositional heterogeneity was evaluated for a series of subsets, each subset having 1,000 sites. It has been known that compositional heterogeneity manifests most strongly in fast-evolving sites38,39. Indeed, our correlation results not only showed that the fast-evolving sites exhibit strongly deviating nucleotide composition, but also the 5,000 fastest-evolving sites are significantly correlated with the most strongly site-compositional heterogeneity (R2 = 0.88) (Fig. 1).

The correlation between the fast-evolving sites and compositional heterogeneity for a series of subsets, each subset having 1,000 sites.
The 5,000 fastest-evolving sites are marked with five solid circles. The large z-score value means strong among-site compositional heterogeneity. The “character similarity” is negatively correlated with evolutionary rate i.e. sites that are incompatible with few other sites are considered rapidly evolving. The x-axis shows the sorted positions from the most highly variable to the most conserved.
Second, we evaluated the fitness between the substitution model and fast-evolving sites. The Conditional Predictive Ordinates (CPO) method was applied to measure how well data for a site can be predicted by the evolutionary model and low (and negative) CPO values for the sites indicate the difficulty to predict the site patterns by the model40. The CPO analyses showed that the fast-evolving sites have the low CPO values, implying the poor fit to the substitution model. Also, the 5,000 fastest-evolving sites are statistically correlated with the lower CPO values (R2 = 0.98) (Fig. 2), thus these fastest-evolving sites are not well described by the evolutionary model. There is also a significant correlation between CPO values and compositional heterogeneity for these 5,000 fastest-evolving sites (R2 = 0.93) (Supplementary Fig. S2), showing that the sites with strongly deviating nucleotide composition have much poorer model fitness.

The correlation between the fast-evolving sites and model fitness for a series of subsets, each subset having 1,000 sites.
The 5,000 fastest-evolving sites are marked with five solid circles. The log(CPO) values are calculated ranges from -infinity (corresponding to CPO value = 0) to 0 (corresponding to CPO value = 1). The large log(CPO) value means the sites can be accurately predicted by the evolutionary model. The x-axis shows the sorted positions from the most highly variable to the most conserved.
Often all third codon positions are excluded for phylogenetic analyses because they exhibit higher substitution rates compared to first and second positions. However some third codon positions are relatively slow-evolving and have the genuine phylogenetic signal41,42 and the first and second codon positions also contain some highly variable sites that should be removed. In our data, there are 3,595 (71.9%) third codon positions and 1,405 (28.1%) first and second positions among the 5,000 fastest-evolving sites. This result shows that not all the third codons are fast-evolving and the site-sorting methods43,44 are helpful to objectively measure variability at each aligned position.
Because the 5,000 fastest-evolving positions significantly correlate most strongly with compositional heterogeneity and have a worse fit to the evolutionary model, we produced five shortened data sets (from 29,240 to 25,240 sites) by sequentially removing the 5,000 fastest-evolving sites in 1,000 increments. The cross-validation tests45 demonstrated that site-heterogeneous CAT+GTR+Γ model had a much better statistical fit than site-homogeneous GTR+Γ model for these shortened data sets (likelihood score difference: 2,797 ± 82 for 30,240 sites; 2,629 ± 115 for 29,240 sites; 2,417 ± 63 for 28,240 sites; 2,225 ± 92 for 27,240 sites; 1,901 ± 39 for 26,240 sites and 1,682 ± 110 for 25,240 sites, in favor of CAT+GTR+Γ model). We also used other shorter datasets (from 24,240 to 20,240 sites) for reconstructing Chlorophyta phylogeny.
To evaluate the robustness and accuracy of these topologies, we calculated the average bootstrap support values (BSVs) for each inferred topology and found that the phylogenies with higher BSVs (>90%) are supported based on the data sets ranging from 30,240 to 25,240 sites. Additional shorter data sets supported the topologies but with lower BSVs (<90%), indicating a poor resolution of phylogenetic inference. Thus we focused on the topologies with >90% BSVs (from 29,240 to 25,240 sites) to discuss the green algae relationships.
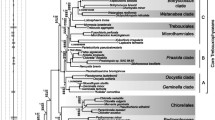
By using a site-heterogeneous CAT+GTR+Γ model and reducing the among-lineage/sites compositional heterogeneity, the Bayesian phylogenetic trees are largely congruent with high support values (Fig. 3; Supplementary Figs S3–S7). They strongly support that Prasinophyceae is paraphyletic and Prasinophyceae and Pedinophyceae are the early branching lineages of the Chlorophyta. In agreement with recent analyses22,46,47, our phylogenomic analyses show that the class Chlorophyceae is monophyletic with full support (1.00 posterior probability) and the classes Ulvophyceae and Trebouxiophyceae are recovered as polyphyletic and paraphyletic groups respectively. It confirms that the widely accepted term “UTC” clade is invalid and the new term “core Chlorophyta” (previous UTC groups plus the Chlorodendrophyceae and Pedinophyceae) is more appropriate.

Bayesian phylogenetic trees under the CAT+GTR+Γ site-heterogeneous model.
(A) The Bayesian tree based on the data set including 30,240 aligned sites from 41 taxa of Chlorophyta. The posterior probability (PP) values are shown on the nodes and values with full support (1.00 PP) are indicated as*. (B) The condensed Bayesian tree of “core Chlorophyta” based on 30,240/29,240/ 28,240/27,240 data sets. The PP values are shown on the nodes from bottom to top corresponding to these 4 data sets and values with full support (1.00 PP) are indicated as*.
In terms of the phylogenetic relationship within Chlorophyta, our chloroplast phylogenomic analyses recovered two separate Ulvophyceae clades. The class Trebouxiopheceae is closest to one Ulvophyceae clade (containing Dasycladales and Bryopsidales) and the class Chlorophyceae is the sister group to a lineage uniting another Ulvophyceae clade (containing Ulotrichales and Oltmannsiellopsidales) and Chlorodendrophyceae (Tetraselmis). A notable difference between our phylogeny and recent genome-scale analyses22,46,47 is the position of Tetraselmis (Chlorodendrophyceae), Pseudendoclonium (Ulotrichales) and Oltmannsiellopsis (Oltmannsiellopsidales). Pseudendoclonium and Oltmannsiellopsis are classified as Ulvophyceae. Tetraselmis is a member of the Chlorodendrophyceae and has been reported as an early branching clade of the core Chlorophyta based on nuclear ribosomal data29,48. However, the recent chloroplast genome-scale analyses recovered Tetraselmis is in the vicinity of the Oltmannsiellopsis-Pseudendoclonium clade47 or a clade uniting Tetraselmis and Oltmannsiellopsis branched early of the core Chlorophyta22. Our suggested phylogeny supported Tetraselmis is close to Oltmannsiellopsis-Pseudendoclonium clade and the Tetraselmis-Oltmannsiellopsis-Pseudendoclonium clade is the sister group to Chlorophyceae. We noted that most of the reported phylogenomic analyses of Chlorophyta have the sparse samples of Ulvophyceae, especially the incomplete sampling of Ulotrichales and Oltmannsiellopsidales. Including more samples from these groups will provide more accurate phylogenetic position of Ulvophyceae.
Conclusions
By assessing the impact from compositional heterogeneity, fast-evolving sites and the model fit in the green algal chloroplast genomic data, the correlation analyses show that the sites with fastest evolutionary rates significantly correlate with strong among-site compositional heterogeneity and have much poorer fit to the evolutionary model. By removing these poor-fitting sites and using a site-heterogeneous substitution model, our chloroplast genomic data consistently indicate that the class Chlorophyceae is a monophyletic group and the classes Ulvophyceae and Trebouxiophyceae are likely not monophyletic. We further provide a proposed phylogenetic tree of Chlorophyta, although the robust relationship needs more investigations. There are still important uncertainties in phylogenetic relationships within Chlorophyta and we anticipate that adding more data from Ulvophyceae and Chlorodendrophyceae will help produce a well-supported relationship and make more conclusive inference on the evolution of green algae.
Materials and Methods
The 53 chloroplast protein-coding genes of 53 taxa (Supplementary Table S1) were collected from three available datasets22,27,49. We first translated the DNA sequences to amino acid using MEGA550 and then aligned them using MUSCLE51. Each aligned protein was back-translated to DNA sequence and was trimmed to exclude poorly aligned positions with complete codons using Gblocks52. These alignments were concatenated to generate a matrix of 30,240 sites.
We applied the TIGER method43 to estimate the relative evolutionary rate for each site by calculating the pairwise character similarity as a proxy. The “character similarity” is negatively correlated with evolutionary rate i.e. sites that are incompatible with few other sites are considered rapidly evolving. The matrix was then sorted from the most highly varied to the most conserved sites and a series of subsets (each having 1,000 sites) were generated for further analysis.
To reduce the potential impact of compositional bias in the genome-scale data, we excluded the taxa with the most highly deviating nucleotide composition and measured the compositional deviation of series of subsets (each having 1,000 sites) by performing the posterior predictive test (z-score as measurement) of compositional homogeneity using the “ppred -comp” command of PhyloBayes53. The z-score is the deviation between the observed value of a given test statistic on the original data and the mean value of the distribution of the test statistic on data replicates under the posterior predictive distribution, divided by the standard deviation of the posterior predictive distribution. A large z-score value indicates the strong compositional heterogeneity.
We evaluated the fitness between the data and evolutionary model using the Conditional Predictive Ordinates (CPO) method40 with 100,000 cycles implemented in Phycas program54. The CPO provides a posterior predictive approach of how well the individual sites fit the model. The high log(CPO) value means the data can be accurately predicted by the evolutionary model.
We performed a cross-validation test in 10 replicates each with 1,100 cycles to evaluate the relative fit of the site-heterogeneous CAT+GTR+Γ model and the standard site-homogeneous GTR+Γ model on the data sets. The Bayesian phylogenetic trees were reconstructed under the CAT+GTR+Γ model that accounts for site-specific heterogeneity using PhyloBayes53. Two independent chains were run for 10,000–20,000 cycles and the convergence was assessed using the maximum bipartition discrepancies across chains.
Additional Information
How to cite this article: Sun, L. et al. Chloroplast Phylogenomic Inference of Green Algae Relationships. Sci. Rep. 6, 20528; doi: 10.1038/srep20528 (2016).
References
Jansen, R. K. et al. Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc. Natl. Acad. Sci. 104, 19369–19374 (2007).
Moore, M. J., Soltis, P. S., Bell, C. D., Burleigh, J. G. & Soltis, D. E. Phylogenetic analysis of 83 plastid genes further resolves the early diversification of eudicots. Proc. Natl. Acad. Sci. 107, 4623–4628 (2010).
Wu, C.-S., Wang, Y.-N., Hsu, C.-Y., Lin, C.-P. & Chaw, S.-M. Loss of different inverted repeat copies from the chloroplast genomes of Pinaceae and cupressophytes and influence of heterotachy on the evaluation of gymnosperm phylogeny. Genome Biol. Evol. 3, 1284–1295 (2011).
Zhong, B. et al. Systematic error in seed plant phylogenomics. Genome Biol. Evol. 3, 1340–1348 (2011).
Philippe, H. et al. Resolving difficult phylogenetic questions: why more sequences are not enough. PLoS-Biol. 9, 402 (2011).
Cooper, E. D. Overly simplistic substitution models obscure green plant phylogeny. Trends Plant Sci. 19, 576–582 (2014).
Mossel, E. & Steel, M. A phase transition for a random cluster model on phylogenetic trees. Math. Biosci. 187, 189–203 (2004).
Lockhart, P. J. et al. Controversy on chloroplast origins. FEBS Lett. 301, 127–131 (1992).
Jeffroy, O., Brinkmann, H., Delsuc, F. & Philippe, H. Phylogenomics: the beginning of incongruence? TRENDS Genet. 22, 225–231 (2006).
Nesnidal, M. P., Helmkampf, M., Bruchhaus, I. & Hausdorf, B. Compositional heterogeneity and phylogenomic inference of metazoan relationships. Mol. Biol. Evol. 27, 2095–2104 (2010).
Liu, Y., Cox, C. J., Wang, W. & Goffinet, B. Mitochondrial phylogenomics of early land plants: mitigating the effects of saturation, compositional heterogeneity and codon-usage bias. Syst. Biol. 63, 862–878 (2014).
Zhong, B. et al. Streptophyte algae and the origin of land plants revisited using heterogeneous models with three new algal chloroplast genomes. Mol. Biol. Evol. 31, 177–183 (2014).
Arbiza, L., Patricio, M., Dopazo, H. & Posada, D. Genome-wide heterogeneity of nucleotide substitution model fit. Genome Biol. Evol. 3, 896 (2011).
Goremykin, V. V. et al. The evolutionary root of flowering plants. Syst. Biol. 62, 50–61 (2013).
Moczydlowska, M., Landing, E. D., Zang, W. & Palacios, T. Proterozoic phytoplankton and timing of chlorophyte algae origins. Palaeontology 54, 721–733 (2011).
Lewis, L. A. & McCourt, R. M. Green algae and the origin of land plants. Am. J. Bot. 91, 1535–1556 (2004).
Leliaert, F., Verbruggen, H. & Zechman, F. W. Into the deep: new discoveries at the base of the green plant phylogeny. Bioessays 33, 683–692 (2011).
Turmel, M., Otis, C. & Lemieux, C. Tracing the evolution of streptophyte algae and their mitochondrial genome. Genome Biol. Evol. 5, 1817–1835 (2013).
Zhong, B., Liu, L., Yan, Z. & Penny, D. Origin of land plants using the multispecies coalescent model. Trends Plant Sci. 18, 492–495 (2013).
Wickett, N. J. et al. Phylotranscriptomic analysis of the origin and early diversification of land plants. Proc. Natl. Acad. Sci. 111, E4859–E4868 (2014).
Leliaert, F. et al. Phylogeny and molecular evolution of the green algae. Crit. Rev. Plant Sci. 31, 1–46 (2012).
Fučíková, K. et al. New phylogenetic hypotheses for the core Chlorophyta based on chloroplast sequence data. Front. Ecol. Evol. 2, 63 (2014).
Mishler, B. D. et al. Phylogenetic Relationships of the‘ Green Algae’ and‘ Bryophytes’. Ann. Mo. Bot. Gard. 451–483 (1994).
Friedl, T. & Zeltner, C. Assessing the relationships of some coccoid green lichen algae and the microthamniales (chlorophyta) with 18s ribosomal rna gene sequence comparisons1. J. Phycol. 30, 500–506 (1994).
Booton, G. C., Floyd, G. L. & Fuerst, P. A. Origins and affinities of the filamentous green algal orders Chaetophorales and Oedogoniales based on 18S rRNA gene sequences. J. Phycol. 34, 312–318 (1998).
Novis, P. M., Smissen, R., Buckley, T. R., Gopalakrishnan, K. & Visnovsky, G. Inclusion of chloroplast genes that have undergone expansion misleads phylogenetic reconstruction in the Chlorophyta. Am. J. Bot. 100, 2194–2209 (2013).
Lemieux, C., Otis, C. & Turmel, M. Chloroplast phylogenomic analysis resolves deep-level relationships within the green algal class Trebouxiophyceae. BMC Evol. Biol. 14, 211 (2014).
Cocquyt, E., Verbruggen, H., Leliaert, F. & De Clerck, O. Evolution and cytological diversification of the green seaweeds (Ulvophyceae). Mol. Biol. Evol. 27, 2052–2061 (2010).
Marin, B. Nested in the Chlorellales or independent class? Phylogeny and classification of the Pedinophyceae (Viridiplantae) revealed by molecular phylogenetic analyses of complete nuclear and plastid-encoded rRNA operons. Protist 163, 778–805 (2012).
Škaloud, P., Kalina, T., Nemjová, K., De Clerck, O. & Leliaert, F. Morphology and phylogenetic position of the freshwater green microalgae Chlorochytrium (Chlorophyceae) and Scotinosphaera (Scotinosphaerales, ord. nov., Ulvophyceae). J. Phycol. 49, 115–129 (2013).
Pombert, J.-F., Otis, C., Lemieux, C. & Turmel, M. The chloroplast genome sequence of the green alga Pseudendoclonium akinetum (Ulvophyceae) reveals unusual structural features and new insights into the branching order of chlorophyte lineages. Mol. Biol. Evol. 22, 1903–1918 (2005).
Turmel, M., Otis, C. & Lemieux, C. The chloroplast genomes of the green algae Pedinomonas minor, Parachlorella kessleri and Oocystis solitaria reveal a shared ancestry between the Pedinomonadales and Chlorellales. Mol. Biol. Evol. 26, 2317–2331 (2009).
Krienitz, L., Ustinova, I., Friedl, T. & Huss, V. A. Traditional generic concepts versus 18S rRNA gene phylogeny in the green algal family Selenastraceae (Chlorophyceae, Chlorophyta). J. Phycol. 37, 852–865 (2001).
López-Bautista, J. M. & Chapman, R. L. Phylogenetic affinities of the Trentepohliales inferred from small-subunit rDNA. Int. J. Syst. Evol. Microbiol. 53, 2099–2106 (2003).
Hendy, M. D. & Penny, D. A Framework for the Quantitative Study of Evolutionary Trees. Syst. Biol. 38, 297–309 (1989).
Parks, M., Cronn, R. & Liston, A. Separating the wheat from the chaff: mitigating the effects of noise in a plastome phylogenomic data set from Pinus L.(Pinaceae). BMC Evol. Biol. 12, 100 (2012).
Raymann, K., Brochier-Armanet, C. & Gribaldo, S. The two-domain tree of life is linked to a new root for the Archaea. Proc. Natl. Acad. Sci. 201420858 (2015).
Muto, A. & Osawa, S. The guanine and cytosine content of genomic DNA and bacterial evolution. Proc. Natl. Acad. Sci. 84, 166–169 (1987).
Sueoka, N. Directional mutation pressure and neutral molecular evolution. Proc. Natl. Acad. Sci. 85, 2653–2657 (1988).
Lewis, P. O., Xie, W., Chen, M.-H., Fan, Y. & Kuo, L. Posterior predictive Bayesian phylogenetic model selection. Syst. Biol. 63, 309–321 (2014).
Stefanović, S., Rice, D. W. & Palmer, J. D. Long branch attraction, taxon sampling and the earliest angiosperms: Amborella or monocots? BMC Evol. Biol. 4, 35 (2004).
Leebens-Mack, J. et al. Identifying the basal angiosperm node in chloroplast genome phylogenies: sampling one’s way out of the Felsenstein zone. Mol. Biol. Evol. 22, 1948–1963 (2005).
Cummins, C. A. & McInerney, J. O. A method for inferring the rate of evolution of homologous characters that can potentially improve phylogenetic inference, resolve deep divergence and correct systematic biases. Syst. Biol. 60, 833–844 (2011).
Goremykin, V. V., Nikiforova, S. V. & Bininda-Emonds, O. R. Automated removal of noisy data in phylogenomic analyses. J. Mol. Evol. 71, 319–331 (2010).
Lartillot, N. & Philippe, H. Improvement of molecular phylogenetic inference and the phylogeny of Bilateria. Philos. Trans. R. Soc. Lond. B Biol. Sci. 363, 1463–1472 (2008).
Leliaert, F. & Lopez-Bautista, J. M. The chloroplast genomes of Bryopsis plumosa and Tydemania expeditiones (Bryopsidales, Chlorophyta): compact genomes and genes of bacterial origin. BMC Genomics 16, 204 (2015).
Melton III, J. T., Leliaert, F., Tronholm, A. & Lopez-Bautista, J. M. The Complete Chloroplast and Mitochondrial Genomes of the Green Macroalga Ulva sp. UNA00071828 (Ulvophyceae, Chlorophyta). PLoS One 10, e0121020 (2015).
Arora, M., Anil, A. C., Leliaert, F., Delany, J. & Mesbahi, E. Tetraselmis indica (Chlorodendrophyceae, Chlorophyta), a new species isolated from salt pans in Goa, India. Eur. J. Phycol. 48, 61–78 (2013).
Lemieux, C., Otis, C. & Turmel, M. Six newly sequenced chloroplast genomes from prasinophyte green algae provide insights into the relationships among prasinophyte lineages and the diversity of streamlined genome architecture in picoplanktonic species. BMC Genomics 15, 857 (2014).
Tamura, K. et al. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739 (2011).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552 (2000).
Lartillot, N., Lepage, T. & Blanquart, S. PhyloBayes 3: a Bayesian software package for phylogenetic reconstruction and molecular dating. Bioinformatics 25, 2286–2288 (2009).
Lewis, P. O., Holder, M. T. & Swofford, D. L. Phycas: Software for Bayesian Phylogenetic Analysis. Syst. Biol. 64, 525–531 (2015).
Acknowledgements
The authors thank C. Lemieux and H. Verbruggen for kindly supplying the data. They also thank reviewers for improving the manuscript. This work was financially supported by the National Natural Science Foundation of China (31570219), the Natural Science Foundation of Jiangsu Province (BK20150971), the Natural Science Foundation of China for Talents Training in Basic Science (J1103507) and the Priority Academic Program Development of Jiangsu Higher Education Institutions.
Author information
Authors and Affiliations
Contributions
B.Z. and D.P. designed the study. L.S., L.F., Z.Z. and X.C. conducted the experiments and analyzed the data. B.Z. and D.P. wrote the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Sun, L., Fang, L., Zhang, Z. et al. Chloroplast Phylogenomic Inference of Green Algae Relationships. Sci Rep 6, 20528 (2016). https://doi.org/10.1038/srep20528
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep20528
- Springer Nature Limited
This article is cited by
-
Phylotranscriptomic insights into a Mesoproterozoic–Neoproterozoic origin and early radiation of green seaweeds (Ulvophyceae)
Nature Communications (2022)
-
The complete chloroplast genome of Stauntonia chinensis and compared analysis revealed adaptive evolution of subfamily Lardizabaloideae species in China
BMC Genomics (2021)
-
Taxonomic scheme of the order Chaetophorales (Chlorophyceae, Chlorophyta) based on chloroplast genomes
BMC Genomics (2020)
-
The genetic diversity and phylogeny of green microalgae in the genus Choricystis (Trebouxiophyceae, Chlorophyta) in Lake Baikal
Limnology (2020)
-
Plastid genomes of Elaeagnus mollis: comparative and phylogenetic analyses
Journal of Genetics (2020)