
Abstract
Two molecules, 1-hydroxypyrene-2-carbaldehyde (HP) and 1-methoxypyrene-2-carbaldehyde (MP) were explored. We investigated their photophysical properties, using experimental transient absorption and theoretical density functional theory/time-dependent density functional theory (DFT/TDDFT). HP and MP have similar geometric conformations but exhibit entirely different photophysical properties upon excitation into the S1 state. In contrast to traditional excited state intramolecular proton transfer (ESIPT) in molecules that exhibit either single or dual fluorescence, HP has an unusual non-fluorescent property. Specifically, the ultrafast ESIPT process occurs in 158 fs and is followed by an intersystem crossing (ISC) component of 11.38 ps. In contrast to HP, MP undergoes only an 8 ps timescale process, which was attributed to interactions between solute and solvent. We concluded that this difference arises from intramolecular hydrogen bonds (IMHBs), which induce drastic structural alterntion upon excitation.
Similar content being viewed by others

Introduction
Compounds that undergo excited state intramolecular proton transfer (ESIPT) have attracted attention1,2,3,4,5,6 as, for fluorescent probes7,8,9, for use in bioimaging10, as light-emitting materials11, as photostabilizers12 and in photophysical studies13,14,15,16,17. The characteristic photophysical feature of a molecule undergoing ESIPT is the sub-picosecond timescale and large Stokes shift, which suggest that the structural geometry of the emissive excited state is significantly displaced from that of the ground state11,17,18,19.
Many functional organic molecules exhibiting ESIPT have been investigated19,20,21,22,23,24. Furthermore, numerous studies have demonstrated that the ESIPT process involves a heterocyclic ring containing a hydroxyl-group and a neighbouring proton acceptor that can form IMHBs22,25. Recently, it was found that intramolecular hydrogen bonding plays a crucial role in the proton transfer process26. The ESIPT process can be facilitated by the excited-state intramolecular hydrogen bond strengthening in the S1 state26,27. The first ESIPT investigation, which was performed on methyl salicylate (MS) by Weller28, demonstrated that MS is a three energy level system containing the ground state, a normal excited state and a tautomer excited state and it is the tautomerism that produces dual fluorescence.
However, despite numerous investigations since that of Weller, the ESIPT process still presents challenges both theoretically and experimentally due to the intrinsically complex physical and chemical nature, such as the cleavage and formation of hydrogen bonds and the subsequent nuclear rearrangement with inversion. More recently, experiments by Mitra et al.25 showed that the ultrafast ESIPT of 4-methyl-2,6-diformyl phenol occurs at approximately 200 fs in cyclohexane followed by an intramolecular vibrational redistribution (IVR) process for approximately 2.8 ps before emitting fluorescence from the ESIPT state. Tang et al.19 investigated the ESIPT process of 7-hydroxy-1-indanone and its derivatives by theoretical and experimental methods, elaborating the details of the synthesis, photophysical properties and OLED application. In our previous report26, we theoretically investigated the salicylaldehyde (SA) molecule, which has exclusively intramolecular O–H⋅⋅⋅O hydrogen bonds that form a strong quasi-aromatic chelating ring. We found that the hydrogen bonded quasi-aromatic chelating ring in the excited state becomes smaller, which facilitates the ESIPT process. We also discovered that SA exhibits single fluorescence, which unlike the mechanism described by Weller28. We believe that the ESIPT system under the effect of IMHBs can exhibit non-fluorescence in addition to single and dual fluorescence. Therefore, we focused on the ESIPT system HP, which was found to be non-fluorescent in the S1 state. We also investigated MP molecule, which cannot form IMHBs, for comparison.
The six-membered, intramolecularly hydrogen-bonded system HP is an ideal model to explore the effect of IMHBs on the ESIPT system. To shed light on the ESIPT dynamics of HP, we report a combined experimental and theoretical method to investigate the HP and MP molecules and to confirm experimental results and discover more detailed information.
Results and Discussion
The structures of the HP (a) and MP (b) molecules are shown in Fig. 1. An IMHB can be formed between the carbonyl group oxygen and the hydroxyl hydrogen in the HP molecule to create a hydrogen-bonded, quasi-aromatic chelating ring. In contrast, the IMHB is not formed in the MP molecule. Figure 1 also depicts the steady-state absorption and fluorescence spectra of HP (a) and MP (b) in cyclohexane (CHX). The absorption spectra show different vibronic structures between HP and MP, which indicates that the maxima of the absorption bands of HP and MP are located near 455 and 417 nm, respectively. The width of the absorption band for HP is broader than for MP and the MP band is split more than that of HP. Because the two molecules have similar geometric conformations, we suggest that the obvious difference in absorption is caused by IMHBs. The fluorescence peak of MP is at 438 nm. Upon 400 nm excitation, HP is non-fluorescent, which is different from dual-fluorescent MS investigated by Weller28.

Shapes of steady-state absorption and fluorescence spectra of HP (a) and MP (b) molecules in CHX.
λex = 400 nm. Structures of the HP and MP molecules are also shown as an inset in (a) and (b), respectively.
We performed femtosecond transient absorption spectroscopy to investigate the excited state properties of HP and MP in CHX. Figure 2 depicts the 3D image plot of transient absorption for HP and MP as a function of wavelength and time delay after excitation. For HP (Fig. 2a), there are two main components, absorption (positive signal) at approximately 520 nm and emission (negative signal) at approximately 450 nm. For MP (Fig. 2b), the absorption is at approximately 500 nm and the emission is at approximately 460 nm. However, in contrast to HP, the lifetime of the absorption at approximately 500 nm is relatively longer and the absorption width is relatively narrower for MP. The 3D plot shows a global overview of the excited dynamics of HP and MP.

3D image plots of the transient absorption for HP (a) and MP (b) in CHX after excitation at 400 nm.
The transient positive and negative differences in absorbance values, which evolve over delay time after excitation at 400 nm, are shown in Fig. 3. Figure 3(a,b) depict the transient absorption spectra of the HP molecule at different delay times. Initially, an emission band at approximately 455 nm appears and ends within hundreds of femtoseconds with a slight blue-shift. In the meantime, a broad absorption band appears and the shape of the transient absorption spectra is changed with time. Before 1.2 ps, the characteristic peak of the absorption band has an obvious red shift from 470 nm to 520 nm and the intensity and the width of the absorption band increase with time. After 1.2 ps, the intensity and width of the absorption band begin to decrease and the characteristic peak has a slight blue-shift from 520 nm to 502 nm that evolves over delay time. In contrast, the time evolution of the transient absorption spectra of MP is shown in Fig. 3(c,d). It takes less time to reach the maximum intensity of absorption and the width is narrower for MP than HP at the same delay time. Moreover, in the range of 0 to 1 ps, the characteristic peak absorption band has a relatively smaller red-shift from 470 nm to 502 nm compared to that of HP. The rate of the absorption intensity decrease is much slower than that of the HP molecule and is without the blue-shift of the characteristic peak.

Femtosecond transient absorption spectra recorded in CHX for HP between −0.2 and 1.2 ps (a), from 1.2 to 50 ps (b) and for MP between −0.2 and 1 ps (c), from 1 to 50 ps (d).
To investigate the lifetime of the dynamics processes for HP and MP, a global fit analysis was used, in which sets of kinetic curves were fit simultaneously using exponential components with common lifetimes (Table 1). The kinetic curves for HP were fitted to two exponentials with lifetimes τ1 ~ 158.6 fs and τ2 ~ 11.38 ps. In contrast to HP, the single index model provided better fits to the experimental data of MP and the lifetime τ1 is 8.42 ps.
Through our experimental investigation, we explored how the HP molecule is different from the MP molecule. The HP molecule exhibits a dual ultrafast process and non-fluorescence emission during excitation at 400 nm, but MP exhibits just one ultrafast process and fluorescence emission. To reveal the mechanism of this phenomenon, we performed theoretical analyses using DFT and TDDFT.
Table 2 shows the calculated absorption and emission peaks of the HP and MP molecules using the TDDFT/B3LYP/TZVP method. The absorption spectrum is calculated by vertical excitation from the ground state to the S1 state and the emission spectrum is calculated by emission from the S1 state equilibrium geometry to the ground state non-equilibrium geometry. The theoretical results are in good agreement with the experimentally measured absorption and emission maxima, which suggests that the method is effective and credible. Moreover, the small Stokes shift of MP suggests that its primary de-excitation process is fluorescence.
To investigate the mechanism of de-excitation for HP, we optimized the geometric conformations of the enol (S0), enol* (S1) and keto* (S1) forms and the results are shown in Fig. 4. These optimized geometries show that the IMHB of HP is formed between C17=O2 and O1–H10 in the enol and enol* form, wherease an IMHB can be formed between C2=O1 and O2–H10 in the keto* form. In addition, the numerical values of the key geometric parameters of different electronic states can be found in supplementary Table S1. Based on the results of the geometric conformations, we found that HP exhibits an ESIPT process corresponding to the fs timescale in our experiment4. Upon excitation to enol*, the distance between O2 and H10 decreases from 1.733 Å to 1.447 Å, whereas the bond length between O1 and H10 increases from 0.988 Å to 1.063 Å, which suggests that H10 has the tendency to approach O2 and depart from O1. Eventually, HP evolves into the keto* state. The intramolecular C17=O2⋅⋅⋅H10–O1 hydrogen bond breaks, concomitantly with the formation of an intramolecular C2=O1⋅⋅⋅H10–O2 hydrogen bond. The keto* state bond angles C17C1C2 and C1C2O1 are both smaller than the corresponding ground state bond angles and the keto* state distance between O1 and O2 becomes shorter than the corresponding ground state distance, which indicates that the hydrogen-bonded quasi-aromatic chelating ring becomes smaller after excitation. Moreover, we found that the IMHB becomes short and that H10 transfers from O1 to O2. The interactions of involved atoms increase when the distances between atoms decrease. Compared to HP, MP cannot form IMHBs in the ground state or in the S1 state due to the lack of a hydrogen donor, such as the hydroxyl hydrogen atom of HP, as shown in Fig. 5. Therefore, we concluded that intramolecular hydrogen bonding in the HP molecule facilitates the ESIPT process. In contrast to the complexity of HP, the geometric conformation of MP is much simpler. There is no obvious structural alteration between S0 and S1, which indicates that MP molecules do not exhibit specific ultrafast processes such as ESIPT and twisted intramolecular charge transfer (TICT), during the de-excitation.

Schematic plots of the HP structure optimized at the S0 and S1 states in the enol (a), enol* (b) and keto* (c) forms; the length shown in the picture is the length of the hydrogen bond.
The numerical values of the key geometric parameters are shown in Table S1. (The golden atom represents C, the light green atom represents H and red atom represents O. The lengths of the hydrogen bonds are also shown.).

Schematic plots of the MP structure optimized at the S0 (a) and S1 states (b).
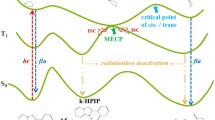
To demonstrate the details of the ESIPT process of HP and to explore the non-fluorescent mechanism of HP upon excitation to the S1 state, we calculated potential energy curves in different electronic states. The potential energy curves of HP were optimized by fixing the distance between O1 and H10 (proton transfer coordinate) at different values, which are recorded in Fig. 6. There is only one stable geometry in the S0 state, which is associated with the enol form. The potential energy curves reveal that the stable points in the S1 state correspond to the coordinates 1.063 Å (enol* form) and 1.540 Å (keto* form), respectively. HP has two stable equilibrium geometries after Frank-Condon transition to the S1 state. To achieve the equilibrium geometry of the S1 state tautomer form, H10 transfers from O1 to O2 and forms the new hydrogen bond C2=O1⋅⋅⋅H10—O2. The existence of the S1 state tautomer equilibrium geometry is direct evidence of an ESIPT process with photoexcitation to the S1 state. The calculation of the electronic excitation energy of HP shown in Table 3 indicates that the energy of the 1st and 2nd triplet states (T1 and T2) are lower than that of the S1 state and the energy of the 3rd triplet state (T3) is higher than that of the S1 state. Therefore, we also calculated the potential energy curves of the T1 and T2 states, as shown in Fig. 6. There is no cross point between S1 and T1. The occurrence of a cross between S1 and T2 is at approximately 1.5 Å to 1.6 Å, which corresponds to the coordinate of the S1 keto* state, i.e., the ISC process occurs between the S1 and T2 states. Therefore, combined with the non-fluorescent excitation to the S1 state, we concluded that the relaxation pathway of HP after ESIPT is the ISC process from the S1 keto* to the T2 state29,30,31. Comparing with the results of Weller’s28, we can find that both HP and MS have two stable equilibrium geometries after Frank-Condon transition to the S1 state. Differently, MS exhibits stepwise dual fluorescence from two stable equilibrium geometries of the S1 state and HP is non-fluorescent due to the ISC process from the S1 keto* state to the T2 state after the ESIPT process.

Calculated potential energy curves along the proton transfer coordinate (O2─H2) of HP in different electronic states.
The vibrational frequencies of the stretching vibrations of C=O and O–H groups that are involved in hydrogen bonds can provide a specific signature of the hydrogen-bonding dynamics32. In the present work, we calculated the IR spectra (B3LYP/TZVP, scaling factor 0.963033) of HP at the S0, S1 states (enol* and keto* forms). The calculated IR spectra of the ground state and S1 state in the spectral range from 1400 to 3500 cm−1 are shown in Fig. 7, which contains not only the characteristic peaks of the O1–H10 group of the S0 and S1 normal forms but also the characteristic peaks of the O2–H10 group of the S1 tautomer form. For the S1 tautomer form, the characteristic peaks at approximately 3213 cm−1, which is assigned as ν(O1–H10) in S0 and at approximately 2113 cm−1, which is assigned as ν(O1–H10) in the enol* form, disappeared, whereas a new characteristic peak appeared at approximately 2492 cm−1 and was assigned as ν(O2–H10). Thus, we can conclude that the O1–H10 bond breaks and a new O2–H10 bond forms, just as the geometry optimization shows in Fig. 4. Consequently, as auxiliary evidence, the IR spectra correlate well with the result that H10 transfers from O1 to O2 in the S1 excited state. Moreover, corresponding IR spectra of HP at the triplet state can be seen in supplementary Figure S6.

Calculated IR spectra of HP in the S0 and S1 states (normal form and tautomer form).
A comparison between the experimental and theoretical results allowed us to understand the properties of the systems more deeply and the theoretical method gave the experimental results a clear interpretation. We show the mechanism of de-excitation for HP in Fig. 8. First, the HP molecule in the S0 enol state is excited to the S1 enol* state; subsequently, an ESIPT process from the S1 enol* state to the S1 keto* state occurs at a timescale of approximately 158.6 fs. The ISC between S1 and T2 in approximately 11.38 ps gives a reasonable explanation for the non-fluorescence of HP upon excitation into the S1 state. In Fig. 3(a,b), the negative signals below 460 nm for HP molecules correspond primarily to ground state depletion. In the range from 0 to 0.1 ps, because the S1 keto* state is not formed, the characteristic peak at approximately 470 nm is attributed to the S1 enol* state. From 0.1 to 1.2 ps, the observed redshift and intensity increment of the absorption band at approximately 520 nm suggest that the population of the S1 keto* state increases with the relaxation from the hot S1 keto* to the cold S1 keto* state. Between 1.2 and 10 ps, the intensity decrement of the absorption peak at approximately 520 nm arises from the decrement of the population in the S1 keto* state due to the ISC process from S1 keto* to T2 on an approximately 10 ps timescale. This process induces the non-fluorescent property of HP upon excitation to the S1 state. After 10 ps, only the intensity and not the shape of the absorption band changes, thus; the characteristic peak at approximately 502 nm corresponds to the T2 state. Compared with the HP molecule, the de-excitation of MP is simpler, as shown in Fig. 3(c,d). Before 1 ps, the observed slight redshift and intensity increment of the absorption at approximately 502 nm suggest that the population of the S1 state increases with the relaxation from the hot S1 to the cold S1 state. After 1 ps, the dominant de-excitation process is fluorescence, which is a ns timescale process. Therefore, the lifetime of S1 for MP is much longer than that for the HP molecule. Furthermore, the lifetime of the HP τ1 was attributed to the ESIPT process and τ2 was associated with the ISC process. The lifetime of the MP τ1 corresponded to interactions between solute and solvent.

Schematic view of the de-excitation mechanism of HP.
Conclusion
In the present exploration, femtosecond transient absorption spectroscopy and DFT/TDDFT were performed to investigate the photoexcitation of HP and MP. We demonstrated that HP exhibits the ESIPT process in the S1 state and discovered the reason for the non-fluorescenec of HP upon excitation to the S1 state in CHX. Specifically, the ESIPT process happens in 158 fs, whereas the ISC process occurs on an 11.38 ps timescale. In contrast to HP, MP undergoes an 8 ps timescale process, which was attributed to interactions between the solute and solvent in the S1 state and it exhibits fluorescence upon excitation to the S1 state. The geometric conformations of HP and MP are similar, but HP differs most from MP in that it is able to form IMHBs. We concluded that the ESIPT and the ISC of HP arise from the existence of IMHBs, which induce drastic structural alterations between the S0 and S1 state.
Methods
In the present work, the ground-state and electronic excited-state geometry optimizations were performed by the DFT and TDDFT34,35,36,37,38,39,40,41,42 methods, respectively. The B3-LYP (Becke’s three-parameter hybrid exchange function with Lee-Yang-Parr gradient-corrected correlation) functional and the TZVP basis sets (the triple-ζ valence quality with one set of polarization functions) were used in our DFT and TDDFT calculations43,44,45,46. In addition, the IR intensities were derived from the gradients of the dipole moment. The electronic structure calculations were performed using the TURBOMOLE program suite45,47.
HP and MP were synthesized according to the literature48. The cyclohexane (CHX) was a super-dry, spectrum-pure, quality reagent purchased from J&K (China) and was used without further purification. The concentration of HP and MP in CHX was 5*10-6 mol/L.
The steady-state absorption and fluorescence spectra were measured with a UV 2550 UV-VIS spectrophotometer and a RF5301 fluorescence spectrophotometer (Shimadzu), respectively.
Transient absorption imaging was based on a femtosecond laser (Coherent Libra, US), which was used as the light source with a power of approximately 4W at 1 kHz repetition rate and a wavelength of 800 nm where the full width at half maximum (FWHM) was 50 fs. The fundamental laser was separated into two beams in a ratio of 9:1. The more intense beam was used for generating the second harmonic (λex = 400 nm) of the fundamental laser using a 0.5 mm BBO (β-BaB2O4, Fujian Castech Crystals Inc., China), which was applied as a pump laser for exciting samples. The energy per pump pulse at the sample was approximately 3 μJ. The other beam passed through a controlled delay line and then focused into a sapphire plate to generate sub-picosecond, super-continuum white light, which served as the probe laser. Two laser beams were incident on the sample in a 0.5 cm quartz cuvette at a small angle (θ ≤ 5°). The fused quartz sample cell was placed in the beam path and the beam diameter was 2 mm. The sensitivity range was 450–750 nm. All experiments were performed at room temperature (22 °C).
Additional Information
How to cite this article: Yin, H. et al. A novel non-fluorescent excited state intramolecular proton transfer phenomenon induced by intramolecular hydrogen bonds: an experimental and theoretical investigation. Sci. Rep. 6, 19774; doi: 10.1038/srep19774 (2016).
References
Abou-Zied, O. K. et al. Detecting local heterogeneity and ionization ability in the head group region of different lipidic phases using modified fluorescent probes. Sci. Rep. 5, 8699 (2015).
Saha, R. et al. Light driven ultrafast electron transfer in oxidative redding of green fluorescent proteins. Sci. Rep. 3, 1580 (2013).
Mutai, T., Tomoda, H., Ohkawa, T., Yabe, Y. & Araki, K. Switching of polymorph-dependent ESIPT luminescence of an imidazo[1,2-a]pyridine derivative. Angew. Chem. Int. Edit. 47, 9522–9524 (2008).
Suzuki, N. et al. A Strap Strategy for construction of an excited-state intramolecular proton transfer (ESIPT) system with dual fluorescence. Angew. Chem. Int. Edit. 53, 8231–8235 (2014).
Higashi, M. & Saito, S. Direct simulation of excited-state intramolecular proton transfer and vibrational coherence of 10-hydroxybenzo[h]quinoline in solution. J. Phys. Chem. Lett. 2, 2366–2371 (2011).
Barbara, P. F., Walsh, P. K. & Brus, L. E. Picosecond kinetic and vibrationally resolved spectroscopic studies of intramolecular excited-state hydrogen atom transfer. J. Phys. Chem. 93, 29–34 (1989).
Shiraishi, Y., Matsunaga, Y., Hongpitakpong, P. & Hirai, T. A phenylbenzoxazole-amide-azacrown linkage as a selective fluorescent receptor for ratiometric sensing of Pb(II) in aqueous media. Chem. Commun. 49, 3434–3436 (2013).
Liu, B. et al. A new ratiometric ESIPT sensor for detection of palladium species in aqueous solution. Chem. Commun. 48, 2867–2869 (2012).
Zhao, J. Z., Ji, S. M., Chen, Y. H., Guo, H. M. & Yang, P. Excited state intramolecular proton transfer (ESIPT): from principal photophysics to the development of new chromophores and applications in fluorescent molecular probes and luminescent materials. Phys. Chem. Chem. Phys. 14, 8803–8817 (2012).
Shynkar, V. V. et al. Fluorescent biomembrane probe for ratiometric detection of apoptosis. J. Am. Chem. Soc. 129, 2187–2193 (2007).
Park, S. et al. A white-light-emitting molecule: Frustrated energy transfer between constituent emitting centers. J. Am. Chem. Soc. 131, 14043–14049 (2009).
Paterson, M. J., Robb, M. A., Blancafort, L. & DeBellis, A. D. Mechanism of an exceptional class of photostabilizers: A seam of conical intersection parallel to excited state intramolecular proton transfer (ESIPT) in o-hydroxyphenyl-(1,3,5)-triazine. J. Phys. Chem. A 109, 7527–7537 (2005).
Corani, A. et al. Excited-state proton-transfer processes of DHICA resolved: From sub-picoseconds to nanoseconds. J. Phys. Chem. Lett. 4, 1383–1388 (2013).
Ma, J. et al. New excited state intramolecular proton transfer (ESIPT) dyes based on naphthalimide and observation of long-lived triplet excited states. Chem. Commun. 48, 9720–9722 (2012).
Cui, G. L., Lan, Z. G. & Thiel, W. Intramolecular hydrogen bonding plays a crucial role in the photophysics and photochemistry of the GFP chromophore. J. Am. Chem. Soc. 134, 1662–1672 (2012).
Fang, X. X. et al. Synthesized blue fluorescent protein analogue with tunable colors from excited-state intramolecular proton transfer through an N-H⋅⋅⋅N hydrogen bond. J. Phys. Chem. Lett. 5, 92–98 (2014).
Yao, H. & Funada, T. Mechanically inducible fluorescence colour switching in the formation of organic nanoparticles of an ESIPT molecule. Chem. Commun. 50, 2748–2750 (2014).
Kim, T. I., Kang, H. J., Han, G., Chung, S. J. & Kim, Y. A highly selective fluorescent ESIPT probe for the dual specificity phosphatase MKP-6. Chem. Commun. 39, 5895–5897 (2009).
Tang, K. C. et al. Fine tuning the energetics of excited-state intramolecular proton transfer (ESIPT): White light generation in a single ESIPT system. J. Am. Chem. Soc. 133, 17738–17745 (2011).
Xu, Y. Q. et al. Switchable and selective detection of Zn2+ or Cd2+ in living cells based on 3′-O-substituted arrangement of benzoxazole-derived fluorescent probes. Chem. Commun. 50, 7514–7516 (2014).
Zhang, J. Y. & Guo, W. A new fluorescent probe for gasotransmitter H2S: high sensitivity, excellent selectivity and a significant fluorescence off-on response. Chem. Commun. 50, 4214–4217 (2014).
Paul, B. K. & Guchhait, N. 1-Hydroxy-2-naphthaldehyde: A prospective excited-state intramolecular proton transfer (ESIPT) probe with multi-faceted applications. J. Lumin. 132, 2194–2208 (2012).
Chevalier, K. et al. ESIPT and photodissociation of 3-hydroxychromone in solution: Photoinduced processes studied by static and time-resolved UV/Vis, fluorescence and IR spectroscopy. J. Phys. Chem. A 117, 11233–11245 (2013).
Chou, P. T. et al. Excited-state intramolecular proton transfer in 10-hydroxybenzo[h]quinoline. J. Phys. Chem. A 105, 1731–1740 (2001).
Mitra, S., Tamai, N. & Mukherjee, S. Intramolecular proton transfer in 4-methyl-2,6-diformyl phenol and its derivative studied by femtosecond transient absorption spectroscopy. J. Photoch. and Photobio. A 178, 76–82 (2006).
Yin, H., Shi, Y. & Wang, Y. Time-dependent density functional theory study on the excited-state intramolecular proton transfer in salicylaldehyde. Spectrochim. Acta, Part A 129, 280–284 (2014).
Lan, X., Yang, D. P., Sui, X. & Wang, D. D. Time-dependent density functional theory (TD-DFT) study on the excited-state intramolecular proton transfer (ESIPT) in 2-hydroxybenzoyl compounds: Significance of the intramolecular hydrogen bonding. Spectrochim. Acta, Part A 102, 281–285 (2013).
Weller, A. Innermolekularer protonenübergang im angeregten zustand. Ber. Bunsenges. Phys. Chem. 60, 1144–1147 (1956).
Zhao, G. J. & Han, K. L. Role of intramolecular and intermolecular hydrogen bonding in both singlet and triplet excited states of aminofluorenones on internal conversion, intersystem crossing and twisted intramolecular charge transfer. J. Phys. Chem. A 113, 14329–14335 (2009).
Menning, S. et al. Bridged tolanes: A twisted tale. J. Org. Chem. 79, 6571–6578 (2014).
Pina, J., Pinheiro, D., Nascimento, B., Pineiro, M. & de Melo, J. S. S. The effect of polyaromatic hydrocarbons on the spectral and photophysical properties of diaryl-pyrrole derivatives: an experimental and theoretical study. Phys. Chem. Chem. Phys. 16, 18319–18326 (2014).
Samant, V. et al. Ultrafast intermolecular hydrogen bond dynamics in the excited state of fluorenone. J. Phys. Chem. A 109, 8693–8704 (2005).
Lukeš, V., Aquino, A. & Lischka, H. Theoretical study of vibrational and optical spectra of methylene-bridged oligofluorenes. J. Phys. Chem. A 109, 10232–10238 (2005).
Cong, L., Yin, H., Shi, Y., Jin, M. X. & Ding, D. J. Different mechanisms of ultrafast excited state deactivation of coumarin 500 in dioxane and methanol solvents: experimental and theoretical study. Rsc. Adv. 5, 1205–1212 (2015).
Wang, Y., Shi, Y., Cong, L. & Li, H. TDDFT study of twisted intramolecular charge transfer and intermolecular double proton transfer in the excited state of 4′-dimethylaminoflavonol in ethanol solvent. Spectrochim. Acta, Part A 137, 913–918 (2015).
Wang, Y., Yin, H., Shi, Y., Jin, M. & Ding, D. Ground-state and excited-state multiple proton transfer via a hydrogen-bonded water wire for 3-hydroxypyridine. New J. Chem. 38, 4458–4464 (2014).
Zhao, G. J. & Han, K. L. Early time hydrogen-bonding dynamics of photoexcited coumarin 102 in hydrogen-donating solvents: Theoretical study. J. Phys. Chem. A 111, 2469–2474 (2007).
Zhao, G. J. & Han, K. L. Effects of hydrogen bonding on tuning photochemistry: Concerted hydrogen-bond strengthening and weakening. ChemPhysChem 9, 1842–1846 (2008).
Song, P., Ding, J. X. & Chu, T. S. TD-DFT study on the excited-state proton transfer in the fluoride sensing of a turn-off type fluorescent chemosensor based on anthracene derivatives. Spectrochim. Acta, Part A 97, 746–752 (2012).
Song, P. & Ma, F. C. Intermolecular hydrogen-bonding effects on photophysics and photochemistry. Int. Rev. Phys. Chem. 32, 589–609 (2013).
Sun, M. et al. Remotely excited Raman optical activity using chiral plasmon propagation in Ag nanowires. Light. Sci. Appl. 2, e112 (2013).
Li, Y., Pullerits, T., Zhao, M. & Sun, M. Theoretical characterization of the PC60BM: PDDTT model for an organic solar cell. J. Phys. Chem. C 115, 21865–21873 (2011).
Whitten, J. L. Coulombic potential energy integrals and approximations. J. Chem. Phys. 58, 4496–4501 (1973).
Dunlap, B. I., Connolly, J. & Sabin, J. On some approximations in applications of Xα theory. J. Chem. Phys. 71, 3396–3402 (1979).
Ahlrichs, R., Bär, M., Häser, M., Horn, H. & Kölmel, C. Electronic structure calculations on workstation computers: The program system turbomole. Chem. Phys. Lett. 162, 165–169 (1989).
Vahtras, O., Almlöf, J. & Feyereisen, M. W. Integral approximations for LCAO-SCF calculations. Chem. Phys. Lett. 213, 514–518 (1993).
Furche, F. & Ahlrichs, R. Adiabatic time-dependent density functional methods for excited state properties. J. Chem. Phys. 117, 7433–7447 (2002).
Zhou, Y., Wang, F., Kim, Y., Kim, S. J. & Yoon, J. Cu2+ -selective ratiometric and “off-on” sensor based on the rhodamine derivative bearing pyrene group. Org. Lett. 11, 4442–4445 (2009).
Acknowledgements
This work was supported by National Basic Research Program of China (973 Program) (2013CB922204) and the National Natural Science Foundation of China (No. 11174106, 10974069). We also acknowledge the High Performance Computing Center of Jilin University, China.
Author information
Authors and Affiliations
Contributions
H.Y. and H.L. carried out the experiment. H.Y. performed the theoretical calculation. Y.S. gave the idea of our investigation. G.X., C.Y.R. and H.M.W. executed sample preparation. M.X.J. gave us the technical support. D.J.D. gave us the investigation guidance and helpful discussion.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Yin, H., Li, H., Xia, G. et al. A novel non-fluorescent excited state intramolecular proton transfer phenomenon induced by intramolecular hydrogen bonds: an experimental and theoretical investigation. Sci Rep 6, 19774 (2016). https://doi.org/10.1038/srep19774
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep19774
- Springer Nature Limited
This article is cited by
-
Theoretically unveiling the effect of solvent polarities on ESDPT mechanisms and photophysical properties of hydroxyanthraquinones
Journal of Molecular Modeling (2022)
-
Elaborating the mechanism of a highly selective fluorescent ‘turn-on’ probe to detect the group IIIA ions: a detailed time-dependent density functional theory study
Theoretical Chemistry Accounts (2022)
-
Tunable electron transfer rate in a CdSe/ZnS-based complex with different anthraquinone chloride substitutes
Scientific Reports (2019)
-
A theoretical prediction about harnessing ESPT process for HBO derivatives
Structural Chemistry (2018)
-
Synthesis, Spectra, and Theoretical Investigations of 1,3,5-Triazines Compounds as Ultraviolet Rays Absorber Based on Time-Dependent Density Functional Calculations and three-Dimensional Quantitative Structure-Property Relationship
Journal of Fluorescence (2018)