
Abstract
Newly exposed volcanic substrates contain negligible amounts of organic materials. Heterotrophic organisms in newly formed ecosystems require bioavailable carbon and nitrogen that are provided from CO2 and N2 fixation by pioneer microbes. However, the knowledge of initial ecosystem developmental mechanisms, especially the association between microbial succession and environmental change, is still limited. This study reports the unique process of microbial succession in fresh basaltic ash, which was affected by long-term exposure to volcanic sulfur dioxide (SO2). Here we compared the microbial ecosystems among deposits affected by SO2 exposure at different levels. The results of metagenomic analysis suggested the importance of autotrophic iron-oxidizing bacteria, particularly those involved in CO2 and N2 fixation, in the heavily SO2 affected site. Changes in the chemical properties of the deposits after the decline of the SO2 impact led to an apparent decrease in the iron-oxidizer abundance and a possible shift in the microbial community structure. Furthermore, the community structure of the deposits that had experienced lower SO2 gas levels showed higher similarity with that of the control forest soil. Our results implied that the effect of SO2 exposure exerted a selective pressure on the pioneer community structure by changing the surrounding environment of the microbes.
Similar content being viewed by others


Introduction
Volcanic eruptions offer numerous opportunities to increase our understanding of terrestrial ecosystem formation. The first key step of the formation process is the input of biologically transformed atmospheric carbon and nitrogen to the ecosystem by pioneer microorganisms1,2,3. Most of the early microbial colonizers probably have CO2 and N2 fixation capabilities and they can grow by utilising photo- or chemolithotrophic energy-generation systems (i.e., by oxidation of inorganic substrates: e.g., S0, Fe2+, CO and H2). As a result, their activity promotes the growth of diverse heterotrophic communities, which are associated with the physicochemical changes of surrounding microenvironments4,5. However, knowledge of the development process of their ecosystems and the mechanisms in the initial stage of the formation is still limited. This study investigated the succession of the early microbial communities in fresh volcanic deposits (<10 years old) which were exposed to volcanic sulfur dioxide (SO2) over long periods. The aim was to develop an understanding of the association between microbial succession and changing environmental conditions.
The island of Miyake (our study site, Miyake-jima; Fig. 1a,b) released a large amount of volcanic SO2 gas from the newly formed crater after the eruption of Mt. Oyama in 2000. The daily average rates of SO2 gas emission peaked at 54 kt d−1 in December 2000 and decreased to 7 kt d−1 by the end of 20026. Although the amount of SO2 emission had further decreased over the past ten years, it was still detectable in 2014 (see details in the Materials section and Supplementary Table S1). The volcanic activity destroyed plant ecosystems through SO2 exposure and volcanic ash deposition and buried mature soil ecosystems7. Likewise, the newly deposited volcanic ash was acidified by exposure to SO2 gas8.

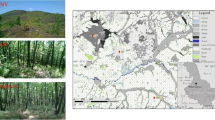
Map of the study site, locations and photographs of sampling sites.
(a) Map showing the location of Miyake-jima on the western rim of the Pacific Ocean. (b) Three study sites on the island. Colour plots show each site (red, OY; blue, IG1; green, CL). (c–e) Photographs of each site (c, site OY in 2005; d, site IG1 in 2009; e, site CL in 2005). (f) SO2 data logger. The map was created using Adobe Illustlater CS6.
Previously, we reported that nitrogen-fixing obligate autotrophic iron-oxidizing bacteria were the dominant group in the bacterial community structure in young deposits of volcanic ash from Miyake-jima (<6.6 years old)9. However, the early colonization of iron oxidizers was not found to be uniform among newly formed substrates in other terrestrial volcanoes. For example, Hawaiian lava and Mt. Pinatubo lahar deposits showed the importance of different types of chemolithotrophs as pioneer organisms (i.e., CO- and H2-oxidizing bacteria) which utilise atmospheric trace gas as energy substrates3,10,11. Additionally, a study of the Fimmvörðuháls lava flow of the Eyjafjallajökull volcano on Iceland reported the dominance of Betaproteobacteria related to non-phototrophic diazotrophs and chemolithotrophs such as Herbaspirillum and Thiobacillus, respectively12. Thus, the dominance of acidophilic iron-oxidizers as pioneer organisms is a unique characteristic of fresh volcanic deposits from Miyake-jima.
The goal of this study was to gain deeper insight into the succession of structure and function of pioneer microbial communities on volcanic ash deposits from Miyake-jima. The present study adopted metagenomic analysis of the microbial ecosystem to examine in-depth entire environmental genomes and thus to obtain more succession results with reduced bias and to incorporate additional data such as microbial functional information.
Results and Discussion
Chemical characteristics of the fresh volcanic ash deposits
We analysed the volcanic ash deposits derived from a site heavily affected by SO2 exposure (site OY; Fig. 1b,c). Time-series samples were taken 3.5, 6.6 and 9.5 years after the eruption (3.5-, 6.6- and 9.5-OY volcanic deposit [VD], respectively). Those datasets were compared with an 8.5-year-old deposit derived from a less-affected site (site IG1; IGVD; Fig. 1b,d) and with forest soil derived from an undamaged site on the island (control sample)13,14 (site CL; CLS; Fig. 1b,e). The impact of long-term SO2 exposure had decelerated the recovery of vegetation and had acidified the deposits7,8. This effect was heavier on the leeward side than on the windward side of Mt. Oyama (OY and IG1 sides, respectively) because the monsoon wind over the island (i.e., westerly and north-easterly winds) affected the direction of the volcanic gas flow7,15. Reference for the volcanic exposure frequency at each site, SO2 gas monitoring data suggested that the detection frequency of atmospheric SO2 gas was the highest in site OY (Supplementary Table S2).
Time-series samples on site OY (OYVDs) were characterised by lower pH (3.4–3.9), as well as by total carbon (TC) content and nitrogen (TN) content (≤0.5 and ≤0.1 g kg−1, respectively) higher than those on sites IGVD and CLS (Table 1). The Pearson’s correlation coefficient (R) for each dataset of chemical properties suggested high correlations between ferrous ion (Fe2+) concentration and pH, as well as SO42− concentration and pH (Fig. 2a). The concentration of Fe2+, which could be considered a critical factor affecting the abundance of iron oxidizers, was negatively correlated with the pH of the deposit (R = 0.8196, p = 0.089); The instability of ferrous iron above pH 4 supported this finding16,17,18. The clear inverse correlation between SO42− concentration and pH (R = 0.9769, p = 0.004) suggested the acidification of the deposits caused by SO42− accumulation due to SO2 exposure, as well as previous reports8,19. Therefore, these relationships suggest that the chemical properties of the OYVD deposits had been affected by SO2 gas exposure.

Relationships among chemical compositions and bacterial numbers.
(a) Relationship between pH and SO42− (blue) or Fe2+ (orange) concentration and (b) Fe2+ concentration and copy number of bacterial 16S rRNA gene (grey) or L. ferrooxidans gyrB gene (red). Error bars, s.d. of triplicate results. Result of the statistical significance test (R, correlation coefficient; R2, determination coefficient of the regression model).
Microbial community succession on the fresh volcanic ash deposits
Prokaryotic 16S rRNA gene (16S rDNA) sequences were obtained from the metagenomic datasets (Supplementary Table S3). The microbial community structure indicates that the most abundant genus was Leptospirillum, which accounted for 22% of the total 16S rDNA gene sequences in 3.5-OYVD (Fig. 3 and Supplementary Table S4). However, this ratio decreased in 6.6-OYVD (14%). A similar tendency to that of Leptospirillum was observed in the second most abundant genus, Acidithiobacillus; the percentages of this group were 17 and 7% in 3.5- and 6.6-OYVDs, respectively. Moreover, we found that Leptospirillum ferrooxidans and Acidithiobacillus ferrooxidans, obligate autotrophic acidophilic nitrogen-fixing bacteria which can utilize ferrous iron as an energy source, were the most abundant species in those genera in the fresh volcanic ash deposits (data not shown). These results were consistent with Fujimura et al. (2012), except for the non-decreasing relative abundance of Acidithiobacillus between 3.5- and 6.6-OYVDs (16 and 20%, respectively). Incidentally, this study revealed diverse communities more than that in our previous report (PCR-clone library analysis), because community analysis using metagenomic dataset enabled detection of many small groups by obtaining a large number of sequences (average: 470) with less bias. Meanwhile, the relative abundances of Leptospirillum and Acidithiobacillus in 9.5-OYVD (3 and 4%, resp.) and IGVD deposits were low (0 and 1%, resp.; Fig. 3 and Supplementary Table S4). Interestingly, IGVD community seems to have the mosaic structure of the 9.5-OYVD and CLS communities; but the dissimilarity index (Fig. 4, discussed below) indicated that the IGVD community was more similar to the CLS community than to the 9.5-OYVD community (Fig. 4 and Supplementary Fig. S1). These results suggested that the initial stage of the microbial community could easily change because of age or changes in the environment. Furthermore, the community structure of CLS showed a high abundance of Acidobacteria and Actinobacteria groups, but Acidithiobacillus and Leptospirillum were undetectable (Fig. 3 and Supplementary Table S4). The CLS community was similar with our previous study in 20059 (CLS of this study was taken in 2007) and it suggests that the community structure of the forest soil is more stable than is the volcanic ash community.

Structure of the prokaryotic microbial community at the genus level.
The relative abundance of each genus was calculated as the ratio of individual 16S rRNA gene assigned sequences to the total number of sequences. Colour legends of major genera are shown; those of minor genera are shown as original data in Supplementary Table S4.

Colour-mapped matrix table of Bray−Curtis dissimilarity indices.
Indices were calculated by using microbial abundance patterns of genus-assigned reads. The dissimilarity indices indicate that the lower number is more similar (i.e., not dissimilar) between samples. The colour indicator is shown in the figure.
Leptospirillum and Acidithiobacillus are commonly dominant in highly acidic environments such as acid mine drainages16,20,21, because such environments are rich in their energy substrates (e.g., sulfide and Fe2+)22. The abundance of these organisms in the OYVDs was also probably affected by environmental conditions such as Fe2+ concentration and acidity; therefore, we compared the relationship between the population size of Leptospirillum (a strict iron oxidizer) and the chemical properties of the deposits (Table 1). Quantitative PCR analysis of the L. ferrooxidans gene encoding for DNA gyrase subunit B (gyrB) showed different population sizes among the volcanic ash deposits. The highest correlation was obtained with the Fe2+ concentration (Fig. 1b; R = 0.990, p = 0.01) and a negative correlation was obtained with pH and SO42− concentration (R = 0.797 and 0.308, p = 0.20 and 0.69, respectively). Meanwhile, the quantity of 16S rDNA showed very weak correlation with Fe2+ concentration (Fig. 1b; R = 0.445, p = 0.56), but it was correlated with SO42− concentration (negative correlation; R = 0.752; p = 0.248). These results imply that the concentrations of inorganic energy substrates for the dominant groups could be a key environmental factor for the microbial community succession in the fresh volcanic ash deposit.
Archaeal contribution to the early ecosystem development in fresh volcanic ash deposits from Miyake-jima seems to be very small because their 16S rDNA sequences in 3.5- and 6.6-OYVDs were undetectable in the community analysis (Fig. 3 and Supplementary Table S4). This result confirms that the PCR assay of the archaeal 16S rDNA amplification products were undetectable despite the use of some archaeal universal primer sets (data not shown). The archaeal 16S rDNA sequence reads were detected in 9.5-OYVD, IGVD and CLS, which respectively accounted for 1.3, 3.6 and 6.5% of the total 16S rDNA (Supplementary Table S4).
Photosynthetic microbes were also not detected in the volcanic ash deposits (Fig. 3 and Supplementary Table S4). Cyanobacterial photosynthesis is one of the important activities in the formation of initial soil ecosystems, which contribute to carbon and nitrogen accumulation and soil aggregate formation in such ecosystems2,5. However, previous reports indicate that sulfite inhibits photosynthetic activity23,24,25; therefore, SO42− accumulation due to SO2 exposure probably repressed the colonization of the photosynthetic microbes and SO42− may therefore be involved in the selection of pioneer communities in the OYVDs.
Bray−Curtis dissimilarities calculated from the relative abundance of each assigned genus (Fig. 4) indicate the similarity of the community composition patterns among samples. The results show the highest similarity between IGVD and CLS and the lowest similarity between 6.6-OYVD and CLS (Fig. 4 and Supplementary Fig. S1; dissimilarity values of 0.36 and 0.78, respectively). Interestingly, the IGVD community was more similar to CLS than to 9.5-OYVD despite the deposit being younger than 9.5-OYVD. If the CLS community was a mature ecosystem, then the IGVD community would had been more developed than the oldest OYVD community. This result suggests that SO2 exposure leads to differences in the community composition by changing the environmental conditions.
Carbon- and nitrogen-fixing microbial communities and their succession
Microbial communities with the selected genes responsible for CO2 fixation were analysed. These genes are related to the Calvin–Benson–Bassham cycle (CBB; rbcL) and the reductive tricarboxylic acid cycle (rTCA; korA, frdA and porA). The relative abundances of these genes, particularly that of the rbcL gene, were higher in OYVDs than in IGVD and CLS (Fig. 5a). Taxonomic assignments of rbcL genes showed the predominance of Acidithiobacillus in OYVDs (24–38% of total rbcL genes; Fig. 5b and Supplementary Table S5). Another abundant iron-oxidizing bacterium, Leptospirillum, lacks the CBB system but possesses the rTCA system. The porA gene sequences derived from Leptospirillum accounted for 70% of the total porA genes in 3.5-OYVD and 30% of genes in 6.6- and 9.5-OYVDs (Fig. 5b). Archaeal rbcL and porA gene abundances showed the highest total percentages in IGVD and 9.5-OYVD, respectively (Supplementary Table S5). Furthermore, cyanobacterial CO2 fixation genes were undetectable in OYVDs but were present in IGVD and CLS.

Relative abundances of carbon-fixation and nitrogen-cycling genes and taxonomic assignments of carbon and nitrogen fixers.
(a) Relative abundances of targeted genes were calculated as the ratio of predicted ORFs in each sample to the total number of ORFs (Supplementary Table S3). Colour legends are shown. (b) Relative abundances of the taxonomic assignments of the rbcL, porA and nifH genes were calculated as the ratio of each assigned gene sequence to the total number of sequences. Colour legends of major genera are shown in this figure; those of minor genera are shown as original data in Supplementary Table S5.
The abundance of the nitrogen fixation gene (nifH) was one of the striking features of OYVDs (Fig. 5a), accounting for ~50% of nitrogen-cycling-related genes. A comparison of the percentage of nifH and the combined abundance of denitrification-related genes (narG, nirK/S, norB and nosZ) showed the relatively low abundance of nifH in IGVD and its undetectable concentrations in CLS. The latter result might had been caused by the relatively low coverage and the high diversity of genes in the CLS metagenome. Taxonomic assignment of the nifH gene indicates that while both Acidithiobacillus and Leptospirillum accounted for as much as 80% of the sequences in 3.5-OYVD, they accounted for lower percentages in 6.6- and 9.5-OYVDs (Fig. 5b and Supplementary Table S5). In these sites, the composition of microbes shifted from an obligate chemolithotrophic community to heterotrophic and/or facultative lithotrophic diazotrophs such as Burkholderia, Beijerinckia and Leptothlix. These results thus supported the significance of iron-oxidizing bacteria in the early ecosystem and confirm that the process of succession differs between nitrogen- and carbon-fixing microbial communities.
The box-plot of Bray−Curtis dissimilarity indices showed high similarity of functional gene compositions among samples (indices <0.5); meanwhile, the community compositions among samples were more diverse (Supplementary Fig. S2). This result implies that the essential housekeeping genes for the maintenance of basic cellular functions in most of the microbes were universally abundant in each metagenome26,27. However, non-housekeeping genes such as those for degradation of organic contaminants were also detectable in OYVDs and IGVD, occurring at ratios similar to those in CLS (data not shown). This detection suggest that most of the potential function, which is the same as for the forest soil, is already present in the early microbial ecosystem even with the lack of a taxonomically diverse community. Nonetheless, the relative abundance of distinguishing non-housekeeping genes (e.g., rbcL) could probably be affected by the abundance of each distinguishing taxonomic group.
Our metagenomic analysis illuminates the process of succession in early microbial ecosystems in acidic fresh volcanic ash deposits, despite the limitations inherent to our study, such as the statistical significance due to the limited number of samples. We observed a unique pioneer community and its succession in less than 10 years on volcanic ash deposits at a fixed site, which seems to reflect the peculiar environment of Miyake-jima. One possible reason for the short-term microbial succession is that environmental factors easily affected the community on the deposit because the volcanic ash deposit had not formed complex structures such as soil aggregates (i.e., smaller buffering effect and less-complex habitats than those of matured soil). Therefore, the change in the volcanic SO2 effect had an important impact on the initial stage of ecosystem formation process in Miyake-jima.
Materials and Methods
Materials
Miyake-jima is a basaltic volcanic island located on the Izu-Mariana arctic ridge in the Pacific Ocean, approximately 180 km south of Tokyo (34°05′ N, 139°31′E; Fig. 1a). Mt. Oyama, the active volcano situated in the centre of the island, last erupted in 2000. This eruption ejected a large amount of volcanic SO2 gas from a newly created summit caldera, contributing a significant amount to the global rate of SO2 gas emission from non-erupting volcanoes (estimated at 26 kt d−1)6. The daily emission of SO2 gas was ~48,000 t d−1 immediately after the eruption19; it then declined slowly after the eruption (Supplementary Table S1). Approximately 283 t d−1 was still being emitted in 2014 (Japan Meteorological Agency, http://www.data.jma.go.jp/svd/vois/data/tokyo/320_Miyakejima/320_So2emission.htm). The SO2 gas had a massive impact on the island ecosystem, destroying ~60% of island vegetation and affecting vegetation recovery7. In addition, the volcanic ash deposits were acidified as a result of SO42− absorption due to SO2 gas exposure8. Kato et al. (2002)8 reported that the fresh volcanic ash showed high content of fine sand (36–76%), acidity [pH (H2O) 3.1–4.0] and high amounts of exchangeable Ca2+ (33.5–115 cmolc kg−1) and Al3+ (0.8–10.2 cmolc kg−1).
The volcanic ash deposits of the 2000 eruption were taken from an unvegetated site, OY (34°04.69′N, 139°31.04′E; 553 m a.s.l.; Fig. 1b), on February 2004 (deposit age, 3.5 years; 3.5-OYVD), March 2007 (6.6 years; 6.6-OYVD) and February 2010 (9.5 years; 9.5-OYVD), as described previously9. Site OY had no vegetation because of exposure to volcanic gas (Fig. 1c). Another study site, IG1 (34°05.37′N, 139°30.84′E; 547 m a.s.l.; Fig. 1b), was less damaged by volcanic gas exposure than was OY. The gas exposure frequency was lower than that of OY (Supplementary Table S2) because of the position of IG1, which is mostly on the windward side of the island7,8. Site IG1 supported a patchy vegetation of a pioneer grass, Miscanthus condensatus and its deposit had higher pH and lower SO42− concentrations than those of the OY deposit (Fig. 1d and Table 1). The volcanic deposits were taken from an unvegetated spot in July 2009 (deposit age, 8.9 years). The forest soil, which had been undisturbed for over 800 years7,13, was sampled from a site at the foot of the mountain in March 2007 (site CL; 34°06.68′N, 139°30.06′E; 97 m a.s.l.; Fig. 1b). The vegetation of site CL was a climax forest13,14 (Fig. 1e). The chemical properties of the soil sample are described in Table 1. Each year, volcanic deposits (approx. 1 kg) were sampled at depths between 0.5 and 50 cm from the surface at almost the same point at sites OY and IG1 each. The deposits were mixed in sterile plastic bags and immediately stored at on ice. The number of sampling points was limited because we needed to consider the direction of SO2 gas flow and because we had limited time for sampling. Samples were divided into two portions and kept at 4 °C and at −20 °C until bacteriological analysis and DNA extraction, respectively.
Volcanic SO2 gas monitoring
Volcanic SO2 gas concentrations, humidity and temperature were monitored and recorded from December 2011 to April 2012 using a Gasman monitor (Crowcon Detection Instruments Ltd, Abingdon, UK) and a Hobo data logger (Onset Computer Corporation, Cape Cod, MA, USA), respectively (Fig. 1f). Recorded datasets were collected from the fields on December 2011, February 2012 and April 2012. Equipments were installed at 34°04.69′N, 139°30.96′E (site OY), 34°05.37′N, 139°30.83′E (site IG1) and 34°06.71′N, 139°30.08′E (site CL). Atmospheric SO2 concentration was measured for one minute every hour. We then converted the dataset to counts data for SO2 detection in each detection range, i.e., 1–2, 2–3, 3–4, 4–5 and ≥5 ppm. However, the total number of recording counts varied (including 0 ppm; Supplementary Table S2) because of gas monitor failure in OY and IG1 due to exposure to high concentrations of SO2 gas. SO2 detection counts data at each concentration were calculated as a percentage of the total number of SO2 detection counts.
Chemical analyses
Measurements of pH, TC content and TN content were performed as described previously9. Total ferrous ion concentration was determined by the 2,2′-dipyridyl method using 1 M sodium acetate trihydrate solution (pH 4.8)28,29. Sulfate was extracted with ultrapure water at a sample to water ratio of 1:2.5 (by mass) in a reciprocal shaker for 1 h at 250 rpm at room temperature. The slurry was centrifuged at 10,000 × g for 3 min and the supernatant was filtered through a cellulose filter paper (No. 5B; Advantec, Tokyo, Japan) and then through a cellulose ester membrane (0.45 μm; Advantec). The concentration of SO42− was determined by the turbidimetric method based on the reaction of sulphate with barium chloride, resulting in the precipitation of barium sulphate30.
Extraction of DNA of the microbial community
Before DNA extraction, duplicate samples containing 5 g of the deposit (a total of 10 g deposit was used for the extraction) of 3.5- 6.6- and 9.5-OYVD and IGVD were washed with 800 mL of sterilised ultrapure water to prevent undesired precipitation after ethanol treatment during DNA extraction. Total genomic DNA was extracted from samples according to the protocol of Zhou et al. (1996)31, which was based on lysis using a high-salt extraction buffer (1.5 M NaCl, pH 8.0) and extended heating (65 °C for 2–3 h) of the sample suspension in the presence of sodium dodecyl sulphate, hexadecyltrimethyl ammonium bromide and proteinase K. Total genomic DNA of CLS (5 g fresh weight, in duplicate) was extracted by using an ISOIL bead-beating kit (Nippon Gene Co. Ltd., Tokyo, Japan) according to the manufacturer’s instructions. We used different extraction methods for CLS because the former method cannot remove humic substances from the solution. Double stranded DNA was quantified by using a Quant-iT PicoGreen assay kit (Invitrogen Life Technologies, Carlsbad, CA, USA).
Quantitative real-time PCR (q-PCR)
The absolute quantification of bacterial 16S rDNA and the L. ferrooxidans gyrB gene encoding the B subunit of DNA gyrase were analysed through the standard curve method of the qPCR assay. The primer sets used for each gene were Qbac-10F (5′-CAGTTTGATCCTGGCTCAG-3′)9 and bac-907R (5′-CCGTCAATTCCTTTRAGTTT-3′)32 for the 16S rDNA and Qlfe-42F (5′-CATCAGGGTTCTGGAGGGTC-3′) and lfe-574R (5′-GGGCAAGAGTGTCAAAAAGG-3′) for the L. ferrooxidans gyrB gene. The Qlfe-42F/lfe-574R primer set was designed in this study on the basis of the complete genome sequence of L. ferrooxidans strain C2-333 by using Primer3Plus software34. The specificity of this primer set was confirmed by in silico PCR assay (Primer-BLAST, National Center for Biotechnology Information (NCBI)) and PCR-clone library analysis9 of the gyrB gene amplification product using the DNA sample of 3.5-OYVD (data not shown). Each forward primer was labelled at the 5′-end with quenching fluorescence dye (Qprobe; J-bio21, Tsukuba, Japan). Details of the Qprobe method and the reaction mixture have been described by Nishizawa et al. (2008)35. Triplicate q-PCR assays for the gyrB gene were performed with an iCycler PCR (Bio-Rad) under the following conditions: 2 min at 95 °C followed by 45 cycles at 95 °C (30 sec), 54 °C (45 sec) and 72 °C (1.5 min) for 16S rDNA; and 2 min at 95 °C followed by 45 cycles at 95 °C (30 sec), 56 °C (45 sec) and 72 °C (1.5 min). The control DNA samples for 16S rDNA and gyrB genes, which were used to generate the standard curves, were 0.1 μg of genomic DNA of Escherichia coli str. K-12 substr. MG1655 and L. ferrooxidans str. C2-3, respectively. Both DNA samples were used in ten-fold dilution series from 104 to 10. The regression of these standard samples showed a high determination coefficient (R2 = 0.996 for 16S; 0.999 for gyrB). Multiple negative controls (nuclease-free water) were also tested in each run to confirm the non-existence of pseudo-positive products. The means of the triplicate data set were used in Fig. 2b.
Sequencing, sequence annotation and functional assignment for metagenomic analysis
Approximately 5 μg of each DNA aliquot was sequenced on a Roche 454 GS FLX Titanium system. Artificially redundant sequences were removed by using a 454 replicate filter.
In the extraction of prokaryotic 16S rDNA sequences for prokaryotic community analysis, we constructed an in-house 16S rDNA database by using the NCBI complete and draft genome sequences for 16S rDNA sorting. The bacterial and archaeal 16S rDNA sequences were assigned using the in-house database by BLASTN programme36,37. The assigned reads were extracted with threshold values which were set to an E value of ≤10−5 and a minimum aligned length of ≥100 bp. Extracted 16S rDNA sequences were annotated by BLASTN by using the Ribosomal Database Project database (RDP, version 10.27)38. Thresholds for the grouping of sequences at phylum and genus levels were set to intra-sequence identities of 76% and 97%, respectively39.
Protein-encoding regions (open reading frames (ORFs)) were predicted by using MetaGeneAnnotator software40. The predicted ORFs were annotated by BLASTP by using the Kyoto Encyclopedia of Genes and Genomes (KEGG) databases41 and an E value of ≤10−5.
Carbon-fixation and nitrogen-cycling gene sequences were chosen from the sequences with the assigned KEGG orthology identifier (K-number). These sequences were normalised by the average complete length of each gene by using data from the NCBI complete genome database, before the relative abundance with respect to the total number of predicted ORFs was calculated.
The members of carbon and nitrogen fixers using the rbcL, porA and nifH genes with assigned sequences were predicted with BLASTP using the NCBI non-redundant database.
Statistical analysis
R, R2 and regression p values of the regression model were calculated by using StatPlus:mac LE (AnalystSoft Inc., CA, USA).
The relative abundance data of the microbial community structure and functional gene annotation for each sample were used in the calculation of the Bray–Curtis dissimilarity index by using R package vegan (http://cc.oulu.fi/~jarioksa/softhelp/vegan/html/vegdist.html). Dissimilarity indices of the microbial community structure were visualised as the distance of hierarchical clusters using by R package pvclust42. A box plot was generated to compare the functional gene dissimilarity indices among different hierarchal functional categories from the SEED-43, COG-44 and KEGG-annotated datasets via the MG-RAST server45 (http://metagenomics.anl.gov/?page=Home). The KEGG functional hierarchy was determined from the KEGG BRITE hierarchy (http://www.genome.jp/kegg/kegg3b.html).
Additional Information
Accession codes: The pyrosequencing reads were deposited in the DDBJ Sequence Read Archive database under accession number DRA001199.
How to cite this article: Fujimura, R. et al. Unique pioneer microbial communities exposed to volcanic sulfur dioxide. Sci. Rep. 6, 19687; doi: 10.1038/srep19687 (2016).
References
Chadwick, O. A., Derry, L. A., Vitousek, P. M., J., H. B. & O., H. L. Changing sources of nutrients during four million years of ecosystem development. Nature 397, 491–497 (1999).
Crews, T. E., Kurina, L. M. & Vitousek, P. M. Organic matter and nitrogen accumulation and nitrogen fixation during early ecosystem development in Hawaii. Biogeochemistry 52, 259–279 (2001).
King, G. M. Contributions of atmospheric CO and hydrogen uptake to microbial dynamics on recent Hawaiian volcanic deposits. Appl. Env. Microbiol. 69, 4067–4075 (2003).
Mackelprang, R. et al. Metagenomic analysis of a permafrost microbial community reveals a rapid response to thaw. Nature 480, 368–371 (2011).
Schmidt, S. K. et al. The earliest stages of ecosystem succession in high-elevation (5000 metres above sea level), recently deglaciated soils. Proc. Biol. Sci. 275, 2793–2802 (2008).
Kazahaya, K. et al. Gigantic SO2 emission from Miyakejima volcano, Japan, caused by caldera collapse. Geology 32, 425–428 (2004).
Kamijo, T. & Hashiba, K. Island ecosystem and vegetation dynamics before and after the 2000-year eruption on Miyake-jima Island, Japan, with implications for conservation of the island’s ecosystem. Glob. Env. Res. 7, 69–78 (2003).
Kato, T., Higashi, H., Kamijo, T. & Tamura, K. Several chemical and mineralogical properties of volcanic ash samples erupted in 2000 from Miyake island. Pedrogist 46, 14–21 (in Japanese with English summary) (2002).
Fujimura, R. et al. Analysis of early bacterial communities on volcanic deposits on the island of Miyake (Miyake-jima), Japan: a 6-year study at a fixed site. Microbes Environ. 27, 19–29 (2012).
Nanba, K., King, G. M. & Dunfield, K. Analysis of facultative lithotroph distribution and diversity on volcanic deposits by use of the large ubunit of ribulose 1,5-bisphosphate carboxylase/oxygenase. Appl. Env. Microbiol. 70, 2245–2253 (2004).
Sato, Y. et al. Occurrence of hydrogen-oxidizing Ralstonia species as primary microorganisms in the Mt. Pinatubo volcanic mudflow deposits. Soil Sci Plant Nutr 50, 855–861 (2004).
Kelly, L. C., Cockell, C. S., Thorsteinsson, T., Marteinsson, V. & Stevenson, J. Pioneer microbial communities of the Fimmvorethuhals lava flow, Eyjafjallajokull, Iceland. Microbiol. Ecol. 68, 504–518 (2014).
Abe, H., Ueno, S., Takahashi, T., Tsumura, Y. & Hasegawa, M. Resilient plant-bird interactions in a volcanic island ecosystem: pollination of Japanese Camellia mediated by the Japanese White-eye. PloS one 8, 10.1371/journal.pone.0062696 (2013).
Kamijo, T. Regeneration process of Castanopsis and Persea forest on Miyake-jima Island, the Izu Islands. Jap. J. Ecol. 47, 1–10 (1997).
Nakada, S., Nagai, M., Kaneko, T., Nozawa, A. & Suzuki-Kamata, K. Chronology and products of the 2000 eruption of Miyakejima Volcano, Japan. B Volcanol 67, 205–218 (2005).
Johnson, D. B. & Hallberg, K. B. Acid mine drainage remediation options: a review. Sci. Total Environ. 338, 3–14 (2005).
Suter, D., Banwart, S. & Stumm, W. Dissolution of Hydrous Iron(III) Oxides by Reductive Mechanisms. Langmuir 7, 809–813 (1991).
Nodvin, S. C., Driscoll, C. T. & Likens, G. E. The effect of pH, sulfate and phosphate concentrations on the adsorption of sulfate by soils. Soil Science 142, 69–75 (1986).
Kazahaya, K. et al. Volcanic Gas Study of the 2000 Miyakejima Volcanic Activity: Degassing Environment Deduced from Adhered Gas Component on Ash and SO2 Emission Rate. J. Geography 110, 271–279 (in Japanese with English summary) (2001).
Baker, B. J. & Banfield, J. F. Microbial communities in acid mine drainage. FEMS Micrbol. Ecol. 44, 139–152 (2003).
Gonzalez-Toril, E., Llobet-Brossa, E., Casamayor, E. O., Amann, R. & Amils, R. Microbial ecology of an extreme acidic environment, the Tinto River. Appl. Env. Microbiol. 69, 4853–4865 (2003).
King, G. M. Chemolithotrophic bacteria: distributions, functions and significance in volcanic environments. Microbes Environ. 22, 309–319 (2007).
Howsley, R. & Pearson, H. W. pH dependent sulphide toxicity to oxygenic photosynthesis in cyanobacteria. FEMS 6, 287–292 (1979).
Wodzinski, R. S., Labeda, D. P. & Alexander, M. Effects of low concentrations of bisulfite-sulfite and nitrite on microorganisms. Appl. Env. Microbiol. 35, 718–723 (1978).
Hallgren, J. E. & Huss, K. Effects of SO2 on photosynthesis and Nitrogen-Fixation. Physiol. Plantarum 34, 171–176 (1975).
Dinsdale, E. A. et al. Functional metagenomic profiling of nine biomes. Nature 452, 629–632 (2008).
Lozupone, C. A. & Knight, R. Global patterns in bacterial diversity. Proc. Natl Acad. Sci. USA 104, 11436–11440 (2007).
Sato, Y. et al. Nitrogenase activity (acetylene reduction) of an iron-oxidizing leptospirillum strain cultured as a pioneer microbe from a recent volcanic deposit on miyake-jima, Japan. Microbes Environ. 24, 291–296 (2009).
Yamanaka, K., Nakamura, H., Matsuo, K. & Motomura, S. Studies on glei formation of soil (Part 1): on the determination of ferrous iron in soil. Jap. J. Soil Sci. Plant Nut. 28, 308–310 (1957).
Blumenthal, P. L. & Guernsey, S. C. In The Determination of Sulfur as Barium Sulfate, 392–436 (Agricultural Experiment Station, Iowa State College of Agriculture and Mechanic Arts, 1915).
Zhou, J., Bruns, M. A. & Tiedje, J. M. DNA recovery from soils of diverse composition. Appl. Env. Microbiol. 62, 316–322 (1996).
Liu, W. T., Marsh, T. L., Cheng, H. & Forney, L. J. Characterization of microbial diversity by determining terminal restriction fragment length polymorphisms of genes encoding 16S rRNA. Appl. Env. Microbiol. 63, 4516–4522 (1997).
Fujimura, R. et al. Complete genome sequence of Leptospirillum ferrooxidans strain C2-3, isolated from a fresh volcanic ash deposit on the island of Miyake, Japan. J. Bacteriol. 194, 4122–4123 (2012).
Untergasser, A. et al. Primer3Plus, an enhanced web interface to Primer3. Nuc. Acid. Res. 35, W71–W74 (2007).
Nishizawa, T., Komatsuzaki, M., Kaneko, N. & Ohta, H. Archaeal Diversity of Upland Rice Field Soils Assessed by the Terminal Restriction Fragment Length Polymorphism Method Combined with Real Time Quantitative-PCR and a Clone Library Analysis. Microbes Environ. 23, 237–243 (2008).
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nuc. Acid. Res. 25, 3389–3402 (1997).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Maidak, B. L. et al. The RDP-II (Ribosomal Database Project). Nuc. Acid. Res. 29, 173–174 (2001).
Yarza, P. et al. Update of the All-Species Living Tree Project based on 16S and 23S rRNA sequence analyses. Sys. Appl. Microbiol. 33, 291–299 (2010).
Noguchi, H., Taniguchi, T. & Itoh, T. MetaGeneAnnotator: detecting species-specific patterns of ribosomal binding site for precise gene prediction in anonymous prokaryotic and phage genomes. DNA research 15, 387–396 (2008).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nuc. Acid. Res. 28, 27–30 (2000).
Suzuki, R. & Shimodaira, H. Pvclust: an R package for assessing the uncertainty in hierarchical clustering. Bioinformatics 22, 1540–1542 (2006).
Overbeek, R. et al. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nuc. Acid. Res. 42, D206–D214 (2014).
Tatusov, R. L., Koonin, E. V. & Lipman, D. J. A genomic perspective on protein families. Science 278, 631–637 (1997).
Meyer, F. et al. The metagenomics RAST server - a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinfo. 9, 386–393 (2008).
Acknowledgements
We thank the staff at the laboratory of M.H. at the University of Tokyo, for their valuable technical help with cloning and sequencing; T. Nishizawa and Y. Guo for their valuable comments on this paper; H. Inoue and the AQUAS Corporation for their chemical analysis; and H. Ozaki for his helpful discussion on bioinformatics analysis. This study was supported in part by the Grants-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (JSPS; Nos. 17310018, 22248038 and 21580173), Environmental Research & Technology Development Fund (D-1106), the JSPS Research Fellowships for Young Scientists (DC2) and the Yuuji Ushiba Research Fund.
Author information
Authors and Affiliations
Contributions
R.F. and H.O. designed this project. All authors contributed to the discussion and provided comments on the manuscript. R.F. performed sampling and computational analysis and wrote the manuscript. Y.S. collected samples and analysed a part of the physicochemical data set. T.K. provided the vegetation samples and the volcanic SO2 monitoring data and assisted substantially in sampling. K.S. extracted 16S rDNA sequences from the metagenomic dataset and annotated the taxonomic information. K.O. and M.H. performed 454-pyrosequence analyses.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Fujimura, R., Kim, SW., Sato, Y. et al. Unique pioneer microbial communities exposed to volcanic sulfur dioxide. Sci Rep 6, 19687 (2016). https://doi.org/10.1038/srep19687
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep19687
- Springer Nature Limited
This article is cited by
-
Young volcanic terrains are windows into early microbial colonization
Communications Earth & Environment (2024)
-
Will free-living microbial community composition drive biogeochemical responses to global change?
Biogeochemistry (2023)
-
Metagenomic analysis reveals rapid development of soil biota on fresh volcanic ash
Scientific Reports (2020)
-
Unexpected fungal communities in the Rehai thermal springs of Tengchong influenced by abiotic factors
Extremophiles (2018)