
Abstract
The acute neurotoxicity of oligomeric forms of amyloid-β 1-42 (Aβ) is implicated in the pathogenesis of Alzheimer’s disease (AD). However, how these oligomers might first impair neuronal function at the onset of pathology is poorly understood. Here we have examined the underlying toxic effects caused by an increase in levels of intracellular Aβ, an event that could be important during the early stages of the disease. We show that oligomerised Aβ induces a rapid enhancement of AMPA receptor-mediated synaptic transmission (EPSCA) when applied intracellularly. This effect is dependent on postsynaptic Ca2+ and PKA. Knockdown of GluA1, but not GluA2, prevents the effect, as does expression of a S845-phosphomutant of GluA1. Significantly, an inhibitor of Ca2+-permeable AMPARs (CP-AMPARs), IEM 1460, reverses the increase in the amplitude of EPSCA. These results suggest that a primary neuronal response to intracellular Aβ oligomers is the rapid synaptic insertion of CP-AMPARs.
Similar content being viewed by others

Introduction
Alzheimer’s disease (AD) is defined by two hallmark pathological features: plaques that are composed of insoluble conjugates of the amyloid precursor protein (APP) cleavage product amyloid beta1-42 (Aβ) and tangles, which are mainly composed of hyperphosphorylated tau1. A large number of studies have now established that Aβ causes neurotoxic effects at the synapse, including the dysregulation of synaptic proteins and degeneration of dendritic spines1,2. However, the cellular events that lead to these pathological changes are poorly characterised, which both limits our understanding of the disease and potentially hampers the development of efficacious therapies.
One approach that has been extensively utilised to probe the mechanism of Aβ toxicity is to apply oligomeric forms of the protein acutely to the hippocampus and study their effects on synaptic transmission and plasticity3,4,5,6. Here, Aβ is applied extracellularly, either by injection into the brain or by the perfusion of hippocampal slices and its toxic effects can take an hour or more to manifest4,5. However, it is unknown whether these noted toxic effects are preceded by other as yet undefined extracellular and intracellular responses to Aβ exposure.
Given the extracellular nature of Aβ exposure in these experiments, it is assumed that the toxic effects observed are mediated by a membrane-bound substrate or event, and/or by the internalization of Aβ by affected neurons. Indeed, evidence suggests that plasma membrane receptors serve as substrates for oligomeric Aβ7. For instance, both metabotropic glutamate receptors and the prion protein receptor interact with Aβ at the synapse and these interactions are known to catalyse synaptic dysfunction and cell death8,9,10,11,12,13. In addition to this, studies now report the capacity for Aβ to form pores in the lipid bilayers of membranes14,15,16,17, which can serve as conduits to induce the aberrant entry of Ca2+ into cells. However, there is some uncertainty about the conditions in which membrane receptors and associated events are responsible for the toxic effects18,19,20, suggesting that additional mechanisms likely also play a role.
A growing number of studies now describe an emerging role of intracellular Aβ accumulation in the pathology of AD21,22. For instance, misprocessed endogenously produced Aβ can accumulate in intracellular compartments as well as the cytosol itself21. Several lines of evidence also suggest that extracellular Aβ can translocate into the cytosol from extracellular spaces23,24,25. Critically, it has been shown that the internalization of Aβ and the presumed increase in the presence of Aβ in intracellular spaces can induce synaptic dysfunction26,27. We have therefore hypothesized that accumulated intracellular Aβ will cause a primary effect on neuronal function. To test this, we have applied Aβ acutely into neurons via a patch electrode and investigated whether intracellular Aβ regulates excitatory synaptic transmission in the CA1-Schaffer collateral synapse in the hippocampus.
Results
Single molecule two-colour fluorescence coincidence detection and analysis of oligomers
Increasing evidence suggests that small, soluble Aβ oligomers are the driving force in Aβ-mediated toxicity and their production leads to synaptic dysfunction3,4,5. Using a protocol whereby synthetic Aβ was aggregated (see Methods), we were able to induce a high population of low-n oligomers, quantified using a single-molecule fluorescence method of confocal two-color coincidence detection (cTCCD) of fluorescently labeled Aβ (Fig. 1a)28. This protocol generated a heterogeneous preparation of Aβ oligomers, which equated to a 1–5 nM component of oligomers (Fig. 1b, c).

Generation of lower-n oligomers of Aβ1-42 (Aβ)
(a) A schematic of the principle of single molecule two-colour fluorescence coincidence detection and analysis of oligomers. The protein is labeled with a red or blue fluorophore and aggregated. The sample is then diluted to picomolar concentrations and analysed using single molecule fluorescence. Monomers passing through the probe volume give rise to non-coincident bursts of fluorescence while oligomers give rise to coincident fluorescent bursts, enabling the fraction of oligomers present in the sample to be determined. The intensity of a coincident burst relative to average monomer bursts was determined, allowing the oligomer size to be estimated. (b) Histogram depicting the proportion of monomers and oligomers. (c) Histogram depicting the size distribution of oligomers present in the preparation of Aβ oligomers.
Intracellular infusion of oligomerised Aβ1-42 (Aβ) causes a rapid increase in the AMPAR-mediated EPSC (EPSCA) in CA1 pyramidal neurons
Since Aβ oligomers are toxic3,4,5, we were interested in determining the intracellular effects of Aβ oligomers on synaptic function. Neurons were injected with oligomerised Aβ via passive diffusion from the patch pipette, whilst basal synaptic transmission was measured. Aβ oligomers caused a rapid increase in the amplitude of the AMPAR-mediated excitatory postsynaptic current (EPSCA) (181 ± 15%, n = 7, Fig. 2a). In contrast, neither the infusion of non-aggregated, monomeric Aβ nor Aβ oligomers that had been pre-incubated with clusterin, a chaperone that sequesters oligomers28, had any significant effect upon EPSCA (81 ± 8%, n = 6, Fig. 2b and 103 ± 9%, n = 7, Fig. 2c, respectively). The effect of Aβ oligomers was independent of the need to evoke EPSCA, since stopping stimulation for 15 min, shortly after obtaining whole-cell configuration, did not prevent the increase in synaptic transmission (closed circle: 192 ± 26%, n = 6, Fig. 2d). In addition, the effect of Aβ oligomers did not require the activation of NMDA receptors (NMDAR), since EPSCA was enhanced in the presence of the NMDAR antagonist D-AP-5 (147 ± 14%, n = 6, Fig. 2e). This effect was also specific for EPSCA since a pharmacologically-isolated NMDAR-mediated EPSC (EPSCN: holding voltage –40 mV, 10 μM NBQX perfusion) was unaffected by Aβ oligomer infusion (99 ± 14%, n = 6, Fig. 2f).

Intracellular infusion of Aβ causes a rapid increase in the AMPAR-mediated EPSC (EPSCA).
(a) The infusion of 1–5 nM oligomeric Aβ into post-synaptic neurons induces a rapid increase in EPSCA (n = 7). (b) Monomeric Aβ did not induce an increase in EPSCA (n = 6). (c) Clusterin (500 nM) prevented the Aβ oligomer-induced facilitation of EPSCA (n = 6). (d) The increase in EPSCA is independent of synaptic activity (n = 6). Filled circles depict Aβ infused neurons and open circles depict control neurons. (e) An NMDAR-antagonist, D-AP5 (50 M) has no effect on the Aβ oligomer-induced facilitation of EPSCA (n = 6). (f) The NMDAR mediated EPSC (EPSCN) is unaffected by infusion of Aβ oligomers (n = 6). In this (and subsequent figures) graphs plot the mean ± S.E.M. of n experiments.
Aβ oligomer-induced increase in EPSCA is dependent on postsynaptic Ca2+ and PKA
We next investigated the signalling cascades that underlie the rapid action of Aβ oligomers on AMPAR-mediated synaptic transmission (Fig. 3). Changes in postsynaptic Ca2+ levels initiate signal cascades involved in the modulation of synaptic transmission29,30,31. Therefore we tested whether blockade of postsynaptic Ca2+ mobilisation affects Aβ-mediated EPSCA regulation. The Aβ oligomer-induced increase was dependent on postsynaptic Ca2+, since it was prevented by postsynaptic infusion of the Ca2+ chelator BAPTA (95 ± 12%, n = 7, Fig. 3a) and relied on Ca2+ release from intracellular stores, since bath applied ryanodine also prevents the Aβ-induced EPSCA increase (108 ± 18%, n = 7, Fig. 3b). We were interested in examining the Ca2+-dependent mechanism responsible for these effects and possible downstream effectors. Ca2+-induced changes in synaptic transmission are known to involve, among other kinases, protein kinase A (PKA)32,33. Accordingly, we tested the involvement of PKA in the observed Aβ-induced EPSCA increase. We found that the effect required the activation of PKA, since it was prevented by either Rp-cAMPS, a cyclic AMP analogue that acts as a competitive antagonist of cAMP-induced activation of PKA (97 ± 8%, n = 6, Fig. 3c) or H89, a PKA inhibitor (94 ± 9%, n = 6, Fig. 3d), but not PKC since it was unaffected by both the PKC inhibitor Ro 32-0432 (171 ± 7%, n = 6, Fig. 3e) or PKC19-31, a pseudosubstrate of PKC which functions to inhibit the kinase (166 ± 13%, n = 6, Fig. 3f).

Aβ oligomer-induced increase in EPSCA is dependent on Ca2+ and PKA.
(a) Neurons were infused with Aβ oligomer in the presence of BAPTA (10 mM) in the filling solution. This prevented the Aβ oligomer facilitated increase in EPSCA (n = 7). (b) Ryanodine infusion via the pipette prevented the Aβ facilitated increase in EPSCA (n = 7). (c) There was no increase in EPSCA following preincubation (30 min) with RP-cAMPS (100 μM) (n = 6). (d) H89 infusion via the pipette prevented the Aβ facilitated increase in EPSCA (n = 6). (e) Ro 32-0432 (10 μM) infusion via the pipette had no effect on the Aβ-mediated increase of EPSCA (n = 6). (f) PKC 19-31 infusion via the pipette did not prevent the Aβ facilitated increase in EPSCA (n = 6). (g) KN-62 (10 μM) preincubation (45 min) and infusion via the pipette prevented the sustained Aβ facilitation of EPSCA (n = 7).
Calcium-calmodulin kinase II (CaMKII) is a Ca2+-sensitive kinase that has also been implicated in the regulation of AMPAR expression34,35. We therefore tested the involvement of CaMKII in Aβ-induced EPSCA increase. When cells were infused with Aβ and the CaMKII inhibitor KN-62, we observed an initial increase in EPSCA that rapidly declined (97 ± 16%, n = 7, Fig. 3g).
Aβ oligomer-induced enhancement of EPSCA is mediated by the GluA1 subunit of AMPARs
Since homomeric forms of GluA1-AMPARs characteristically display greater conductance than GluA2 containing AMPARs36,37, we hypothesized that infusion of Aβ facilitates EPSCA through an increase in synaptic homomeric GluA1 AMPARs. Indeed, the activation of PKA can lead to the insertion of GluA1-containing, GluA2-lacking AMPARs, known as Ca2+-permeable AMPARs (CP-AMPARs)38,39. To test this, we knocked down either GluA1 or GluA2 in neurons of organotypic hippocampal slices using biolistic shRNA transfection. GluA1-shRNA transfected cells did not show the rapid effect of Aβ oligomer infusion on EPSCA (90 ± 7%, n = 7, Fig. 4a). In comparison, a rapid increase of EPSCA was observed in GluA2-shRNA transfected cells (176 ± 18%, n = 8, Fig. 4b). The synaptic expression of GluA1 involves the PKA-dependent phosphorylation of the serine 845 residue (s845) of GluA140. Therefore, we knocked down endogenous GluA1 whilst simultaneously expressing an shRNA resistant form of GluA1 that cannot be phosphorylated at s845 (s845-phosphomutant). Infusion of Aβ oligomers in s845-phosphomutated cells had no effect on EPSCA (89 ± 8%, n = 7, Fig. 4c).

Aβ oligomer-induces expression of CP-AMPARs
(a) Aβ failed to increase EPSCA in GluA1-shRNA transfected cells (n = 7). (b) Aβ oligomers infusion increases EPSCA in GluA2-shRNA transfected cells (n = 8). (c) Aβ failed to increase EPSCA in GluA1-S845 phosphomutant transfected cells (n = 7). (d) Bath application of IEM 1460 (100 μM) has no effect on basal transmission EPSCA (n = 6). (e) The Aβ oligomer-mediated increase in EPSCA is reduced by bath application of IEM (n = 8).
Collectively, these data suggest that the rapid Aβ oligomer-induced changes in EPSCA may be due to a PKA-dependent synaptic insertion of CP-AMPARs. To test this directly, we bath-applied IEM 1460 (IEM), a compound that selectively blocks CP-AMPARs41. IEM had no effect on EPSCA when using control pipette solution (99 ± 8%: 30 min after IEM treatment, n = 6, Fig. 4d), which is consistent with a negligible contribution by CP-AMPARs to basal AMPAR-mediated transmission. However, IEM dramatically reduced the EPSCA following infusion with Aβ oligomers (159 ± 11%: 10 min after infusion of Aβ oligomers; 77 ± 10%: 30 min after the start of IEM treatment, n = 8, Fig. 4e). This suggests that Aβ oligomer infusion causes a rapid increase in the synaptic expression of CP-AMPARs, resulting in the observed facilitation of EPSCA amplitude.
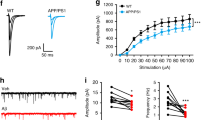
Using biotinylation assays from hippocampal slices, we found that the surface expression of GluA1 was significantly increased with exogenous Aβ treatment but that there was no change in GluA2/3 expression (Fig. 5A). This suggests that exogenously applied Aβ also induces the insertion of CP-AMPARs. To support these findings, we measured EPSCA during the extracellular perfusion of Aβ. We found that there was an increase of EPSCA on application of Aβ (145 ± 7%, n = 6, Fig. 5B), which was prevented when slices were continually perfused with IEM (89 ± 6%, n = 6, Fig. 5C).

Exogenous application of Aβ induces GluA1 surface expression.
(a) Aβ treatment caused an increase in the surface expression of GluA1, but not GluA2/3 as shown through a biotinylation assay. (b) Exogenous application of Aβ caused an increase in EPSCA (n = 6), (c) which was prevented when slices were perfused with IEM (n = 6).
Discussion
Here we have revealed a rapid synaptic response to intracellular accumulation of Aβ oligomers. Several lines of evidence suggest that extracellular Aβ oligomers are taken up into neurons where they impair synaptic function22. By studying the effects of intracellularly applied Aβ oligomers we have found a rapid action: the insertion of CP-AMPARs via a PKA-dependent phosphorylation of s845 of GluA1. These effects, occurring as a primary response to the emergence of cytosolic Aβ oligomers, could contribute to a key catalyzing mechanism of subsequent aberrant synaptic transmission. This finding therefore highlights a surprising discrepancy in our current understanding of the effects of Aβ on synaptic receptors. Whereas previous studies have shown that Aβ can drive the downregulation of synaptic transmission, in some cases mediated by the internalization of AMPARs and NMDARs42,43,44,45, our findings and those of others seem to indicate, in contrast, that Aβ can actually facilitate synaptic transmission, possibly through inducing the expression of receptors46,47,48,49. Presumably this is due to different time courses of Aβ-mediated toxic effects (the above studies, for example, range in treatment times from minutes to hours) and/or CP-AMPARs mediated secondary toxic insults50,51.
The mechanisms regulating the trafficking of GluA1-containing AMPARs have previously been characterised and generally converge on C-terminus phosphorylation events52,53,54. One canonical mechanism is the phosphorylation of the s845 residue on GluA1, priming its expression at the synapse52. Our finding that the expression of S845A, a mutant form of GluA1 which cannot be phosphorylated at s845, blocks the Aβ-induced enhancement of EPSCA, suggests that Aβ operates this rapid effect via a regulated physiological mechanism; the PKA-mediated phosphorylation of GluA1-s845. CaMKII has previously been implicated in AMPAR regulation34,35. Consistent with this role, we found that inhibiting CaMKII blocks the Aβ-induced enhancement of EPSCA. Interestingly, we observed a delayed effect under these conditions; whilst there was an initial increase in EPSCA, this rapidly declined. This might be explained by previously reported roles for CaMKII in the synaptic stabilization of AMPARs55. Therefore, PKA and CaMKII may act in concert in this mechanism, promoting the expression and then stabilization of synaptic AMPARs, respectively. Together, these data raise the interesting question as to whether Aβ might actually operate physiologically to regulate synaptic glutamate receptor expression and whether its aberrant cytosolic presence leads to a dysregulated physiological process. Clearly, more work is required to further understand a possible non-pathological role of Aβ.
CP-AMPARs are expressed at an early postnatal age and are replaced with GluA2-containing Ca2+-impermeable AMPARs during development56,57,58. CP-AMPARs are critically involved in physiological34,59,60 and pathological plasticity in the matured synapse61,62,63. Furthermore, growing evidence suggests CP-AMPARs prime neurodegenerative diseases including stroke, ischaemia and amyotrophic lateral sclerosis62,64. We found that blocking CP-AMPARs prior to exposure to exogenous Aβ prevented the facilitation of synaptic transmission. Therefore, our findings support the hypothesis that progressive Aβ-mediated CP-AMPAR expression is a pivotal catalyst for the onset of pathology.
The early accumulation of intracellular Aβ has been shown to be neurotoxic65 and transgenic models have shown it to be sufficient for cognitive impairments prior to the increase in extracellular Aβ21,66. Indeed, the accumulation of intracellular Aβ has previously been shown to be prevalent in the brains of AD patients67,68,69 and this is thought to be one of the earliest events in the pathology, preceding Aβ plaques and neurofibrillary tangles67,70. A recent report has shown that the infusion and accumulation of Aβ into neurons can have significant impairing effects on synaptic function71. Accounting for these findings and our data, the accumulation of intracellular Aβ will likely prove to be a catalyzing event in the pathogenesis of the disease. Given that the primary response to an increase in intracellular Aβ appears to be the expression of CP-AMPARs at the synapse, targeting CP-AMPARs may provide a means of restoring synaptic function in AD.
Methods
Amyloid-β preparation
The amyloid-β 1-42 peptide (Aβ; Millipore, UK) was first dissolved at a concentration of 1 mg / ml in 100% HFIP (1,1,1,3,3,3-hexafluoro-2-propanol [Sigma-Aldrich]). This solution was incubated at room temperature for 1 h with occasional vortexing. Next, the solution was sonicated for 10 min in a water bath sonicator. The solution was then dried under a gentle stream of nitrogen gas. 100% DMSO was then used to resuspend the peptide, which was then incubated at room temperature for 12 min with occasional vortexing. This solution was finally aliquoted into smaller volumes and stored at –80 °C. For a working solution, D-PBS (Invitrogen, UK) was added to the peptide stock solution and incubated for 2 h at room temperature to allow for peptide aggregation. To prepare monomeric Aβ, the same proceedure outlined above was followed, with the exception of the 2 h room temperature aggregation step.
Electrophysiology
All animal experiments were carried out in accordance with the UK Scientific Procedures Act, 1986 and associated guidelines. The methods were carried out in accordance with the approved guidelines. All experimental protocols were approved by the University of Bristol Animal Welfare & Ethical Review Body. Acute hippocampal slices were prepared from 26 - to 32 - day-old male Wistar rats. Animals were sacrificed by dislocation of the neck and then decapitated. The brain was rapidly removed and placed in ice-cold artificial CSF (aCSF) containing (in mM): 124 NaCl, 3 KCl, 26 NaHCO3, 1.25 NaH2PO4, 2 CaCl2, 1 MgSO4, 10 D-glucose and 0.1 picrotoxin (bubbled with 95% O2 / 5% CO2). Transverse hippocampal slices (400 μm thick) were prepared using a McIllwain tissue chopper (Mickle Laboratory Engineering Co. Ltd., Gomshall, UK). Hippocampal slices were stored in aCSF (20–25 °C) for 1–2 h before transferring to the recording chamber, in which they were submerged in aCSF (~30 °C) flowing at 2 ml / min. Stimulating electrodes were placed in the CA2 (Schaffer Collateral pathway). Single stimuli (constant voltage) were delivered to the Schaffer Collateral input at 30 sec intervals (0.016 Hz).
For whole-cell recordings, recording pipette (4–6 MΩ) solutions (280 mOsm [pH 7.2]) comprised (mM) CsMeSO4, 130; NaCl, 8; Mg-ATP, 4; Na-GTP, 0.3; EGTA, 0.5; HEPES 10; QX-314, 6. CA1 neurons were voltage clamped at −70 mV. Recordings were carried out using a MultiClamp 700B amplifier (Axon Instruments, Foster City, CA). EPSC amplitude, series resistance, input resistance and DC were monitored and analyzed online and offline using the WinLTP software (http://www.ltp-program.com). Only cells with series resistance <25 MΩ with a change in series resistance <10% from the start were included in this study. The amplitude of the excitatory postsynaptic currents (EPSCs) was measured and expressed relative the normalized baseline (first 5 min of recording).
Hippocampal Slice Culture and Whole-Cell Patch Recording
Organotypic hippocampal slice cultures were prepared from 6–8 days old Wistar rats. Rats were decapitated and brains were rapidly removed and placed in cold cutting solution that contained (mM) sucrose, 238; KCl, 2.5; NaHCO3, 26; NaH2PO4, 1; D-glucose, 11; MgCl2, 5 and CaCl2, 1. Hippocampal slices (350 μm) were cut using a McIlwain tissue chopper and cultured on semi-permeable membrane inserts (Millipore Corporation, Bedford, MA, USA) in a six-well plate containing culture medium (78.8% minimum essential medium, 20% heat-inactivated horse serum, 30 mM HEPES, 26 mM D-glucose, 5.8 mM NaHCO3, 2 mM CaCl2, 2 mM MgSO4, 70 μM Ascorbic Acid, 0.1% 1 mg / ml Insulin, pH adjusted to 7.3 and 320–330 mOsm). Slices were cultured for 7 – 9 days in vitro (DIV) with a change of medium every 2 days, without antibiotics. Neurons were transfected using a biolistic gene gun (Helios Gene-gun system, Bio Rad, U.S.A.) at DIV 3–4 (100 μg DNA; 90% of the construct to test; 10% pEGFP-C1). Electrophysiological recordings were performed at 3–4 days after transfection. Recordings were carried out in solution containing (mM) NaCl, 119; KCl, 2.5; NaHCO3, 26; NaH2PO4, 1; D-glucose, 11; CaCl2, 4; MgCl2, 4; picrotoxin, 0.02; 2-chloroadenosine,0.01 and gassed with 5% CO2 / 95% O2.
GluA1 shRNA
The following complementary oligonucleotide sequences were annealed and ligated into the EcoRI / ApaI sites of the pSilencer v1.0 vector (Ambicom): (Forward) 5’-GAACTGGCAGGTAACGGCTTTCAAGAGAAGCCGTTACCTGCCAGTTCTTTTTT-3’ and (Reverse) 5’-AATTAAAAAAGAACTGGCAGGTAACGGCTTCTCTTGAA AGCCGTTACCTGCCAGTTCGGCC-3’. The plasmid was then amplified in DH5α competent cells and the purified DNA was qualitatively analysed and sequenced to determine satisfactory plasmid ligation.
GluA2 shRNA
The following complementary oligonucleotide sequences were annealed and ligated into the EcoRI / ApaI sites of the pSilencer v1.0 vector (Ambicom): (Forward) 5’-CCATCGAAAGTGCTGAGGATTCAAGAGATCCTCAGCACT TTCGATGGAATTT TTT-3’ and (Reverse) 5’-AATTAAAAAATTCCATCGAAAGTGCTGAGGATCTCTTGAATCCTCAGCACTTTCGATGGGCC-3’. The plasmid was then amplified in DH5α competent cells and the purified DNA was qualitatively analysed and sequenced to determine satisfactory plasmid ligation.
GluA1 S845 mutant
The GluA1 S845A construct was a generous gift from Jeff Bernhardt. Briefly, site-directed mutagenesis was performed with Chameleon (Stratagene) on the pRK5_GluA1i construct mutating serine 845 residue to alanine.
Slice biotinylation and NeutraAvidin pull-down. Surface biotinylation of acute slices was performed as described previously with some modifications72. Briefly, slices were initially washed twice in aCSF and subsequently incubated in aCSF containing 1 mg / ml Sulfo-NHS-SS-Biotin (Thermo Scientific, Rockford, USA) for 45 min at 4 °C to allow for labelling of all surface membrane proteins. Excess biotin was removed by washes in aCSF containing NH4Cl. Tissue was then homogenised in lysis buffer containing 25 mM Tris (pH 7.5), 150 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 10m M NaF and a cocktail of protease inhibitors (Sigma, St Louis, USA) and incubated for 30 min prior to centrifugation at 1,000 g to remove cellular debris. The total protein concentration was determined using the Pierce BCA kit. Subsequently, 100 μl of StreptaAvidin beads (Upstate, USA) were added to 500 μg of protein lysate and placed on a rotator at 4 °C for 2 hr. Samples were then washed five times in lysis buffer; beads were pulled-down after each wash by gentle centrifugation. Bound proteins were eluted by adding 2 X SDS reducing buffer and moderate heating at 60 °C for 30 min. The resulting supernatant was transferred to new tubes and heated at 90 °C for 5 min prior to gel loading.
Statistical Analyses
Data were analyzed from one slice per rat (i.e., n = number of slices = number of rats). Data pooled across slices are expressed as the mean ± s.e.m. Significance (p < 0.05) was tested using two-tailed t-tests. For electrophysiology experiments, mean ± s.e.m. data from the 40 min timepoint are described.
Additional Information
How to cite this article: Whitcomb, D. J. et al. Intracellular oligomeric amyloid-beta rapidly regulates GluA1 subunit of AMPA receptor in the hippocampus. Sci. Rep. 5, 10934; doi: 10.1038/srep10934 (2015).
References
Spires-Jones, T. L. & Hyman, B.T. The intersection of amyloid beta and tau at synapses in Alzheimer’s disease. Neuron 82, 756–771 (2014).
Crimins, J. L, Pooler, A., Polydoro, M., Luebke, J. I. & Spires-Jones, T. L. The intersection of amyloid beta and tau in glutamatergic synaptic dysfunction and collapse in Alzheimer’s disease. Ageing Res Rev 12, 757–763 (2013).
Cullen, W. K., Suh, Y. H., Anwyl, R. & Rowan, M. J. Block of LTP in rat hippocampus in vivo by beta-amyloid precursor protein fragments. Neuroreport 8, 3213–3217 (1997).
Wang, Q., Walsh, D.M., Rowan, M.J ., Selkoe, D. J . & Anwyl, R. Block of long-term potentiation by naturally secreted and synthetic amyloid beta-peptide in hippocampal slices is mediated via activation of the kinases c-Jun N-terminal kinase, cyclin-dependent kinase 5 and p38 mitogen-activated protein kinase as well as metabotropic glutamate receptor type 5. J Neurosci 24, 3370–3378 (2004).
Jo. J . et al. Abeta(1-42) inhibition of LTP is mediated by a signaling pathway involving caspase-3, Akt1 and GSK-3beta. Nat Neurosci 14, 545–547 (2011).
Walsh, D. M. et al. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416, 535–539 (2002).
Dinamarca, M. C., Rios, J. A. & Inestrosa N. C. Postsynaptic Receptors for Amyloid-beta Oligomers as Mediators of Neuronal Damage in Alzheimer’s Disease. Front Physiol 3, 464 (2012).
Lauren, J., Gimbel, D. A., Nygaard, H. B., Gilbert, J. W. & Strittmatter, S. M. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 457, 1128–1132 (2009).
Chen, S., Yadav, S. P. & Surewicz, W. K. Interaction between human prion protein and amyloid-beta (Abeta) oligomers: role OF N-terminal residues. J Biol Chem 285, 26377–26383 (2010).
Larson, M . et al. The complex PrP(c)-Fyn couples human oligomeric Abeta with pathological tau changes in Alzheimer’s disease. J Neurosci 32, 16857–16871a (2012).
Kudo, W . et al. Cellular prion protein is essential for oligomeric amyloid-beta-induced neuronal cell death. Hum Mol Genet 21, 1138–1144 (2012).
Um, J. W. et al. Metabotropic glutamate receptor 5 is a coreceptor for Alzheimer abeta oligomer bound to cellular prion protein. Neuron 79, 887–902 (2013).
Nicoll, A. J. et al. Amyloid-beta nanotubes are associated with prion protein-dependent synaptotoxicity. Nat Commun 4, 2416 (2013).
Lin, H ., Bhatia, R. & Lal, R. Amyloid beta protein forms ion channels: implications for Alzheimer’s disease pathophysiology. FASEB J 15, 2433–2444 (2001).
Demuro, A., Mina, E., Kayed, R., Milton, S. C., Parker, I & Glabe, C. G. Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J Biol Chem 280, 17294–17300 (2005).
Jang, H . et al. Truncated beta-amyloid peptide channels provide an alternative mechanism for Alzheimer’s Disease and Down syndrome. Proc Natl Acad Sci U S A 107, 6538–6543 (2010).
Lin, H. & Arispe, N. J. Single-cell screening of cytosolic [Ca] reveals cell-selective action by the Alzheimer’s Abeta peptide ion channel. Cell Stress Chaperones, 20, 333–342 (2015).
Kessels, H. W, Nguyen, L. N ., Nabavi, S. & Malinow, R. The prion protein as a receptor for amyloid-beta. Nature 466, E3-4; discussion E4-5 (2010).
Balducci, C . et al. Synthetic amyloid-beta oligomers impair long-term memory independently of cellular prion protein. Proc Natl Acad Sci U S A 107, 2295–2300 (2010).
Cisse, M., Sanchez, P.E., Kim, D.H., Ho., K., Yu., GQ. & Mucke, L. Ablation of cellular prion protein does not ameliorate abnormal neural network activity or cognitive dysfunction in the J20 line of human amyloid precursor protein transgenic mice. J Neurosci 31, 10427–10431 (2011).
LaFerla, F. M., Green, K. N. & Oddo, S. Intracellular amyloid-beta in Alzheimer’s disease. Nat Rev Neurosci 8, 499–509 (2007).
Mohamed, A . & Posse de Chaves, E. Abeta internalization by neurons and glia. Int J Alzheimers Dis 2011, 127984 (2011).
Yazawa, H et al. Beta amyloid peptide (Abeta42) is internalized via the G-protein-coupled receptor FPRL1 and forms fibrillar aggregates in macrophages. FASEB J 15, 2454–2462 (2001).
Nagele, R. G., D’Andrea, M. R., Anderson, W. J. & Wang, H. Y. Intracellular accumulation of beta-amyloid (1-42) in neurons is facilitated by the alpha 7 nicotinic acetylcholine receptor in Alzheimer’s disease. Neuroscience 110, 199–211 (2002).
Clifford, P. M. et al. Abeta peptides can enter the brain through a defective blood-brain barrier and bind selectively to neurons. Brain Res 1142, 223–236 (2007).
Yankner, B. A., Duffy, L. K. & Kirschner, D. A. Neurotrophic and neurotoxic effects of amyloid beta protein: reversal by tachykinin neuropeptides. Science 250, 279–282 (1990).
Takadera, T ., Sakura, N., Mohri, T. & Hashimoto, T. Toxic effect of a beta-amyloid peptide (beta 22–35) on the hippocampal neuron and its prevention. Neurosci Lett 161, 41–44 (1993).
Narayan, P . et al. The extracellular chaperone clusterin sequesters oligomeric forms of the amyloid-beta(1-40) peptide. Nat Struct Mol Biol 19, 79–83 (2012).
McGlade-McCulloh, E., Yamamoto, H., Tan, S. E., Brickey, D. A. & Soderling, T. R. Phosphorylation and regulation of glutamate receptors by calcium/calmodulin-dependent protein kinase II. Nature 362, 640–642 (1993).
Lledo, P. M., Hjelmstad, G. O., Mukherji, S., Soderling, T. R., Malenka, R. C. & Nicoll, R. A. Calcium/calmodulin-dependent kinase II and long-term potentiation enhance synaptic transmission by the same mechanism. Proc Natl Acad Sci U S A 92, 11175–11179 (1995).
Mulkey, R. M., Endo, S., Shenolikar, S. & Malenka, R. C. Involvement of a calcineurin/inhibitor-1 phosphatase cascade in hippocampal long-term depression. Nature 369, 486–488 (1994).
Frey, U ., Huang, Y. Y . & Kandel, E. R. Effects of cAMP simulate a late stage of LTP in hippocampal CA1 neurons. Science 260, 1661–1664 (1993).
Nayak, A., Zastrow, D. J., Lickteig, R., Zahniser, N. R., Browning, M. D. Maintenance of late-phase LTP is accompanied by PKA-dependent increase in AMPA receptor synthesis. Nature 394, 680–683 (1998).
Hayashi, Y., Shi, S. H., Esteban, J. A., Piccini, A., Poncer, J. C. & Malinow, R. Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science 287, 2262–2267 (2000).
Wang, J. H. & Kelly, P. T. Postsynaptic injection of CA2+/CaM induces synaptic potentiation requiring CaMKII and PKC activity. Neuron 15, 443–452 (1995).
Clem, R. L., Anggono, V. & Huganir, R. L. PICK1 regulates incorporation of calcium-permeable AMPA receptors during cortical synaptic strengthening. J Neurosci 30, 6360–6366 (2010).
Swanson, G.T. & Kamboj, S. K., Cull-Candy, SG. Single-channel properties of recombinant AMPA receptors depend on RNA editing, splice variation and subunit composition. J Neurosci 17, 58–69 (1997).
Man, H. Y ., Sekine-Aizawa, Y . & Huganir, R. L. Regulation of {alpha}-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor trafficking through PKA phosphorylation of the Glu receptor 1 subunit. Proc Natl Acad Sci U S A 104, 3579–3584 (2007).
Roche, K. W ., O’Brien, R. J., Mammen, A. L., Bernhardt, J . & Huganir, R. L. Characterization of multiple phosphorylation sites on the AMPA receptor GluR1 subunit. Neuron 16, 1179–1188 (1996).
He, K., Song, L., Cummings, L. W ., Goldman, J ., Huganir, R. L. & Lee, H. K. Stabilization of Ca2+-permeable AMPA receptors at perisynaptic sites by GluR1-S845 phosphorylation. Proc Natl Acad Sci U S A 106, 20033–20038 (2009).
Samoilova, M. V., Buldakova, S. L ., Vorobjev, V. S., Sharonova, I. N . & Magazanik, L. G. The open channel blocking drug, IEM-1460, reveals functionally distinct alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionate receptors in rat brain neurons. Neuroscience 94, 261–268 (1999).
Almeida, C. G. et al. Beta-amyloid accumulation in APP mutant neurons reduces PSD-95 and GluR1 in synapses. Neurobiol Dis 20, 187–198 (2005).
Hsieh, H. et al. AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron 52, 831–843 (2006).
Gu, Z, Liu, W & Yan, Z. {beta}-Amyloid impairs AMPA receptor trafficking and function by reducing Ca2+/calmodulin-dependent protein kinase II synaptic distribution. J Biol Chem 284, 10639–10649 (2009).
Monfort, P. & Felipo, V. Amyloid-beta impairs and ibuprofen restores, the cGMP pathway, synaptic expression of AMPA receptors and long-term potentiation in the hippocampus. J Alzheimers Dis 22, 795–809 (2010).
Hartley, DM et al. Protofibrillar intermediates of amyloid beta-protein induce acute electrophysiological changes and progressive neurotoxicity in cortical neurons. J Neurosci 19, 8876–8884 (1999).
Tozaki, H . et al. The inhibitory and facilitatory actions of amyloid-beta peptides on nicotinic ACh receptors and AMPA receptors. Biochem Biophys Res Commun 294, 42–45 (2002).
Ye, C., Walsh, D. M., Selkoe, D. J & Hartley, D. M. Amyloid beta-protein induced electrophysiological changes are dependent on aggregation state: N-methyl-D-aspartate (NMDA) versus non-NMDA receptor/channel activation. Neurosci Lett 366, 320–325 (2004).
Wang, D., Govindaiah, G ., Liu, R., De Arcangelis, V,. Cox, C. L. & Xiang, Y. K. Binding of amyloid beta peptide to beta2 adrenergic receptor induces PKA-dependent AMPA receptor hyperactivity. FASEB J 24, 3511–3521 (2010).
Carriedo, S. G., Yin, H. Z., Sensi, S. L. & Weiss, J. H. Rapid Ca2+ entry through Ca2+-permeable AMPA/Kainate channels triggers marked intracellular Ca2+ rises and consequent oxygen radical production. J Neurosci 18, 7727–7738 (1998).
Sensi, S. L., Yin, H. Z., Carriedo, S. G., Rao, S. S. & Weiss, J. H. Preferential Zn2+ influx through Ca2+-permeable AMPA/kainate channels triggers prolonged mitochondrial superoxide production. Proc Natl Acad Sci U S A 96, 2414–2419 (1999).
Oh, M. C., Derkach, V. A. & Guire, E. S., Soderling, T. R. Extrasynaptic membrane trafficking regulated by GluR1 serine 845 phosphorylation primes AMPA receptors for long-term potentiation. J Biol Chem 281, 752–758 (2006).
Lee, H. K., Takamiya, K., He, K., Song, L. & Huganir, R. L. Specific roles of AMPA receptor subunit GluR1 (GluA1) phosphorylation sites in regulating synaptic plasticity in the CA1 region of hippocampus. J Neurophysiol 103, 479–489 (2010).
He, K., Lee, A ., Song, L., Kanold, P. O. & Lee, H. K. AMPA receptor subunit GluR1 (GluA1) serine-845 site is involved in synaptic depression but not in spine shrinkage associated with chemical long-term depression. J Neurophysiol 105, 1897–1907 (2011).
Opazo, P., Sainlos, M. & Choquet, D. Regulation of AMPA receptor surface diffusion by PSD-95 slots. Curr Opin Neurobiol 22, 453–460 (2012).
Pickard, L., Noel, J., Henley, J. M., Collingridge, G. L. & Molnar, E. Developmental changes in synaptic AMPA and NMDA receptor distribution and AMPA receptor subunit composition in living hippocampal neurons. J Neurosci 20, 7922–7931 (2000).
Kumar, S. S., Bacci, A., Kharazia, V . & Huguenard, J. R. A developmental switch of AMPA receptor subunits in neocortical pyramidal neurons. J Neurosci 22, 3005–3015 (2002).
Brill, J. & Huguenard, J. R. Sequential changes in AMPA receptor targeting in the developing neocortical excitatory circuit. J Neurosci 28, 13918–13928 (2008).
Cull-Candy, S., Kelly, L. & Farrant, M. Regulation of Ca2+-permeable AMPA receptors: synaptic plasticity and beyond. Curr Opin Neurobiol 16, 288–297 (2006).
Derkach, V. A., Oh, M. C., Guire, E. S. & Soderling, T. R. Regulatory mechanisms of AMPA receptors in synaptic plasticity. Nat Rev Neurosci 8, 101–113 (2007).
Feldmeyer, D . et al. Neurological dysfunctions in mice expressing different levels of the Q/R site-unedited AMPAR subunit GluR-B. Nat Neurosci 2, 57–64 (1999).
Liu, B. et al. Ischemic insults direct glutamate receptor subunit 2-lacking AMPA receptors to synaptic sites. J Neurosci 26, 5309–5319 (2006).
Spaethling, J. M, Klein, D. M., Singh, P . & Meaney, D. F. Calcium-permeable AMPA receptors appear in cortical neurons after traumatic mechanical injury and contribute to neuronal fate. J Neurotrauma 25, 1207–1216 (2008).
Kwak, S . & Weiss, J. H. Calcium-permeable AMPA channels in neurodegenerative disease and ischemia. Curr Opin Neurobiol 16, 281–287 (2006).
Zhang, Y., McLaughlin, R., Goodye,r C., LeBlanc, A. Selective cytotoxicity of intracellular amyloid beta peptide1-42 through p53 and Bax in cultured primary human neurons. J Cell Biol 156, 519–529 (2002).
Oddo, S., Caccamo, A ., Kitazawa, M., Tseng, BP, LaFerla, F. M. Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer’s disease. Neurobiol Aging 24, 1063–1070 (2003).
Gouras, G. K. et al. Testosterone reduces neuronal secretion of Alzheimer’s beta-amyloid peptides. Proc Natl Acad Sci U S A 97, 1202–1205 (2000).
Wegiel, J. et al. Intraneuronal Abeta immunoreactivity is not a predictor of brain amyloidosis-beta or neurofibrillary degeneration. Acta Neuropathol 113, 389–402 (2007).
Aoki, M., Volkmann, I., Tjernberg, L. O ., Winblad, B . & Bogdanovic, N. Amyloid beta-peptide levels in laser capture microdissected cornu ammonis 1 pyramidal neurons of Alzheimer’s brain. Neuroreport 19, 1085–1089 (2008).
D’Andrea, M. R., Nagele, R.G., Wang, H. Y., Peterson, P. A. & Lee, D. H. Evidence that neurones accumulating amyloid can undergo lysis to form amyloid plaques in Alzheimer’s disease. Histopathology 38, 120–134 (2001).
Ripoli, C . et al. Intracellular accumulation of amyloid-beta (Abeta) protein plays a major role in Abeta-induced alterations of glutamatergic synaptic transmission and plasticity. J Neurosci 34, 12893–12903 (2014).
Thomas-Crusells, J ., Vieira, A ., Saarma, M. & Rivera, C. A. novel method for monitoring surface membrane trafficking on hippocampal acute slice preparation. J Neurosci Methods 125, 159–166 (2003).
Acknowledgements
This study was supported by UK Wellcome Trust-MRC Neurodegenerative Disease Initiative Programme to K.C., G.L.C., D.K. and P.G.H. The Korea-UK Alzheimer’s disease Research Consortium grant to D-H.K. (Korean Ministry of Health and Welfare). E.L.H. and P.R. were supported by MRC PhD studentships. K.C. was supported by the Wolfson Research Merit Award and Royal Society, London.
Author information
Authors and Affiliations
Contributions
The study was conceived and designed by K.C. Electrophysiological studies were conducted by D.J.W., E.L.H., P.R., D-H.K., J.J. and biochemical assays were conducted by T.P., G.W. and P.N. The manuscript was written by D.J.W., E.J.K., P.G.H., D.K., G.L.C. and K.C.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Whitcomb, D., Hogg, E., Regan, P. et al. Intracellular oligomeric amyloid-beta rapidly regulates GluA1 subunit of AMPA receptor in the hippocampus. Sci Rep 5, 10934 (2015). https://doi.org/10.1038/srep10934
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep10934
- Springer Nature Limited
This article is cited by
-
The Q/R editing site of AMPA receptor GluA2 subunit acts as an epigenetic switch regulating dendritic spines, neurodegeneration and cognitive deficits in Alzheimer’s disease
Molecular Neurodegeneration (2023)
-
Aß Pathology and Neuron–Glia Interactions: A Synaptocentric View
Neurochemical Research (2023)
-
Aβ42 oligomers trigger synaptic loss through CAMKK2-AMPK-dependent effectors coordinating mitochondrial fission and mitophagy
Nature Communications (2022)
-
Bioactive human Alzheimer brain soluble Aβ: pathophysiology and therapeutic opportunities
Molecular Psychiatry (2022)
-
Amyloid-β oligomers in the nucleus accumbens decrease motivation via insertion of calcium-permeable AMPA receptors
Molecular Psychiatry (2022)