
Abstract
Using a variable-composition ab initio evolutionary algorithm implemented in the USPEX code, we have performed a systematic search for stable compounds in the Ca-Bi system at different pressures. In addition to the well-known tI12-Ca2Bi and oS12-CaBi2, a few more structures were found by our calculations, among which phase transitions were also predicted in Ca2Bi (tI12 → oI12 → hP6), Ca3Bi2 (hP5 → mC20 → aP5) and CaBi (tI2 → tI8), as well as a new phase (Ca3Bi) with a cF4 structure. All the newly predicted structures can be both dynamically and thermodynamically stable with increasing pressure. The superconductive properties of cF4-CaBi3, tI2-CaBi and cF4-Ca3Bi were studied and the superconducting critical temperature Tc can be as high as 5.16, 2.27 and 5.25 K, respectively. Different superconductivity behaviors with pressure increasing have been observed by further investigations.
Similar content being viewed by others

Introduction
Superconductivity has been deeply studied and developed very quickly since its discovery in 1911, but its origin remains enigmatic. The copper oxide family is of enduring interest for initiating energetic activities of high-temperature superconductivity1,2 and has been applied in a variety of fields. The discovery of the iron-based superconductors3, which is unconventional, has attracted great attention and aroused extensive research with the intention of finding new superconductors. Iron-based superconductors have been extended to various material groups, such as the so called 11113,4, 1225, 1116, 117 compounds, etc. Moreover, it is observed that compounds in which iron is completely substituted by other 3d, 4d, or 5d transition metals8,9, exhibit superconductivity. Since a large variety of phosphide, arsenide and antimonide superconductors have been found, attention is now focusing on bismuthides and related pnictide systems. So bismuth has been a part of various superconducting compounds, such as NiBi310, Bi4O4S311,12, CsBi4Te613 and LaO1-xFxBiS214. Recently, Sturza et al. reported a new complex alkaline earth intermetallic compound superconductor Ca11Bi10-x15 with Tc ~ 2.2 K, which stimulates our interests to find new superconductors in the Ca-Bi system.
A large number of BCS superconductors have been theoretically proposed16,17,18 as the development of computational crystal structure prediction tools19,20 and methods. In this paper, we perform a systematic search for thermodynamically stable calcium bismuth at ambient and high pressure by using the variable-composition ab initio evolutionary algorithm21,22,23 and density functional theory (DFT). Here we predict several new structures at different pressures that were never reported and discuss their structures, electronic structures and superconductivity properties of the selected structures.
Results
Crystal structure and structural properties of calcium bismuthides
Some reported experimental crystal structures in the Ca-Bi system are summarized by H. Kim et. al.24 and other similar Sr-Bi compounds have also been studied by first-principles calculations25. In this paper, we use the variable-composition evolutionary algorithm, which is very effective, to predict stable compositions and their structures. We have performed structure searches with up to 16 atoms in the unit cell at different pressures for the Ca-Bi system of all possible compositions.
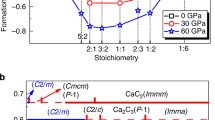
Fig. 1 shows the enthalpies of formation of the predicted structures. At ambient pressure, it can be clearly seen that there are three stable structures on the convex hull, i.e., tI12-Ca2Bi (Fig. 2a), hP5-Ca3Bi2 (Fig. 2b) and oS12-CaBi2 (Fig. 2c). The previously reported oP32-Ca5Bi326 and tI84-Ca11Bi1027, although not found by our prediction possibly limited by the computational resources, are also on the convex hull, suggesting that they are thermodynamically stable. However, these two structures lie above the convex hull curve at 30 GPa, which implies that they will become metastable phases under high-pressure conditions.

Enthalpy differences (top) and convex hull for the Ca-Bi system (bottom).
Calculated enthalpy differences as a function of pressure relative to tI12 of Ca2Bi (top left), hP5 of Ca3Bi2 (top middle) and tI2 of CaBi (top right) and convex hull (bottom) for the Ca-Bi system at ambient pressure(red) and 30 GPa (black).

Crystal structures for (a): tI12-Ca2Bi, (b): hP5-Ca3Bi2, (c): oS12-CaBi2, (d): cF4-Ca3Bi and (e): cF4-CaBi3.
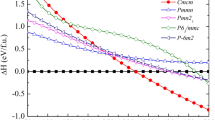
The Ca3Bi2 phase has been reported in the literature without structural information. Our prediction indicates that it has a hexagonal structure of the La2O3 type and belongs to the space group of  (164), with Pearson symbol hP5. Our calculations reveal that Ca3Bi2 undergoes a series phase transitions, i.e., from hP5 to mC20 (space group C2/m, 12) at 4 GPa and mC20 to aP5 (space group
(164), with Pearson symbol hP5. Our calculations reveal that Ca3Bi2 undergoes a series phase transitions, i.e., from hP5 to mC20 (space group C2/m, 12) at 4 GPa and mC20 to aP5 (space group  166) at about 59 GPa. The calculated phonon dispersion of hP5-Ca3Bi2 indicates that the phonon mode along the A-H direction is imaginary at 0 GPa and it will be dynamically stable at high pressure, which can be seen in Fig. 3 (further study shows that the imaginary phonon mode along the A-H direction will disappear at above 0.5 ~ 0.6 GPa, which can be seen in Figure S1). We also observed a high-pressure phase transition in Ca2Bi, which is from tI1228 (space group I4/mmm, 139) to oI12 (space group Cmmm, 65) at 14 GPa and from oI12 to hP6 (space group P63/mmc, 194) at 33GPa. For CaBi2, we predict an oS12 structure (space group Cmcm, 63) with lattice constants a = 4.782 Å, b = 17.160 Å, c = 4.598 Å at 0 GPa compared with the previously reported a = 4.701 Å, b = 17.053 Å, c = 4.613 Å29, which lies on the convex hull at ambient and high pressure and is both dynamically and thermodynamically stable.
166) at about 59 GPa. The calculated phonon dispersion of hP5-Ca3Bi2 indicates that the phonon mode along the A-H direction is imaginary at 0 GPa and it will be dynamically stable at high pressure, which can be seen in Fig. 3 (further study shows that the imaginary phonon mode along the A-H direction will disappear at above 0.5 ~ 0.6 GPa, which can be seen in Figure S1). We also observed a high-pressure phase transition in Ca2Bi, which is from tI1228 (space group I4/mmm, 139) to oI12 (space group Cmmm, 65) at 14 GPa and from oI12 to hP6 (space group P63/mmc, 194) at 33GPa. For CaBi2, we predict an oS12 structure (space group Cmcm, 63) with lattice constants a = 4.782 Å, b = 17.160 Å, c = 4.598 Å at 0 GPa compared with the previously reported a = 4.701 Å, b = 17.053 Å, c = 4.613 Å29, which lies on the convex hull at ambient and high pressure and is both dynamically and thermodynamically stable.

Phonon dispersion curves for the hP5-Ca3Bi2 at 0 GPa and 5 GPa.
The previously reported Ca-Bi phase diagrams30 mentioned two phases, CaBi3 and CaBi, without structural information. This work suggests a cubic cF4 structure for CaBi3 (space group  , 221) and a tetragonal tI2 structure for CaBi (space group P4/mmm, 123), which are thermodynamically unstable at ambient pressure and can be stable at high pressure. The cF4-CaBi3 is very close to the convex hull curve but lies a little above it. In addition, another tetragonal tI8 structure of CaBi (space group I41/amd, 141) was also predicted, which is proved to be thermodynamically more stable at above 45 GPa. Besides, our research also predicted a new phase of Ca-Bi system, i.e., cF4-Ca3Bi, the space group of which is the same with cF4-CaBi3 and can be thermodynamically stable at high pressure. In cF4-Ca3Bi, the Ca and Bi atoms occupy Wyckoff 3c and 1a positions, respectively. However, the situation of the occupancy is just the opposite in cF4-CaBi3, which can be seen in Figs. 2d and 2e. The structural parameters for the predicted structures are listed in Table 1.
, 221) and a tetragonal tI2 structure for CaBi (space group P4/mmm, 123), which are thermodynamically unstable at ambient pressure and can be stable at high pressure. The cF4-CaBi3 is very close to the convex hull curve but lies a little above it. In addition, another tetragonal tI8 structure of CaBi (space group I41/amd, 141) was also predicted, which is proved to be thermodynamically more stable at above 45 GPa. Besides, our research also predicted a new phase of Ca-Bi system, i.e., cF4-Ca3Bi, the space group of which is the same with cF4-CaBi3 and can be thermodynamically stable at high pressure. In cF4-Ca3Bi, the Ca and Bi atoms occupy Wyckoff 3c and 1a positions, respectively. However, the situation of the occupancy is just the opposite in cF4-CaBi3, which can be seen in Figs. 2d and 2e. The structural parameters for the predicted structures are listed in Table 1.
Electronic structures
We calculated the band structures and density of states of all the predicted structures. All the calcium bismuthides are metallic except for the hP5-Ca3Bi2 structure, which is semiconductive at 0 GPa, with a narrow band gap of 0.42 eV and becomes metallic at around 20 GPa. Through the calculations we can conclude that the ratio of Ca and Bi will affect their contributions to the DOS at the Fermi level (EF). For example, if a calcium rich structure of CaxBi1-x, i.e., x > 0.5, the density of states at EF comes mainly from the calcium atoms, in particular Ca d states and bismuth p orbital contributes most to bismuth states. On the contrary, the density of states at EF comes mainly from the bismuth atoms in a bismuth rich structure, in particular Bi p states and calcium d orbital contributes most to calcium states at the Fermi level. Fig. 4 shows the band structures and DOS of the cF4-Ca3Bi and cF4-CaBi3 structures. The density of states at EF of these two structures is 0.32 and 0.20 states/eV, respectively. A careful examination of the band structures show multiple steep bands crossing the Fermi level as well as flat bands, which is considered to be a necessary condition for superconductivity to occur31. As bismuth based materials may have strong spin-orbital coupling (SOC) effect32,33, the electronic structures of cF4-Ca3Bi and cF4-CaBi3 have been calculated by including the SOC effect and compared with those without SOC effect, as shown in Figure S2 and S3.

Band structure and partial density of states for cF4-Ca3Bi and cF4-CaBi3.
Superconductivity properties
The superconductivity of the selected structures can be conveniently studied by EPC calculation. The calculated Eliashberg spectral function and the electron-phonon coupling strength λ are shown in Fig. 5 for cF4-Ca3Bi and cF4-CaBi3 at 60 and 30 GPa, respectively. The superconducting critical temperature can be estimated from the Allen-Dynes modified McMillan equation34

where the electron-phonon coupling constant is calculated as

the logarithmic frequency average is

and a typical value of the Coulomb pseudopotential μ* = 0.10 is used.

Total and projected phonon density of states (PHDOS) and Eliashberg function α2F(ω) for cF4-Ca3Bi at 60 GPa and cF4-CaBi3 at 30 GPa and the corresponding integrated electron-phonon coupling constant λ(ω).
Our calculations suggest that cF4-Ca3Bi shows no superconductivity behavior below 25 GPa. Its superconducting transition temperature rises as the pressure increases. The dynamic instability of cF4-Ca3Bi is confirmed as the phonon spectrum displays imaginary frequencies above 60 GPa, at which we predict that cF4-Ca3Bi will be superconductor with a Tc of 5.25 K, total λ of 0.96 and ωlog of 146.1 cm−1. 72% of the total λ results from modes below 140 cm−1, which are mainly displacements of bismuth atoms. The decomposition of the phonon density of states into contributions from the atoms (Fig. 5a) shows that the calcium atoms (below 140 cm−1) has a slight contribution to the electron-phonon coupling in this compound. On the contrary, cF4-CaBi3 can be a superconductor at 0 GPa with a Tc of 5.16 K, total λ of 1.23 and ωlog of 56.2 cm−1, which will accordingly turn into 0.61 K, 0.41 and 127.8 cm−1 at 30 GPa. 84% of the total λ results from modes below 125 cm−1, which are mainly displacements of bismuth atoms, the same with cF4-Ca3Bi. A further study of the phonon density of states of cF4-CaBi3 indicates that the calcium atoms (below 180 cm−1) has a negligible contribution to the electron-phonon coupling in this compound (Fig. 5b). The calculated λ, ωlog and Tc at different pressure for cF4-Ca3Bi and cF4-CaBi3 are listed in Table 2 and Table 3, respectively. The tI2-CaBi exhibits different superconductivity behavior through a brief study of our calculation as the Tc will reach a maximum value of 2.27 K at around 10 GPa, as is shown in Fig. 6. A further analysis of the result shows that bismuth phonon mode is thought to play a large role in the superconductivity of cF4-Ca3Bi and cF4-CaBi3.

The calculated logarithmic average phonon frequency (ωlog), EPC (λ) and critical temperature Tc for tI2-CaBi as a function of pressure.
Discussion
In summary, by using the variable-composition evolutionary algorithm, we performed a systematic search for all possible compositions in the Ca-Bi system at different pressures. Except the previously reported oP32-Ca5Bi3 and tI84-Ca11Bi10, we found 10 novel structures either totally unreported or only mentioned but no detail information. In addition, we predicted a series of phase transitions in Ca2Bi, Ca3Bi2 and CaBi and also one stoichiometry (Ca3Bi) with a cF4 structure. All the newly predicted structures can be both dynamically and thermodynamically stable as the pressure increases. Based on conventional BCS theory, cF4-CaBi3 is superconductor with a Tc of 5.16 K at 0 GPa and will drop with pressure increases. While cF4-Ca3Bi shows no superconductive behavior below 25 GPa and the Tc value is enhanced with increasing pressure and reaches 5.25 K at 60 GPa. Compared to the above, tI2-CaBi is much different as the Tc will reach a maximum value of 2.27 K at around 10 GPa. The newly predicted structures of calcium bismuthides and superconductivity behavior of cF4-CaBi3, tI2-CaBi and cF4-Ca3Bi would stimulate further experimental and theoretical studies on alkaline earth metal bismuthides and pnictide.
Methods
We used the evolutionary algorithm USPEX to search for low-enthalpy stable structures as implemented in the USPEX code35,36, which has been widely used to predict stable high-pressure crystal structures without requiring any experimental information.
The underlying structural relaxations and electronic structure calculations of Ca-Bi over a wide range of the pressure presented here were performed within the density functional theory (DFT), using the all electron projector augmented wave (PAW) method37 as implemented in the Vienna ab initio simulation package (VASP)38. The 3s23p64s2 and 5d106s26p3 electrons are treated as valence electrons for Ca and Bi atoms, respectively. The exchange-correlation energy was treated within the generalized gradient approximation (GGA), using the functional of Perdew-Burke-Ernzerhof39 for both Ca and Bi. A plane-wave cutoff energy of 500 eV and dense Monkhorst-Pack k-point meshes40 with the reciprocal space resolution of 2π × 0.03 Å−1 were used for all structures to ensure that the enthalpy calculations are converged to better than 1 meV/atom.
The calculation of electron-phonon coupling (EPC) parameter λ are performed using the pseudopotential plane-wave method within the density functional perturbation theory (DFPT) as implemented in the Quantum Espresso package41 using Martins Troullier-type norm-conserving pseudopotential with cutoff energies of 80 and 360 Ry for the wave functions and the charge density, respectively. In order to interpolate the interatomic force constant matrix for the phonon dispersions, 4 × 4 × 4, 4 × 4 × 4 and 4 × 4 × 3 q-meshes in the first Brillouin zone (BZ) were used for interpolation for cF4-CaBi3, cF4-Ca3Bi and tI2-CaBi, respectively. The denser 24 × 24 × 24, 24 × 24 × 24 and 24 × 24 × 18 grids were sufficient to ensure the convergence needed for the EPC calculations for the three calcium bismuthides, respectively.
To ensure the dynamical stability of the newly predicted structures, the phonon dispersion curves were calculated throughout the Brillouin zone using the finite-displacement approach as implemented in the PHONOPY code42.
References
Orenstein, J. Advances in the Physics of High-Temperature Superconductivity. Science 288, 468–474 (2000).
Schilling, A., Cantoni, M., Guo, J. D. & Ott, H. R. Superconductivity above 130 K in the Hg–Ba–Ca–Cu–O system. Nature 363, 56–58 (1993).
Kamihara, Y., Watanabe, T., Hirano, M. & Hosono, H. Iron-based layered superconductor La[O1-xFx]FeAs (x = 0.05–0.12) with Tc = 26 K. J. Am. Chem. Soc. 130, 3296–3297 (2008).
Han, F. et al. SrFeAsF as a parent compound for iron pnictide superconductors. Phys. Rev. B 78, 180503 (2008).
Christianson, A. D. et al. Unconventional superconductivity in Ba0.6K0.4Fe2As2 from inelastic neutron scattering. Nature 456, 930–932 (2008).
Tapp, J. H. et al. LiFeAs: An intrinsic FeAs-based superconductor with Tc = 18 K. Phys. Rev. B 78, 060505 (2008).
Hsu, F. C. et al. Superconductivity in the PbO-type structure alpha-FeSe. Proc. Natl. Acad. Sci. 105, 14262–14264 (2008).
Bauer, E. D., Ronning, F., Scott, B. L. & Thompson, J. D. Superconductivity in SrNi2As2 single crystals. Phys. Rev. B 78, 172504 (2008).
Imai, M. et al. Superconductivity in 122 antimonide SrPt2Sb2. Supercond. Sci. Technol. 26, 075001 (2013).
Kumar, J. et al. Physical property and electronic structure characterization of bulk superconducting Bi3Ni. Supercond. Sci. Technol. 24, 085002 (2011).
Mizuguchi, Y. et al. BiS2-based layered superconductor Bi4O4S3 . Phys. Rev. B 86, 220510 (2012).
Singh, S. K. et al. Bulk superconductivity in bismuth oxysulfide Bi4O4S3 . J. Am. Chem. Soc. 134, 16504–16507 (2012).
Malliakas, C. D., Chung, D. Y., Claus, H. & Kanatzidis, M. G. Superconductivity in the narrow-gap semiconductor CsBi4Te6 . J. Am. Chem. Soc. 135, 14540–14543 (2013).
Lee, J. et al. Crystal structure, lattice vibrations and superconductivity of LaO1−xFxBiS2 . Phys. Rev. B 87, 205134 (2013).
Sturza, M. et al. Superconductivity in the intermetallic pnictide compound Ca11Bi10-x . Phys. Rev. B 89, 054512 (2014).
Zhou, X.-F. et al. Superconducting high-pressure phase of platinum hydride from first principles. Phys. Rev. B 84, 054543 (2011).
Li, Y. L. et al. Formation of Nanofoam carbon and re-emergence of Superconductivity in compressed CaC6 . Sci. Rep. 3, 3331 (2013).
Raza, Z., Errea, I., Oganov, A. R. & Saitta, A. M. Novel superconducting skutterudite-type phosphorus nitride at high pressure from first-principles calculations. Sci. Rep. 4, 5889 (2014).
Hautier, G., Jain, A. & Ong, S. P. From the computer to the laboratory: materials discovery and design using first-principles calculations. J. Mater. Sci. 47, 7317–7340 (2012).
Curtarolo, S. et al. The high-throughput highway to computational materials design. Nat. Mater. 12, 191–201 (2013).
Lyakhov, A. O., Oganov, A. R. & Valle, M. How to predict very large and complex crystal structures. Comput. Phys. Commun. 181, 1623–1632 (2010).
Oganov, A. R. et al. Evolutionary Crystal Structure Prediction as a Method for the Discovery of Minerals and Materials. Rev. Mineral. Geochem. 71, 271–298 (2010).
Oganov, A. R., Lyakhov, A. O. & Valle, M. How evolutionary crystal structure prediction works-and why. Acc. Chem. Res. 44, 227–237 (2011).
Kim, H. et al. Thermodynamic properties of calcium–bismuth alloys determined by emf measurements. Electrochim. Acta 60, 154–162 (2012).
Wang, Y. et al. Thermodynamic assessment of the Sr–In and Sr–Bi systems supported by first-principles calculations. Calphad 45, 49–54 (2014).
Martinez-Ripoll, M., Haase, A. & Brauer, G. The crystal structure of Ca5Bi3 . Acta Crystallogr. B 30, 2004–2006 (1974).
Deller, K. & Eisenmann, B. On the Intermetallic Compounds Ca11Sb10 and Ca11Bi10 . Z. Naturforsch 31b, 29–34 (1976).
Eisenmann, B. & Schafer, H. The Crystal Structures of Ca2Sb and Ca2Bi. Z. Naturforsch 29, 13–15 (1974).
Merlo, F. & Fornasini, M. L. Crystal structure of some phases and alloying behaviour in alkaline earths, europium and ytterbium pnictides. Mater. Res. Bull. 29, 149–154 (1994).
Notin, M. et al. The thermodynamic properties of calcium intermetallic compounds. J. Alloys Compd. 220, 62–75 (1995).
Deng, S., Köhler, J. & Simon, A. A Chemist Approach to Superconductivity. J. Supercond. 15, 635–638 (2002).
Wang, Z. et al. Dirac semimetal and topological phase transitions in A3Bi (A = Na, K, Rb). Phys. Rev. B 85,195320 (2012).
Liu, Z. K. et al. Discovery of a Three-Dimensional Topological Dirac Semimetal, Na3Bi. Science 343, 864–867 (2014).
Allen, P. B. Transition temperature of strong-coupled superconductors reanalyzed. Phys. Rev. B 12, 905–922 (1975).
Oganov, A. R. & Glass, C. W. Crystal structure prediction using ab initio evolutionary techniques: principles and applications. J. Chem. Phys. 124, 244704 (2006).
Lyakhov, A. O., Oganov, A. R., Stokes, H. T. & Zhu, Q. New developments in evolutionary structure prediction algorithm USPEX. Comput. Phys. Commun. 184, 1172–1182 (2013).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Kresse, G. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Monkhorst, H. J. & Pack, J. D. Special points for Brillouin-zone integrations. Phys. Rev. B 13, 5188–5192 (1976).
Giannozzi, P. et al. QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials. J. Phys.: Condens. Matter 21, 395502 (2009).
Togo, A., Oba, F. & Tanaka, I. First-principles calculations of the ferroelastic transition between rutile-type and CaCl2-type SiO2 at high pressures. Phys. Rev. B 78, 134106 (2008).
Acknowledgements
This work was supported by the Research Foundation of Education Bureau of Hebei Province (ZD20131039), the Natural Science Foundation of Hebei Province (E2014203243) and the NSFC (Grant No. 51121061), which is gratefully acknowledged.
Author information
Authors and Affiliations
Contributions
C.Z.F. conceived the idea. X.D. performed the ab initio evolutionary simulations and the superconductivity properties calculations. C.Z.F.and X.D. wrote the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Dong, X., Fan, C. Rich stoichiometries of stable Ca-Bi system: Structure prediction and superconductivity. Sci Rep 5, 9326 (2015). https://doi.org/10.1038/srep09326
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep09326
- Springer Nature Limited
This article is cited by
-
Structural and electronic properties of amorphous bismuth calcium borate from first-principle calculations
Structural Chemistry (2021)