

Abstract
The ever-increasing demand for fluorinated molecules due to their widespread applications has raised substantial interest in the development of new synthetic methodologies that selectively introduce fluorine into molecular scaffolds. While transition-metal-catalysed fluorination reactions in principle provide a direct means to convert inert C–H bonds into C–F bonds, fundamental challenges such as the high energetic barriers associated with the formation of C–F bonds by reductive elimination, among others, remain to be systematically addressed. Carboxylic acids, owing to their versatile synthetic utility in organic synthesis, serve as ideal model substrates in this context. Here we report a protocol that enables the β-C(sp3)–H fluorination of free carboxylic acids, giving access to a wide range of fluorinated carboxylic acids. The rational design of the oxidizing reagent proved to be crucial in establishing the protocol and introduces an additional design dimension to the field of C–H activation.

Similar content being viewed by others


Main
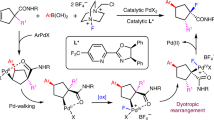
Fluorine, owing to its small size and high electronegativity, can markedly influence chemical, physical and biological properties, such as the lipophilicity, solubility, conformational flexibility and metabolic stability of organic molecules. Therefore, the incorporation of fluorine into molecular scaffolds is of great interest in various fields, including medicinal chemistry1,2, agricultural chemistry3,4, material science5 and positron emission tomography6,7. Generally, two fundamentally distinct classes of fluorination reagents exist in the chemical toolbox: electrophilic (F+ or F.) and nucleophilic (F−) fluorine sources that offer the possibility of forging C–F bonds with orthogonal applicability profiles (substrate structures, functional group compatibilities and so on). Nucleophilic sources of fluorine that are often readily accessible, inexpensive and bench-stable have been widely exploited to construct C–F bonds using functional group interconversion approaches8,9,10. However, these methods heavily rely on the availability of pre-installed reactive handles or native functional groups, which limits the applications of such protocols and implies the need to develop complementary synthetic methods that enable the direct conversion of inert C–H bonds to C–F bonds. In this regard, among other existing strategies11,12,13, transition-metal-catalysed fluorination reactions have emerged as a very promising technique to synthesize fluorinated building blocks in an atom- and step-economical manner. Despite the importance of C(sp3)–F motifs in various fields, transition-metal-catalysed C(sp3)–H fluorination reactions remain underdeveloped in comparison with analogous C(sp2)–H fluorinations11,12. The development of transition-metal-catalysed C(sp3)–H fluorination reactions faces several intrinsic challenges. The formation of C–F bonds by reductive elimination (RE) from low-valent transition metal centres, such as Pd(II) complexes, is very challenging14,15. Consequently, oxidants with a very high oxidation potential are used, to generate high-valent transition-metal species, from which the RE becomes relatively facile. The RE thus constitutes the main obstacle to C–F bond formation. Additional challenges arise from the poor reactivity of inert C(sp3)–H bonds, caused by their low acidity, high bond energies and increased entropic penalty for cyclopalladation resulting from the larger conformational flexibility in the starting material (compared with arene ortho-C–H activation)16,17. To circumvent the challenging RE from Pd(II), a limited number of transition-metal-catalysed C(sp3)–H fluorination reactions have been reported for carboxylic acid derivatives using strongly oxidizing electrophilic fluorinating reagents capable of generating Pd(IV), all of which required the covalent attachment of exogenous directing groups18 on the substrate19,20,21,22. Commercially available electrophilic fluorinating reagents often feature N–F bonds that, besides serving as a stoichiometric oxidant to produce Pd (IV), also transfer the required fluorine atom to palladium alongside the nitrogen-based fragment. From a conceptual point of view, the presence of this (sometimes strongly coordinating) N-containing moiety on palladium may be detrimental for the activation and functionalization of substrates where the entire catalytic cycle relies on designed external ligands. In such cases, the identification of suitable electrophilic fluorinating reagents by structural fine-tuning may not be feasible. In 2012, Sanford and co-workers established the first example of a transition-metal-catalysed C(sp3)–H fluorination reaction using a nucleophilic fluoride source in combination with an exogenous oxidant and accomplished a benzylic C(sp3)–H fluorination23. Despite this seminal work showing the potential of this strategy as an alternative to electrophilic fluorination reagents, this approach has not been exploited for the fluorination of other substrate classes so far. The transition-metal-catalysed direct C(sp3)–H activation and functionalization of free carboxylic acids constitutes a highly attractive goal, as the carboxylic acid moiety is one of the most fundamental and synthetically versatile functional groups in organic chemistry that occurs in the plethora of natural products and pharmacologically relevant compounds24,25. However, the development of such methods remains very challenging due to the low directing ability of carboxylate unit and prevalence of competing coordination modes between palladium and carboxylate moiety, among other reasons26. Over the past years, the area of palladium-catalysed C(sp3)–H activation and functionalization of free carboxylic acids has witnessed remarkable advancements through contributions by various research groups including our laboratory. Most of these studies focused on developing new C–C bond forming reactions26,27,28. Among the C-heteroatom bond forming reactions, the C–O bond formation received substantial attention29,30,31,32. Recently, Yu and co-workers have reported the bromination and chlorination of free carboxylic acids33. Despite the notable progress in this field, the particularly attractive fluorination of free carboxylic acids has remained out of reach. When aiming to develop such a transformation, several important factors have been considered. Due to the weak directing ability of the carboxylate moiety, the use of designed external ligands has proven crucial to accelerate the rate of C–H activation–functionalization reaction, influencing not only the challenging C–H activation step but also the other steps of a catalytic cycle34,35,36. Consequently, progress in this field has hitherto always relied on the extensive screening of ligands, in some cases including the design of novel ligand motifs, to identify a suitable ligand scaffold, followed by extensive structural fine-tuning of the ligand structure and the reaction conditions. In this Article, we set out to develop a direct C(sp3)–H fluorination of carboxylic acids using a nucleophilic fluoride source. As discussed above, the C–F bond formation should rely on a Pd (II)/Pd (IV) catalytic cycle and require the use of a strong external oxidant. So far, studies relying on a Pd (II)/Pd (IV) catalytic cycle have tested a limited number of commercially available oxidants and, after discovering initial promising reactivity, focused on optimizing the remaining reaction parameters, a strategy that proved insufficient for the envisaged reaction. We realized that the typically employed oxidants transfer organic species onto the catalyst during the oxidation process that remain on the catalyst during critical steps of the catalytic cycle. We envisioned that the design of a suitable oxidant (YOOZ) might be key to enable the desired reactivity (Fig. 1). Such peroxide-based oxidants would transfer two components (YO and OZ) onto the catalyst during oxidation, each of them having different roles in the catalytic cycle. The C–H activated palladacycle Int-1 formed via C–H activation of the carboxylic acid would first undergo oxidative addition (OA) with the oxidant to generate Int-2. The generation of this Pd(IV) species is expected to be influenced heavily by the groups (OY and OZ), which define the redox potential of the reagent. Within Int-2, the properties of the oxidant’s component (OZ) must ensure it undergoes fast ligand exchange with the incoming fluoride, thereby enabling the generation of Int-3. The other component of the oxidant (OY), together with the external ligand, is expected to influence the C–F RE process from Int-3. Furthermore, both groups OY and OZ would have to be chosen such as to suppress undesired side reactions in Int-2 or Int-3, in particular the possible C–O RE to give C–OY, C–OZ or a β-lactone. We thus envisaged that the design of oxidants could serve as a new dimension in reaction optimization to complement ligand design and the choice of reaction conditions and enable highly challenging transformations such as the direct C(sp3)–H fluorination of carboxylic acids reported herein.

A proposed catalytic cycle consisting of a C–H activation giving intermediate 1, an OA to give intermediate 2, a ligand exchange giving intermediate 3 and a RE giving the product and regenerating the catalyst is shown, as well as a potential alternate mechanism with an electrophilic fluorinating agent going directly from Int-1 to Int-3 (ref. 23). Challenges associated with the target transformation are shown, and the concept of oxidant design to address these challenges is introduced. Int, intermediate; LG, leaving group; OX/OY, fragments of the designed oxidant.
Results and discussion
Reaction design and optimization
With this design element in hand, we initiated our optimization studies with pivalic acid (1a) as a model substrate and silver fluoride as a fluorine source. To gain initial information regarding the optimal oxidant structure, we evaluated common organic peroxides owing to their well-known ability of oxidizing Pd(II) to Pd(IV)37,38,39. Initially, BzOOt-Bu showed an encouraging reactivity with 20% yield (see Supplementary Information for more details). Due to the propensity of t-BuO− to undergo facile ligand exchange with the incoming nucleophile40, we expected that a structural fine-tuning of the benzoyl motif would influence the steric and electronic properties of Pd(IV) in both Int-2 and Int-3. After an extensive systematic structural variation of the oxidant (for detailed yields of various oxidants see Supplementary Information), the oxidant OX1 in conditions A (Fig. 2) provided the desired product in 55% yield, while the commercially available oxidants (OX3, OX4 and OX6) performed substantially worse, highlighting that ligand design alone was not sufficient to unlock the desired reactivity. Notably, the presence of an electron-donating methyl group in OX5 turned out be detrimental for the reaction, suggesting that the electronic properties of the benzoyl group play a crucial role in this process. We also observed that the reaction benefitted from strong electron-withdrawing groups, such as –CF3, or –NO2, on the oxidant and was sensitive to the position of the substituent on the arene ring, with a substitution at the para position giving maximal yields. Although the commercially available ligand Ac-β-Ala-OH gave considerable reactivity, a further optimization of ligand structure revealed L1 to be the best-performing ligand under conditions A. With these conditions in hand, we became interested in expanding our protocol to α-non-quaternary substrates, which are considered to be inherently more challenging substrates owing to the lack of a favourable Thorpe–Ingold effect and the reduced stability of the intermediately formed palladacycle. As commercially available oxidants (OX3, OX4 and OX6) offered little reactivity, a structural fine-tuning of the oxidant led us to discover the benzoyl peroxide-based oxidant OX2, which provided 51% yield of the desired product 2b alongside 40% of recovered starting material under conditions B (Fig. 2). Once more, an extensive structural fine-tuning of the Ac-β-Ala-OH motif led us to identify L2 as a suitable ligand. It is important to note that the commercially available oxidants performed substantially worse than OX2 even under the optimized conditions (Fig. 2), which again highlights how crucial the strategy of oxidant design was in developing this transformation.

Reactions were conducted on a 0.1 mmol scale. Yields were determined by 1H NMR spectroscopic analysis of the crude reaction mixture using 1,3,5-trimethoxybenzene as an internal standard. L, ligand; OX, oxidant.
Scope studies of the fluorination reaction
Having identified the suitable reaction conditions, we proceeded to study the scope of our transformation. The substrate scope of α-quaternary substrates was evaluated first. As expected from the optimization studies, the product 2a derived from pivalic acid 1a was obtained in 55% yield and a mono:di ratio of 5:1 under conditions A (Fig. 3). Variation in the aliphatic chain length provided satisfactory yields for 2c (65%, mono:di ratio of 13:1) and 2d (53%). Substrates having trifluoromethyl substituent 2e (62%, mono:di ratio of 16:1) as well as fluorine substituent 2f (62%, mono:di ratio of 9:1) on the side chain were well tolerated. We were furthermore interested in examining various aryl-containing substrates to check whether the benzylic C–H bonds present in these substrates, which could cause side reactions due to their low bond dissociation energies, are compatible with our reaction conditions. A range of functional groups was well tolerated, including an ester 2h (42%), nitrile 2i (44%), sulfur-containing motifs in 2j (60%) and 2k (52%), a halogen-substituent in 2l (57%), a nitro group in 2m (53%, mono:di ratio of 19:1), a ketone in 2n (57%) and a trifluoromethyl group in 2o (54%). To further probe the synthetic utility of our protocol, a representative reaction was performed on a 1 mmol scale giving 2c in 55% yield, thereby demonstrating that our protocol can be conducted on a preparatively useful scale. We proceeded to explore the scope of α-non-quaternary substrates under conditions B. Substrates with gradual increase in the chain length (2p, 2b, 2q, 2r and 2s) as well as a branched-chain 2t provided satisfactory yields, while propionic acid provided 16% NMR spectroscopic yield (see Supplementary Information for details). A series of substrates bearing heteroatom-containing functional groups (2u–2x) afforded the respective fluorinated products in moderate to good yield. Electron-poor arene groups, such as in ester 2z (43%), sulfonate 2aa (43%), haloarene 2ab (45%), nitro-substituted 2ac (45%), ketone 2ae (41%) and trifluoromethyl-substituted 2af (54%), were well tolerated under our reaction conditions. Electron-rich arenes, on the other hand, gave poor yields under our optimized reaction conditions, presumably due to side reactions via the highly reactive benzylic C(sp3)–H bonds. Considering that such side reactions would consume some of the oxidant, we reasoned that a slight increase in the oxidant loading could help to drive our desired reaction to completion. To our delight, this proved to be effective, allowing us to obtain good yield for substrates bearing electron-rich arenes. Overall, a considerable functional group tolerance could once more be demonstrated even for the challenging α-non-quaternary substrates.

Reactions were conducted on a 0.2 mmol scale. Mono:di ratios larger than 20:1 are not reported. aIsolated after conversion to naphthyl ester. bIsolated after conversion to the benzyl ester. cThe 1H NMR spectroscopic yield using CH2Br2 as internal standard. d2.25 equivalents of OX2 were used. eOX1 was used as limiting reagent with 2.5 equivalents of pivalic acid.
Mechanistic insights
Considering the challenges associated with the key C–F RE, we became interested in gaining initial mechanistic insights, concerning this part of the reaction mechanism. As depicted in Fig. 4a, there are two plausible pathways that could lead to the product formation: Int-2 could first undergo ligand exchange with the incoming nucleophile F− to give Int-3, from where a C–F RE would form the desired product (path A; cf. Fig. 4). Alternatively, either Int-2 and/or Int-3 could undergo a C–O RE to form a β-lactone, which would then undergo a subsequent SN2 reaction with F− to afford 2 (path B). Interestingly, during the scope investigations, we could in some cases detect traces of β-lactone (<3% 1H NMR spectroscopic yield), which could either constitute a side product formed by a competing RE from Int-2 and Int-3 (path A) or a reaction intermediate (path B). We thus performed control experiments to elucidate which one of these pathways is operative. When β-lactone 3a was subjected to conditions A, the corresponding fluorinated product 2c was not detected by 1H NMR and 19F NMR spectroscopic analyses, while more than 95% of 3a could still be detected after the reaction, implying that the SN2 reaction of β-lactone with F− does not occur to a relevant degree under our reaction conditions. To further corroborate this result, we considered the scenario that a reactive intermediate formed during our C–H fluorination reactions could be necessary to trigger the ring-opening process of β-lactone. To probe this, 1a and 3a were subjected to conditions A together, which resulted in the formation of 2a in 55% yield (proving that the β-lactone has no detrimental effect on the reaction) while 2c was not detected. Again, more than 95% of β-lactone 3a was recovered after the reaction. Overall, these results allow us to rule out path B and, thus, constitute strong support for the involvement of a C–F RE in the direct fluorination of carboxylic acids.

a, Possible pathways for the generation of fluorinated acids 2 from intermediate 2. Path A denotes the direct fluorination via a C–F reductive elimination from Int-3 as shown in more detail in Fig. 1. Path B consists of a C–O RE forming a β-lactone 3, followed by a nucleophilic substitution with fluoride to give fluorinated acid 2. b, A control experiment shows that an independently synthesized β-lactone 3a is not converted to the respective fluorinated acid 2c under the reaction conditions. A second control experiment shows that, when substrate 1a is present alongside 3a, it is converted to 2a in 55% yield as expected from the scope studies. β-Lactone 3a is recovered in both control experiments, thereby excluding product formation via β-lactones as intermediates as potential mechanistic scenario and corroborating to a mechanism involving a direct C-F RE. Yields were determined by 1H NMR spectroscopic analysis of the crude reaction mixture using 1,3,5-trimethoxybenzene as an internal standard.
Conclusion
In summary, we have developed a protocol that enables the direct β-C(sp3)–H fluorination of free carboxylic acids. The reaction constitutes a rare example in C–H fluorination, where a nucleophilic fluoride source is combined with an external oxidant. The reported synthetic method employs the free carboxylic acid as the limiting reagent and uses an operationally simple source of fluoride. It was shown to be suitable for challenging α-non-quaternary carboxylic acids and displays considerable functional group tolerance, giving access to a broad range of fluorinated compounds. The key to success in this study has been the rational design of the oxidants. We identify oxidant design as a new design dimension that complements ligand design and can be adopted broadly in organic synthesis to develop synthetic methods that require high-valent transition-metal species. The further use of these oxidants to unlock new reactivities is currently ongoing in our laboratory.
Methods
Fluorination of α-quaternary carboxylic acids
An oven-dried 10 ml Schlenk tube was charged with Pd(OAc)2 (4.5 mg, 0.020 mmol, 10 mol%), L1 (5.7 mg, 0.020 mmol, 10 mol%), NaHCO3 (8.4 mg, 0.10 mmol, 0.5 equiv.), OX1 (59.8 mg, 0.250 mmol, 1.25 equiv.), AgF (63.4 mg, 0.500 mmol, 2.5 equiv.), carboxylic acid (0.2 mmol, 1.0 equiv.) and HFIP (3.5 ml). The reaction mixture was stirred (stirring speed 900 rpm) at 60 °C for 24 h in a pre-heated metal block. The reaction mixture was allowed to cool to room temperature, and formic acid (0.2 ml) was added. The mixture was filtered through a pad of Celite using CH2Cl2 (35 ml) to complete the elution, and the volatiles were removed under reduced pressure. K2CO3 (138 mg, 1.00 mmol, 5.0 equiv.), NaI (4.5 mg, 0.060 mmol, 0.3 equiv.), 2-(bromomethyl)naphthalene (220 mg, 1.00 mmol, 5.0 equiv.) or benzyl bromide (118 µl, 1.00 mmol, 5.0 equiv.) and acetone (5 ml) were added, and the resulting mixture was stirred at room temperature for 24 h. The mixture was filtered through a pad of Celite using CH2Cl2 (25 ml) to complete the elution, all volatiles were removed under reduced pressure and the residue was purified by silica gel column chromatography to afford the desired fluorinated product as an ester, with the yields given in Fig. 3.
Fluorination of α-non-quaternary carboxylic acids
An oven-dried 10 ml Schlenk tube was charged with Pd(OAc)2 (4.5 mg, 0.020 mmol, 10 mol%), L2 (17.2 mg, 50.0 µmol, 25 mol%), KHCO3 (20 mg, 0.20 mmol, 1.0 equiv.), OX2 (132.4 mg, 350.0 µmol, 1.75 equiv.), AgF (63.4 mg, 0.500 mmol, 2.5 equiv.), carboxylic acid (0.2 mmol, 1.0 equiv.) and HFIP (2.2 ml). The reaction mixture was stirred (stirring speed 900 rpm) at 60 °C for 24 h in a pre-heated metal block. The reaction mixture was allowed to cool to room temperature, and formic acid (0.2 ml) was added. The mixture was filtered through a pad of Celite using CH2Cl2 (35 ml) to complete the elution, and the volatiles were removed under reduced pressure. Cs2CO3 (425 mg, 1.00 mmol, 6.5 equiv.), NaI (75 mg, 0.050 mmol, 2.5 equiv.), 2-(bromomethyl)naphthalene (220 mg, 1.00 mmol, 5.0 equiv.) or benzyl bromide (118 µl, 1.00 mmol, 5.0 equiv.) and acetone (5 ml) were added, and the resulting mixture was stirred at room temperature for 24 h. The mixture was filtered through a pad of Celite using CH2Cl2 (25 ml) to complete the elution, all volatiles were removed under reduced pressure and the residue was purified by silica gel column chromatography to afford the desired fluorinated product as an ester, with the yields given in Fig. 3.
Data availability
The data supporting the findings reported in this study, description of experiments, analytical data and NMR spectra are available within the paper and its Supplementary Information files.
References
Hagmann, W. K. The many roles for fluorine in medicinal chemistry. J. Med. Chem. 51, 4359–4369 (2008).
Müller, K., Faeh, C. & Diederich, F. Fluorine in pharmaceuticals: looking beyond intuition. Science 317, 1881–1886 (2007).
Jeschke, P. Recent developments in fluorine-containing pesticides. Pest Manag. Sci. 80, 3065–3087 (2024).
Pazenok, S. et al. Modern fluorine-containing agrochemicals. in Patai’s Chemistry of Functional Groups 1–77 (Wiley, 2022).
Okazoe, T. Overview on the history of organofluorine chemistry from the viewpoint of material industry. Proc. Jpn Acad. Ser. B 85, 276–289 (2009).
van der Born, D. et al. Fluorine-18 labelled building blocks for PET tracer synthesis. Chem. Soc. Rev. 46, 4709–4773 (2017).
Campbell, E., Jordan, C. & Gilmour, R. Fluorinated carbohydrates for 18F-positron emission tomography (PET). Chem. Soc. Rev. 52, 3599–3626 (2023).
Britton, R. et al. Contemporary synthetic strategies in organofluorine chemistry. Nat. Rev. Methods Primers 1, 47 (2021).
Campbell, M. G. & Ritter, T. Modern carbon–fluorine bond forming reactions for aryl fluoride synthesis. Chem. Rev. 115, 612–633 (2015).
Liang, T., Neumann, C. N. & Ritter, T. Introduction of fluorine and fluorine-containing functional groups. Angew. Chem. Int. Ed. 52, 8214–8264 (2013).
Leibler, I. N.-M., Gandhi, S. S., Tekle-Smith, M. A. & Doyle, A. G. Strategies for nucleophilic C(sp3)–(radio)fluorination. J. Am. Chem. Soc. 145, 9928–9950 (2023).
Szpera, R., Moseley, D. F. J., Smith, L. B., Sterling, A. J. & Gouverneur, V. The fluorination of C–H bonds: developments and perspectives. Angew. Chem. Int. Ed. 58, 14824–14848 (2019).
Cheng, Q. & Ritter, T. New directions in C–H fluorination. Trends Chem. 1, 461–470 (2019).
Grushin, V. V. & Marshall, W. J. Ar–F reductive elimination from palladium(II) revisited. Organometallics 26, 4997–5002 (2007).
Watson, D. A. et al. Formation of ArF from LPdAr(F): catalytic conversion of aryl triflates to aryl fluorides. Science 325, 1661–1664 (2009).
Arndtsen, B. A., Bergman, R. G., Mobley, T. A. & Peterson, T. H. Selective intermolecular carbon–hydrogen bond activation by synthetic metal complexes in homogeneous solution. Acc. Chem. Res. 28, 154–162 (1995).
Meng, G. et al. Achieving site-selectivity for C–H activation processes based on distance and geometry: a Carpenter’s approach. J. Am. Chem. Soc. 142, 10571–10591 (2020).
Sambiagio, C. et al. A comprehensive overview of directing groups applied in metal-catalysed C–H functionalisation chemistry. Chem. Soc. Rev. 47, 6603–6743 (2018).
Zhang, Q., Yin, X.-S., Chen, K., Zhang, S.-Q. & Shi, B.-F. Stereoselective synthesis of chiral β-fluoro α-amino acids via Pd(II)-catalyzed fluorination of unactivated methylene C(sp3)–H bonds: scope and mechanistic studies. J. Am. Chem. Soc. 137, 8219–8226 (2015).
Zhu, Q., Ji, D., Liang, T., Wang, X. & Xu, Y. Efficient palladium-catalyzed C–H fluorination of C(sp3)–H bonds: synthesis of β-fluorinated carboxylic acids. Org. Lett. 17, 3798–3801 (2015).
Miao, J., Yang, K., Kurek, M. & Ge, H. Palladium-catalyzed site-selective fluorination of unactivated C(sp3)–H bonds. Org. Lett. 17, 3738–3741 (2015).
Zhu, R.-Y. et al. Ligand-enabled stereoselective β-C(sp3)–H fluorination: synthesis of unnatural enantiopure anti-β-fluoro-α-amino acids. J. Am. Chem. Soc. 137, 7067–7070 (2015).
McMurtrey, K. B., Racowski, J. M. & Sanford, M. S. Pd-catalyzed C–H fluorination with nucleophilic fluoride. Org. Lett. 14, 4094–4097 (2012).
Gooßen, L. J. et al. New catalytic transformations of carboxylic acids. Pure Appl. Chem. 80, 1725–1733 (2008).
Gooßen, L. J., Rodríguez, N. & Gooßen, K. Carboxylic acids as substrates in homogeneous catalysis. Angew. Chem. Int. Ed. 47, 3100–3120 (2008).
Uttry, A. & van Gemmeren, M. Direct C(sp3)–H activation of carboxylic acids. Synthesis 52, 479–488 (2020).
Dutta, S. et al. functionalisation of free carboxylic acids. Chem. Sci. 13, 2551–2573 (2022).
Das, A. & Maji, B. The emergence of palladium-catalyzed C(sp3)–H functionalization of free carboxylic acids. Chem. Asian J. 16, 397–408 (2021).
Ghosh, K. K., Uttry, A., Koldemir, A., Ong, M. & van Gemmeren, M. Direct β-C(sp3)–H acetoxylation of aliphatic carboxylic acids. Org. Lett. 21, 7154–7157 (2019).
Zhuang, Z. & Yu, J.-Q. Lactonization as a general route to β-C(sp3)–H functionalization. Nature 577, 656–659 (2020).
Chan, H. S. S., Yang, J.-M. & Yu, J.-Q. Catalyst-controlled site-selective methylene C–H lactonization of dicarboxylic acids. Science 376, 1481–1487 (2022).
Kao, L.-C. & Sen, A. Platinum(II) catalysed selective remote oxidation of unactivated C–H bonds in aliphatic carboxylic acids. J. Chem. Soc. Chem. Commun. 1242–1243 (1991).
Hu, L. et al. Enhancing substrate–metal catalyst affinity via hydrogen bonding: Pd(II)-catalyzed β-C(sp3)–H bromination of free carboxylic acids. J. Am. Chem. Soc. 145, 16297–16304 (2023).
Engle, K. M., Wang, D.-H. & Yu, J.-Q. Ligand-accelerated C–H activation reactions: evidence for a switch of mechanism. J. Am. Chem. Soc. 132, 14137–14151 (2010).
Ghosh, K. K. & van Gemmeren, M. Pd-catalyzed β-C(sp3)–H arylation of propionic acid and related aliphatic acids. Chem. Eur. J. 23, 17697–17700 (2017).
Uttry, A., Mal, S. & van Gemmeren, M. Late-Stage β-C(sp3)–H deuteration of carboxylic acids. J. Am. Chem. Soc. 143, 10895–10901 (2021).
Koten, G., Alsters, P. L., Teunissen, H. T., Boersma, J. & Spek, A. L. Oxygenation of cyclopalladated N,N-dimethylbenzylamine complexes by inorganic and organic peroxides: oxygen insertion into the palladium–carbon bond. Organometallics 12, 4691–4696 (1993).
Canty, A. J., Jin, H., Skelton, B. W. & White, A. H. Oxidation of complexes by (O2CPh)2 and (ER)2 (E=S, Se), including structures of Pd(CH2CH2CH2CH2)(SePh)2(bpy) (bpy = 2,2′-bipyridine) and MMe2(SePh)2(L2) (M = Pd, Pt; L2 = bpy, 1,10-phenanthroline) and C.O and C.E bond formation at palladium(IV). Inorg. Chem. 37, 3975–3981 (1998).
Giri, R. et al. Pd-catalyzed stereoselective oxidation of methyl groups by inexpensive oxidants under mild conditions: a dual role for carboxylic anhydrides in catalytic C–H bond oxidation. Angew. Chem. Int. Ed. 44, 7420–7424 (2005).
Zhuang, Z., Herron, A. N., Fan, Z. & Yu, J.-Q. Ligand-enabled monoselective β-C(sp3)–H acyloxylation of free carboxylic acids using a practical oxidant. J. Am. Chem. Soc. 142, 6769–6776 (2020).
Acknowledgements
We thank Kiel University and the Deutsche Forschungsgemeinschaft (DFG; Emmy-Noether-Programme GE 2945/2-1 to M.v.G. and Walter Benjamin Programme HI 2351/1-1 to K.H.) for generous financial support. We thank the technical staff of our institute for their excellent service. F. Deufel, G. Kohlmeyer-Yilmaz, F. Sönnichsen and G. Ayoub Agha are acknowledged for helpful scientific discussions. M. Luft is acknowledged for measuring IR spectra and melting points.
Funding
Open access funding provided by Christian-Albrechts-Universität zu Kiel.
Author information
Authors and Affiliations
Contributions
S.M., F.J. and K.H. performed the experiments and analysed the data. S.M. and M.v.G. conceived and designed the experiments and wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Synthesis thanks Biplab Maji, Michael Schnürch and Bing-Feng Shi for their contribution to the peer review of this work. Primary Handling Editor: Thomas West, in collaboration with the Nature Synthesis team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Description of experiments, analytical data and NMR spectra.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mal, S., Jurk, F., Hiesinger, K. et al. Pd-catalysed direct β-C(sp3)–H fluorination of aliphatic carboxylic acids. Nat. Synth (2024). https://doi.org/10.1038/s44160-024-00578-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s44160-024-00578-6
- Springer Nature Limited