
Abstract
Carbon is considered to exist in three basic forms: diamond, graphite/graphene/fullerenes, and carbyne, which differ in a type of atomic orbitals hybridization. Since several decades the existence of the fourth basic carbon allotropic form with the face-centered cubic (fcc) crystal lattice has been a matter of discussion despite clear evidence for its laboratory synthesis and presence in nature. Here, we obtain this carbon allotrope in form of epitaxial films on diamond in a quantity sufficient to perform their comprehensive studies. The carbon material has an fcc crystal structure, shows a negative electron affinity, and is characterized by a peculiar hybridization of the valence atomic orbitals. Its bandgap (~6 eV) is typical for insulators, whereas the noticeable electrical conductivity (~0.1 S m−1) increases with temperature, which is typical for semiconductors. Ab initio calculations explain this apparent contradiction by noncovalent sharing p-electrons present in the uncommon valence band structure comprising an intraband gap. This carbon allotrope can create a new pathway to ‘carbon electronics’ as the first intrinsic semiconductor with an ultra-wide bandgap.
Similar content being viewed by others


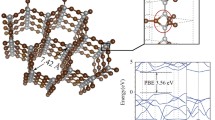
Introduction
For more than 60 years scientists have been aware of an unusual allotropic form of carbon with the face-centered cubic (fcc) structure but could not elucidate it and recognize it as an individual phase. Up to date a great number of studies indicating the existence of this carbon allotrope emerged1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22, nevertheless, according to ref. 1 its crystal structure “…remains a mystery”.
First crystallographic evidence for the presence of fcc-carbon in synthetic diamonds was reported in the classical work of ref. 2. The X-ray diffraction (XRD) from diamond grits obtained at high temperatures and ultra-high pressures showed, apart from conventional for diamond diffraction pattern, also strong (200)-, (222)-, and (420)-reflexes which are forbidden for the diamond crystal lattice. Besides that, there were side satellite diffraction maxima found for all diamond reflexes. These additional maxima form the pattern of a face-centered cubic crystal structure.
Subsequently, an initially amorphous film, which was produced via carbon condensation assisted by the bombardment with Ar+-ions and annealed at a low temperature, contained nanoparticles of fcc-carbon3. According to electron diffraction results the lattice constant of the fcc-carbon particles was found to be 3.57 Å, which is close to that of diamond (3.567095 ± 0.000017 Å according to ref. 23). The authors of ref. 3 assumed that this carbon allotrope is a metallic form of carbon based on the crystal structure itself and closeness of the lattice parameter to that of typical fcc-metals. However, as it was shown in ref. 4, the metallic nature of fcc-carbon is out of the question due to the similarity of its crystal lattice parameter to that of typical fcc metals (e.g., Cu and Ni) at a significantly lower atomic radius of carbon compared to that of such fcc metals.
Later, this carbon allotrope was also synthesized at ultra-high pressures4,5,6,7. In 1991 Hirai and Kondo obtained nanocrystals of fcc-carbon by rapid cooling following the shock compression of graphite sheets5. The authors introduced for the first time the term “n-diamond“ (new-diamond) to designate this carbon allotrope, when considering the closeness of its lattice parameter to that of diamond but ignoring the occurrence of the reflexes forbidden for the diamond crystal lattice. In 1994 the uncommon carbon allotrope was obtained by the dynamic shock compression of a diamond/amorphous carbon mixture6. In the same year fcc-carbon was reported to be produced from natural graphite at high temperatures and static ultra-high pressures7.
Chemical vapor deposition was also employed for obtaining this form of carbon8,9. Nanoparticles of fcc-carbon were produced in the early 2000s by etching a diamond single crystal in a hydrogen plasma and also by plasma assisted chemical vapor deposition10,11,12. In particular, experimental evidence based on the density of electron states and electron energy loss spectroscopy (EELS) was obtained in ref. 12 clearly indicating that fcc-carbon fundamentally differs from diamond with respect to the nature of its chemical bonds12.
Chlorination of SiC and other carbides also resulted in obtaining this carbon allotropic form in the early 2000s13,14. The authors of refs. 13,14 designated it as “diamond-structured carbon” and, later, the term “carbide-derived carbon” was introduced and is presently employed in the literature.
The first evidence for the existence of fcc-carbon in nature, particularly as nanoparticles in the asphaltenes precipitated from a crude oil from Mexico, was reported in 200415. Afterwards, in the late 2000s—early 2010s, a number of publications16,17,18,19,20,21 provided evidence for the presence of fcc-carbon nanoparticles in nature (soils, rocks, Greenland ice, etc.). It was suggested that such nanoparticles are likely to form in Earth’s mantle and are transferred to Earth’s surface during volcanos’ eruptions, so they have a terrestrial origin4.
According to the results of electron diffraction reported in the literature this carbon allotrope has a space group Fm \(\bar{3}\) m (group number 225) with a lattice parameter of about 3.54–3.57 Å. Nevertheless, because of the closeness of its lattice parameter to that of diamond, in many publications on its synthesis1,5,6,7,13,14 and existence in nature15,16,17,18,19,20,21, the carbon allotrope was considered as a diamond polymorph and designated as “n-diamond”, so its crystal structure is still a matter of discussions22.
The issue of the existence of fcc-carbon was also a subject of several publications on ab initio calculations24,25,26,27,28. This structure was recognized as less stable than that of diamond, and the experimentally established crystal lattice parameter could not be confirmed by calculations from the first principles24,25,26,27. In one published work27 the occurrence of fcc-carbon was explained by hydrogen doping of diamond; however, this assumption predicted the XRD pattern different from that obtained experimentally. In contrast to refs. 24,25,26,27, the basic theoretical results of ab initio calculations reported in ref. 28 justified the existence of the carbon allotrope with the face-centered cubic structure and predicted its lattice constant matching experimental data.
The major objective of the present work was to obtain fcc-carbon in a quantity sufficient to perform its comprehensive study by different methods and unambiguously establish its crystal and electronic structure. To explain the established peculiar electronic properties and electronic structure of this carbon allotrope ab initio calculations were carried out.
Results
Synthesis of fcc-carbon films
Thin films of fcc-carbon were obtained on single-crystalline diamond substrates by plasma-assisted chemical vapor deposition (PACVD) from CH4-H2 gas mixtures at very special conditions given in Methods. An exclusive formation of fcc-carbon instead of diamond is explained by faster etching rate of the latter. This happens at insufficient for the diamond deposition presence of metastable C-H species combined with unusually low substrate temperature. Still, the decomposition of such carbon species occurs with a deposition rate of ~0.14 nm/min. In a very narrow window of deposition conditions this decomposition results in the formation of the fcc-carbon phase, which presumably has lower susceptibility to etching by atomic hydrogen compared to that of diamond. The sample surface is shown in Supplementary Information, Fig. S1.
Crystal structure
Figure 1a and Table 1 give the electron diffraction pattern from nm-sized fragments obtained from the single-crystalline film by the mechanical detachment (scratching) with a Cu grid. The pattern clearly indicates that the carbon film has a face-centered cubic crystal lattice with the space group Fm \(\bar{3}\) m, as it comprises all the reflections forbidden for the diamond crystal lattice. It is worth noticing that the material revealed a remarkable plasticity when it was mechanically detached from the film.

a Electron diffraction pattern from nm-sized fragments obtained from the single-crystalline film by the mechanical detachment (scratching) with a Cu grid. b–d X-ray diffraction maxima obtained from the film at different tilting angles Psi. Note that the (200)- and (222)-reflexes are forbidden for the diamond crystal lattice. The (111)- and (220)-reflexes exist for both phases; the peaks pointed by starlets and having significantly higher intensity correspond to the diamond substrate.
The XRD pattern also shows reflexes forbidden for the diamond crystal lattice (Fig. 1b, c and Table 1). The fact that some reflexes forbidden for the diamond crystal lattice are split (for example, the (200)-reflex shown in Fig. 1b) can be explained by the nonuniform distortion of the cubic crystal lattice. This phenomenon is discussed and explained in Supplementary Information, Fig. S2 and Fig. S3).
As one can see from all the acquired diffraction patterns (Fig. 1b–d), the fcc-carbon film grows on the (100)-diamond substrate epitaxially. No additional peaks were detected in the classical grazing incidence procedure (incident angle varied in the range 0.5–5°) or Bragg-Brentano XRD experiments. We also point out that no diffraction maxima in the 10°…30° 2θ-range, which could arise from graphite, graphene/carbon nanotubes or oxidized graphene, were detected. A comparison of the Phi-scans and rocking curves (see Supplementary Information, Fig. S4 and Fig. S5) for the diffraction maxima of the same system of planes in diamond and in fcc-carbon uniquely corroborates the epitaxial relation between the film and the substrate.
The high-resolution transmission electron microscopy (HRTEM) image (Fig. 2a, left) also confirms the face-centered cubic crystal structure of the film. It provides, as it is shown in Fig. 2a, the interplanar distance of roughly 2 Å, corresponding to the distance between (200) atomic planes in the face-centered cubic lattice schematically illustrated by the (110)-projection of an elementary cell (Fig. 2a, right). The fast Fourier transform (FFT) and inverse FFT images shown in Fig. 2b indicate a similar distance (~1.8 Å), which is even closer to the expected one. The determined from interatomic distances {111} and {200} planes build the angle of ~55°, that well corresponds to the face-centered crystal structure. A combination of the interatomic distances and angles determined from the HRTEM image unambiguously rules out the diamond structure.

a HRTEM image of the fcc-carbon crystal lattice in the (110) orientation (left) and schematic (110)-projection of the fcc-elementary cell (right); b fast Fourier transform (FFT) image with the indicated angle between the {111} and {200} planes (left) and inverse FFT image (right).
The lattice spacing values calculated from our experimental diffraction patterns are summarized and compared with the literature data in Table 1. The free-standing fcc-carbon particles reveal the lattice spacings close to those reported in literature. On the contrary, the epitaxial film on diamond shows relatively large deviations of the lattice spacings, and they cannot be consistently explained by the existence of strains, which supports the abovementioned plasticity of the material.
Spectroscopic characteristics
The Raman spectrum recorded from mechanically detached fragments of the film is shown in Fig. 3a. As one can see, the Raman spectrum does not comprise any signals. It is well known that materials with a face-centered cubic crystal lattice, the primitive unit cell of which contains only a single atom, have only acoustic phonons and no optical phonons29,30. It should be noted that the presence of nanodiamonds in the film would manifest a couple of broad vibrational modes and their presence can therefore be discarded. Accordingly, the Raman spectrum from the carbon film on the diamond substrate (Fig. 3b) comprises no material related signals; only the strong peak from the diamond substrate is visible due to the transparency of the fcc-carbon film. Some increase of FWHM after film deposition is consistent with the dramatic change of the substrate surface geometry and roughness. Diamond substrate reveals apart from the main, characteristic for diamond signal, some additional modes that have several orders of magnitude lower intensity and can be ascribed to the presence of structural defects at the diamond surface.

a Raman spectrum obtained for mechanically detached fragments of the film located in a cell of a Cu grid; the dashed circle marks the area of analysis. b A comparison of Raman spectra obtained for the as-deposited epitaxial film on diamond substrate and the bare diamond substrate. Insets show the corresponding optical images. c Vacuum ultraviolet (VUV) reflection spectrum presented as measured for the film (inset) and as a Tauc-plot reconstructed on the basis of the Kubelka-Munk theory (see Methods). d X-ray photoelectron spectrum (XPS) obtained for the as-deposited carbon film after its fitting (the fitting procedure is described in Methods).
Vacuum ultraviolet reflection spectrum presented as measured for the film and as a Tauc-plot reconstructed based on the Kubelka-Munk theory is shown in Fig. 3b. Based on these results a value of bandgap of the carbon film is found to be nearly 6.1 eV. As the transmittance data were not affordable the magnitude of absorption coefficient is not given here. We detected a nonzero background which is explained by a depleted occupation of the valence band (see Ab initio calculations).
The analysis of the XPS spectrum presented in Fig. 3d is nontrivial. The spectrum shows a significant degree of surface contamination since the sample was exposed to air. No sp2 contribution was detected that allows excluding any influence of graphitized fraction on the film properties, particularly on the electrical conductivity. The major peak C1s is present at an untypically low value of 283.9 eV providing evidence that there are electrons in the system, which are weaker bound than if any kind of covalent bonds would exist. The contributions at 286.2 and 288.7 eV are attributed to the carbon chemical states respectively in the C-O-C and O-C=O species. The peak at ~284.5 eV which we attributed to an adventitious carbon (C-C, C-H) has almost the same position as the sp2-contribution, which was used for the calibration (see Methods). In the latter case, however, the (π−π)-satellite would show up and the good match of the binding energy (BE) seems to be incidental. Indeed, the BE measured for adventitious carbon was found to vary from 284.08 to 286.74 eV depending on the substrate31. The peak, which we ascribe as originating from fcc-carbon (~283.9 eV), was fit with the asymmetric line shape because of the material electrical conductivity (see, e.g., ref. 32). Such an asymmetry can arise from the multi-electron excitations and electron–hole pairs creation in the delocalized sub-valence band, which is believed to comprise nearly unhybridized s and p electrons (see Ab initio calculations). Note that the symmetric peak at 284.8 eV, which is typical for diamond, and signals from π-π* bonds in the range between ~290 and 293 eV, which are typical for sp2-hybridized carbon33, are absent in the spectrum.
The results of Fourier-transform infrared spectroscopy (FTIRS) also clearly indicate a difference in the nature of chemical bonds between diamond and fcc-carbon and provide evidence for the absence of carbon-hydrogen bonds in the electronic structure of fcc-carbon (see Supplementary Information, Fig. S6). Thus, the explanation of the crystal structure of fcc-carbon due to the presence of hydrogen atoms between adjacent carbon atoms suggested in ref. 27 is confirmed to be incorrect.
Electrical conductivity and electronic structure
It was reported in ref. 22 that fcc-carbon is characterized by electrical conductivity. Based on its crystal structure it was suggested to have a metallic character of conductance. Our comprehensive measurements clearly demonstrated that the material is a semiconductor (Fig. 4). The conductivity at room temperature is reasonably high (~10−3 S cm−1). As noted in Methods the Hall voltage could not be measured reliably. Besides the unfavorable for this method film shape, the concentration of free charge carriers in fcc-carbon is expected to be fairly low. Therefore, its reliable determination is the matter of a special sample preparation which is beyond the scope of this study.

Activation energy was determined for the linear correlation range. The film thickness was set to 1 μm.
The extrapolated value of the activation energy (~230 meV) is unusually high if compared to classical semiconductors. It is known that both chemical state and size of crystallites (flakes) affect this energy. In polycrystalline and composite carbon materials, the activation energy is characterized by the hoping mechanism and electrons’ tunneling34. The high value of Ea may be caused by a poor percolation of electron conducting domains35,36. Comparable magnitudes of Ea are demonstrated also by organic semiconductors, in which the ionization of molecules is a crucial stage of the conductivity thermal activation37; this is assumed to indicate the similarity of the nature of chemical bonds in fcc-carbon and inter-molecular bonds in organic semiconductors (see below).
The optical bandgap established for the carbon film is nearly 6.1 eV. The estimation based on the EELS published in our previous work12 gives the bandgap value close to 7 eV. It is known that an electronic bandgap is usually larger than an optical gap due to the need to overcome the Coulomb repulsion energy when electrons are injected into the material. In our case the difference may also result from a crystal orientation which was (100) in case of the optical measurements and uncertain in electronic measurements. The established discrepancy between the values of bandgap obtained by different methods may also relate to the weird shape of the film (see Supplementary Information for details). In fact, the bandgap is strongly dependent on the interatomic distance and increases if atoms are remote from each other and the degree of electrons’ delocalization decreases. In that sense, fcc-carbon shows consistently larger bandgap than that of diamond.
A precise determination of the work function (ϕ) is challenging for the diamond surface owing to several factors described in refs. 38,39,40. Among them possible O- or H-termination, presence of graphitic carbon and carbon-containing adsorbates. Moreover, the crystallinity (crystal size) and crystallographic orientation also have an impact on the ionization energy40. To avoid surface adsorbates originated from air and consequent changes of electronic properties, the Kelvin probe (KP) was performed and photoelectron yield spectroscopy (PYS) measurements according to ref. 39 in a dry nitrogen atmosphere were carried out. Prior to the KP measurements the surface of the samples were acid cleaned to eliminate dangling hydrogen atoms and weakly bound adsorbates from ambient air as described in Methods.
The results of the work function (ϕ) measurements by using the Kelvin probe method show quite a large difference between the measured curves for fcc-carbon and diamond (Fig. 5a). While a value of 4.74 eV is obtained for the diamond substrate, which agrees well with the published data (between 4.2 and 5.4 eV for the (100)-surface of diamond39), a significantly lower work function of 4.60 eV is determined for the fcc-carbon film. Note that no difference was observed between measurements in the dark and under illumination (Fig. 5a), namely, no surface photovoltage (SPV) related to the surface band bending is observed. This can be explained by the flat band conditions or Fermi level pinning at the film surface41.

a Work function measured by Kelvin probe (KP) in darkness and under illumination at λ= 172 nm. b Photoemission spectra PY(hν)1/3 measured by the photoelectron yield spectroscopy (PYS), in which the thin dashed lines represent the linear fits for the determination of the ionization energy. c Experimentally established electronic density of states (DOS) spectra that were acquired from PYS measurements (the energy of the valence band maximum (VBM) is set to 0 eV).
The ionization energy, which determines the valence band maximum (VBM), was found from PYS spectra shown in Fig. 5b, which was obtained due to photoexcitation of electrons by photons in the energy range of 3.8–7.4 eV. Near the emission onset, the photoelectron yield PY(hν) can be approximated by PY(hν) ∝ (hν−Ei)3, where hν is the energy of the incident photon and Ei is the ionization energy of the surface42. The ionization energy of the surface is thus determined by a linear fit of the PY(ℎν)1/3 onset, as shown by the dashed lines in Fig. 5b; the results are summarized in Table 2. As it can be seen, compared to the VBM position of diamond, such a position for the carbon film is higher by 350 meV. Note that the work function of the carbon film is lower by 140 meV. Consequently, the calculated VBM position of the carbon film relatively to the Fermi level, according to EF − EVBM = ϕ − Ei, is shifted downwards by about 500 meV, laying at 0.54 eV below the Fermi level. In contrast, the VBM of diamond lays only 0.05 eV below the Fermi level.
The electron affinity EA, which defines the conduction band maximum, is determined as EA = Ei − Eg, where Eg is the material bandgap. Considering the diamond bandgap of about 5.5 eV according to ref. 43 we obtained for diamond the value of EA = −0.68 eV. This negative value is in between the values of −0.82 eV and −0.45 eV measured on the same (100) crystal plane of diamond with H- and O-terminations respectively according to ref. 39. Other studies showed for H-terminated diamonds a strongly negative EA value in vacuum of up to −1.3 eV (see, e.g., ref. 44). On the other hand, O-terminated diamonds often showed a positive EA value due to the strong electronegativity of the carbonyl groups involved45. A negative EA value was also established for the fcc-carbon film by considering its bandgap of 6.1 eV determined from the optical measurements (see above). Thus, due to the negative electron affinity an easy emission of electrons from the fcc-carbon films is thought to be possible.
In addition to the ionization energy, the PYS spectra contain information on the electronic density of states (DOS) spreading from the Fermi level (EF) down to hv − Φ below EF. The DOS spectra were obtained from the PY(hv)–hv plots as the first derivative of the photoelectron yield PY(hν) with respect to the photon energy (hν), thus as d(PY)/d(hν). The experimentally obtained DOS are presented in Fig. 5c, which shows a higher density of states near the VBM and by about five times higher DOS at 1.5 eV below VBM for the fcc-carbon film. This can be an indication for higher holes effective mass in the carbon film compared to that of diamond. As it is shown in the next section, this is consistent with the calculated electronic band structure of the diamond and fcc-carbon structures.
Ab initio calculations
The major objective of this part is to obtain a computational model of fcc-carbon describing its electronic structure and explaining its electrical conductivity at the large value of bandgap.
Our computational model was built within the framework of the Density Functional Theory. The goal of the model is to explain some properties of fcc-carbon that were experimentally observed. An extensive benchmark of different energy functionals was conducted in order to reproduce the electronic properties of the fcc-carbon (see Methods, Computational Models), and the revPBE DFT functional46 was finally selected, which reproduces our preliminary experimental findings.
The electronic band dispersion of fcc-carbon resembles that of one of pure diamond (Fig. 6a–d).

a–c Diamond and b–d fcc-carbon are compared. The red and green lines in the top and bottom panels show the s- and p- character of the bands along the k-path respectively, the thicker the colored line, the larger the character. The purple arrow indicates the predicted optical band gap with its value reported on the side. The blue shadowed area indicates the filling of the valence band. e Isosurface (0.018 e/Å3) of the fcc-carbon electron density generated by the upper part of the valence band; the shape of the isosurface shows the electron delocalization across the crystal lattice.
The indirect band gap of diamond is found to be 4.0 eV, which is smaller than the experimental value of nearly 5.5 eV (see above). It is known that DFT functionals tend to underestimate the gap width47; however, here the focus is on the comparison of the valence band dispersion between the two materials and, therefore, such difference does not affect the forthcoming discussion.
The valence band of fcc-carbon is separated from the conduction band by a wide band gap (about 7.1 eV), which is consistent with the value determined experimentally (see above). The empty valence states closest to the Fermi level are found to generate a very low density of delocalized electrons only in the region located between carbon atoms, that is, such electrons are free to move throughout the whole crystal lattice (Fig. 6e). For this reason, despite the large band gap value, fcc-carbon is expected to have properties of an intrinsic semiconductor that is confirmed by the conductivity measurements (Fig. 4).
Unlike diamond, the valence band in fcc-carbon is split into a lower and an upper part by an intraband gap of about 0.8 eV. Those sub-bands are occupied by s- and p-electrons respectively. It can therefore be expected that the photoelectron spectroscopy resolves two signals for fcc-carbon. The recorded XPS spectra (see Fig. 3d), show a broad asymmetric peak which can be explained as a result of the convolution of two peaks occurring at 283.9 and 284.5 eV, respectively.
According to the theoretical model, the specific constitution of the s- and p-electrons’ hybridization leads to the formation of low-energy states in the range [−19.4, −11.2] eV with the predominant s-character, which is quantified as 93% contribution to the total density of states (see Fig. 7b). Moreover, this energy range is separated from the rest of the valence band by a small gap of 0.8 eV. It is known that a large symmetric distribution of the valence s-electrons induces a smaller binding energy of the core s-electrons48. For this reason, we detect the C1s peak with unusually low binding energy of 283.9 eV. The high energy (284.5 eV) peak represents then the impact of the shared (conducting) predominantly p-electrons from the upper valence sub-band. As mentioned above, this peak contains the signal from an adventitious carbon as well.

DOS of (a, c) diamond and (b, d) fcc-carbon. In (b) and (d), the curves corresponding to the px and py orbitals overlay. The VBM is set to 0 eV and the Fermi level as defined for intrinsic semiconductors is shown by a dashed line. The blue shadowed area indicates the filling of the valence band as in Fig. 6. If the Bloch states of the valence band are projected on the hydrogen-like functions centered at the carbon site, it is observed that the lowest band below the intraband gap has a prevalent s-character (Fig. 6b), while the remaining three bands have a predominant p-character (Fig. 6d).
This leads to a peculiar type of |sp3> hybridization, which is derived from the analysis of the density of states (Fig. 7). The lowest band can be represented by the superposition |sp3 > lowest = 0.93|s > +0.01|px > +0.01|py > +0.05|pz >, while the remaining occupied states above the intraband gap can be described by the linear combination |sp3 > bond = 0.16|s > +0.24|px > +0.24|py > +0.36|pz >. The electronic configuration of fcc-carbon is thus characterized by a very low rate of hybridization of s and p electrons, in which each electron occupies one orbital in the configuration [2s12p3]. The overall hybridization for the valence band of fcc-carbon is |sp3> = 0.26|s > +0.25|px>+0.25|py>+0.24|pz>, while in the case of diamond is |sp3> = 0.31|s > +0.23|px > +0.23|py > +0.23|pz>; equivalently, in fcc-carbon the character of the valence band is 26% of s-kind and 74% of p-kind, while in the case of diamond is 31% and 69%, respectively. This is consistent with the fact that the C-C distance in fcc-carbon (2.51 Å) is larger than the interatomic distance in diamond (1.54 Å). In fact, it is known that the degree of hybridization, which is the percent contribution of the s and p orbitals to the hybridized ones, determines the C–C distance; in general, the larger the s character, the shorter and stronger the bond49,50,51. In other words, the unusual s-p hybridization is responsible for the larger C-C distances in fcc-carbon. Eventually, exactly the excess of s character in the hybridization (sp3 →sp2 → sp) generates a nonisotropic spatial orientation of the electronic clouds.
One should recall here that the onset of a PYS signal is caused by the presence of electrons in occupied orbitals, and its magnitude is proportional to the density of states. In our systems, while the valence band of the diamond system is completely occupied, the valence band of the fcc-carbon is partially filled. It is thought that the measured 0 eV energy level (Fig. 5c) corresponds to the calculated energy levels of about 0 eV for diamond and −6 eV for fcc-carbon DOS (Fig. 7). Accordingly, it can be assumed that the DOS trends extracted from the PYS data (Fig. 5c) are fairly reproduced by the calculated DOS in the energy ranges [−2.5; 0.0] eV and [−8; −6] eV of the diamond and fcc-carbon systems, respectively (Fig. 7c, d). No perfect agreement is to expect here, since the computational model does not take into account the effect of the temperature on the electronic band structure and its occupancy.
To better understand how in fcc-carbon the 3D electronic cloud about each atom looks like, the Electron Localization Function (ELF)52 was calculated. The ELF gives information on possible local accumulation of electrons. On the basis the Electron Localization Function sections shown in Fig. 8 it can be supposed that the distribution of electronic density around carbon atoms is inhomogeneous. Yet the maximum value of the ELF is found to be about 0.3, which indicates a large degree of delocalization of the overall electron density. Such a large rate of delocalization accounts for the electron sharing in the regions of overlapping the electronic clouds, where there is a non-negligible concentration of delocalized electrons (Fig. 6e). The same delocalization allows the accommodation of each carbon atom at the center of a regular polyhedron formed by 12 nearest neighboring carbon atoms, thus forming the Fm \(\bar{3}\) m face-centered cubic crystal lattice with coordination number of 12.

a ELF corresponding to the (001)-; b – to the (010)-; and c – to the (100)-planes containing carbon atoms. The blue-green-red gradient represents ELF values in the range [0; 0.3]. The conventional unit cell is indicated by a thin black square, while the position of each carbon atom is at the center of each circular feature of the section.
The X-ray diffraction pattern of the fcc-carbon was simulated by means of the calculated electronic density. The simulated pattern well reproduces the experimental electron and X-ray diffraction data obtained from the fcc-carbon film (see Table 1), which justifies our theoretical model.
Discussion
A formation of chemical bonds in fcc-carbon can be clarified in light of two problems
Firstly, the coordination number of carbon atoms in fcc-structure equal to 12, which cannot be obtained by the four valence electrons of carbon atoms. The peculiar hybridization of the s and p orbitals in the valence band established by the ab initio calculations generates a spherically symmetric electronic density distribution with a large degree of delocalization; this allows for a symmetric distribution of 12 carbon atoms near a reference one.
Secondly, the shortest interatomic distance in fcc-carbon was determined as ~2.5 Å that corresponds to a carbon atomic radius significantly larger than the covalent radius (0.77 Å), but smaller than the van der Waals radius (3.4 Å). Thus, the interaction among carbon atoms in fcc-carbon resembles the so-called “van der Waals gap” corresponding to unconventional chemical bonds, like π–π stacking53.
A hypothesis suggested in ref. 4 explains the nature of chemical bonds in fcc-carbon based on their similarity to the well-known noncovalent intermolecular π–π interactions, or π‐stacking, in organic compounds and carbon-based materials. This kind of noncovalent interaction is typical for organic compounds, in particular, organic semiconductors which usually contain aromatic rings or another system of conjugated double-bonds (see, e.g., refs. 37,54,55,56,57). Apparently, some degree of electron sharing between individual molecules must exist to ensure electrical conductivity in these materials. The parallel π‐stacking interactions, also known as “π‐stacking pancake bonding”, are likely to contain a component of electron sharing that can be viewed as noncovalent or “covalent‐like”56. The intermolecular C–C distance in this case can be as short as 2.7 Å57, which is close to that in fcc-carbon.
In organic semiconductors the configuration of molecules and their mutual arrangement provide a 3D-system of conductive interrelated channels. It is also known that the triplet state of such molecules with nonhybridized p-states is often more stable than the singlet one58,59. This phenomenon stipulates a long-lived charge transfer event59, which is in turn responsible for the conductivity and other features of organic semiconductors37. The triplet states are also known to exist in carbon nanotubes and other carbon materials (see, e.g., ref. 58). It can be assumed that carbon atoms in fcc-carbon might be trapped in the triplet state that results in a valence band splitting and a charge separation; as a result, noncovalent electron sharing can take place among carbon atoms. Further theoretical investigations and ultrafast transient spectroscopic measurements are needed to confirm this hypothesis.
Intriguingly, other chemical elements of the IV group (Si and Ge) in the periodic table form allotropes with the fcc-structure. Indeed, as it was found in ref. 60, germanium crystallizes into a metastable phase with the face-centered cubic crystal lattice having a lattice parameter close to that of the common germanium allotrope with the diamond structure. Silicon was found to exist in the face-centered crystal structure at normal conditions so far only in the form of nanoparticles61. As it was established in this work the fcc-carbon films remain stable at ambient conditions for months and survives heating up to at least 100 °C in inert atmosphere. The reason of the formation and relative stability of fcc-carbon is still unclear and can be addressed to the following studies.
It is worth giving the outlook for promising applications of fcc-carbon
Despite an existing variety of conductive carbon materials, the attractive area of “carbon electronics” remains unavailable. Several reasons can be named: dependency of properties on crystal lattice orientation (graphite), challenges with up-scaling and patterning (graphene), high sensitivity of electronic properties with respect to the substrate and impurities (graphene), instability/irreproducibility of doping and lack of shallow donors/acceptors (diamond). Diamond though has an exceptionally high thermal conductivity and wide bandgap making diamond an ideal material for its employment as wafers for microelectronic devices62. Technological progress in the past years made the mass production of diamond supports economically affordable.
Wide bandgap semiconductors are of great interest for high power/high frequency applications implying power electronics63. Such materials also open the area of high-temperature electronics which is required in automotive, aerospace, and energy production industries64. Transistors based on these materials should basically reveal larger breakdown field and electron saturation velocity65. Science and engineering were focused so far on such materials as SiC and GaN with the band-gap energies (Eg) somewhat above 3 eV. Conductors with Eg beyond these values, namely ultrawide-bandgap (UWBG) semiconductors, extend application fields to RF electronics, deep-UV optoelectronics, quantum devices and others66. Such materials (e.g., β-Ga2O3, diamond and AlGaN) would allow increasing the Schottky barrier height and/or set up an energy barrier for traversing electrons65. There are still serious technological barriers to cope, which are the control of doping and of epitaxial growth itself. Despite a persistent need and efforts invested, p-type doping of diamond remains very challenging67. Speaking about conventional doping with boron, these acceptors are only weakly activated at room temperature due to the high ionization energy (0.36 eV). Therefore, higher boron concentration is needed that leads to undesirable effects and hinders the use of boron-doped diamond in microelectronics68.
In contrast, fcc-carbon possesses relatively high conductivity in its undoped state. Beyond that, it has an ultra-wide bandgap. It was proposed in ref. 69 to designate this carbon allotrope as “metametallic carbon”, as it possesses the crystal structure, that is typical for metals, but manifests itself as semiconductor with respect to the temperature dependence of electrical conductivity. As it was comprehensively studied and discussed in this work, fcc-carbon is presumably characterized by an uncommon type of conductivity which was not reported in the literature so far. Therefore, we propose to classify this material, together with the other ones possessing similar noncovalent sharing of the generally localized valent electrons, into a separate class of quasivalent solids. So fcc-carbon can be designated as quasivalent carbon.
The nature of chemical bonds in this carbon allotrope stipulates negative electron affinity and allows its high polarizability and very low thermal conductivity to be expected. These properties open a novel pathway to “carbon electronics” encompassing such high-end fields as high power/high-frequency electronics, transparent electronics, electron multipliers, field emitters, quantum memory and others70,71,72,73,74.
Methods
Deposition of carbon films
The diamond single-crystal substrates (plates) employed for the deposition of fcc-carbon films were produced with a microwave PACVD reactor operating at a frequency of 2.45 GHz. Such thin diamond plates were made from the initial diamond single crystal; they have the (100) orientation with <100> edges and dimensions of 3.8 × 3.8 × 0.3 mm with a polished surface subjected to acid cleaning with an RMS roughness <20 nm.
The fcc-carbon films were obtained of these substrates with the aid of PACVD by use of the following deposition parameters: temperature—around 600 °C, hydrogen content—98 vol%, methane content—2 vol%, total pressure—29.33 kPa, power—nearly 2.2 kW, deposition time—120 h. The plasma had the shape of ball of roughly 5 cm in diameter. Owing to simultaneous etching of diamond by plasma its surface was relatively rough (Fig. S1) and the film thickness was inhomogeneous. According to numerous SEM studies, the film thickness varies in the range of 100 nm–3 mm being on average ~1 μm. The appearance of the films in an optical microscope indicates that the films are transparent.
After the deposition process, the coated samples were subjected to the treatment in diluted HNO3 for 24 hours to remove hydrogen typically adsorbed on the diamond film surface in the form of dangling bonds leading to a noticeable surface conductivity75.
Examination of the crystal structure
The carbon films on the diamond substrates were examined by different methods.
Obtaining electron diffraction patterns and high-resolution transmission electron microscopy (HRTEM) images were performed by use of a Tecnai TF20 X-Twin microscope (FEI). The FFT and inverse FFT images were obtained based on the HRTEM image by use of the ImageJ software76,77.
The atomic force microscopy (AFM) maps were acquired on the C3000 instrument (Nanosurv, Switzerland) using Tap190Al-G probes at 160 kHz frequency. Linear profiles were extracted in the free Gwyddion software. Optical images were obtained with the help of the LEXT OLS4100 confocal laser microscope (Olympus, Japan). The scanning electron microscopy (SEM) images were obtained on the GeminiSEM 500 instrument (Carl Zeiss GmbH, Germany) using 5 kV accelerating voltage, 20 mm aperture and 4 mm working distance.
The X-ray diffraction (XRD) measurements were performed on the D8 Discover system (Bruker, Germany) using Cu Ka radiation. From the primary side Göbel mirror, 0.2 mm slit, 2-bounce 220-Ge monochromator and a 0.5 mm collimator were used. From the secondary side a combination of 1 mm, 0.6 mm and 0.2 mm slits was set. A 1D Lynxeye XE-T detector in a 0D mode and in a high-count rate for the alignment and in a high-resolution mode for actual measurement was used. Symmetric scans were conducted with 0.01°/2” steps. Rocking curves and Phi-scan were acquired with 0.005°/2” and 0.05°/0.5” steps respectively. The intensity of each diffraction maximum was optimized based on the following principal alignment steps: (1) setting detector to a maximum intensity of the beam; (2) parallelizing beamline to the sample surface in combination with Z-positioning; (3) fine-tilting around X/Y axes and linear X/Y translations for each series of diffraction maxima. Fitting of diffraction maxima was performed with the aid of a PeakFit program using pseudo-Voigt function and based on the area approximation. To establish the lattice constant, two standards were used: (1) stoichiometric, cubic single crystalline (100)-SrTiO3 and (2) diamond substrate itself. In the former case the positions of reflexes within relevant 2 Theta-ranges for each tilting angle of interest (Psi = 0°, 45° and 55°) were determined. Comparing these data with the standard (PDF 01-070-8508) from the ICDD database, the instrumental error for each necessary Psi-2 Theta combination was determined. Using those errors, the lattice constant of diamond was calculated from the positions of the (111)-, (220)-, and (400)-maxima determined. A comparison of the obtained values with the standard (PDF 00-006-0675) provided the error of the lattice constant determination, which is given in Table 1.
Computational models
Since the fcc-carbon is not widely accepted as an individual phase, we performed an extensive screening of as many Hamiltonian descriptions as possible, trying to obtain a model, which reproduces most of the experimental data at our disposal. To this aim, we fixed the lattice parameter to the experimental value of nearly 3.6 Å as detailed below, and do not perform any structural relaxation.
The model geometry of fcc-carbon consists of only one atom in the primitive unit cell (Supplementary Information, Fig. S7), which we locate at the origin of the cell. The space group is Fm \(\bar{3}\) m. The lattice parameters of the primitive unit cell are a = b = c = 2.511 Å, realizing the lattice angles α = β = γ = 60°; the corresponding conventional unit cell has lattice parameters a = b = c = 3.551 Å and angles α = β =γ = 90°
To model the interatomic interactions, we tested several DFT functionals within the LDA and GGA approximation46,78,79,80,81,82,83,84,85,86,87 (Table 3), also including van der Waals corrections implicitly considered in the meta-GGA description88,89. Concerning the calculations with the meta-GGA energy functionals, we were able to achieve the SCF convergence at lattice parameters different than the experimental ones; since we aim to create a model with the atom geometry consistent with our experimental data, we did not consider the meta-GGA functionals in the comparison. We also considered the presence of electron screening effect within the GW description90, however, as expected, they were not showing any particular feature in the electronic structure which has not been already reproduced by some of the standard DFT functionals. The DOS and band structures generated with the considered DFT functionals showed similar features. After taking also into account the computational load, we finally selected the revPBE46 DFT functional with no van der Waals corrections, which reproduces most of our preliminary experimental findings as discussed in the main text of the present manuscript. We perform our simulations by means of the ABINIT package with the PAW formalism90. We set the energy cut-off at 2000 eV and the PAW supplementary grid cut-off at 4600 eV, while we sample the Brillouin zone with an 87x87x87 Monkhorst-Pack mesh. The SCF solution of the Schrödinger equation is considered converged when the difference between two consecutive cycles is less than 3 × 10−10 eV. Since the electronic energy levels display a band gap about the Fermi level right from the beginning of the SCF iterations, we did not experience any technical issue for the SCF convergence; for this reason, there has been no need to use any smearing method and the band occupancy is an integer number at any sampled point of the reciprocal space. We include the ABINIT input file “Supplementary Data 1” as supporting information in order to facilitate the reader in reproducing the results.
We calculated the electronic band dispersion of pure diamond for comparison. The space group of the diamond structure is Fd3m. The lattice parameters of the primitive unit cell are a = b = c = 2.518 Å, realizing the lattice angles α = β = γ = 60°; the corresponding conventional unit cell has lattice parameters a = b = c = 3.561 Å and angles α = β = γ = 90°. We use the same computational settings as described above.
The atom-projected DOS provides us quantitative information on the s and p character of the valence band, that is the relative contribution of the s and p orbitals to the total dos. The character of each orbital is obtained by integrating the projected DOS from the bottom of the valence band up to the Fermi level, normalized by the integral of the total DOS. For example, the s character is obtained as:
where s(E) and g(E) are the s-projected and total density of states, respectively.
Spectroscopic and electrical measurements
Raman spectroscopy was performed with the aid of a Horiba Xplora spectrometer equipped with a 532 nm laser (Horiba Scientific). The measurements were conducted using ~6 μW power, 1800 mm−1 grating, ×100 lenses and 2 min acquisition time. Carbon particles of several microns in size were detached by a tiny diamond blade from the film’s surface and placed into the cells of a Cu grid. The operation was performed under a light microscope.
X-ray photoelectron spectroscopy (XPS) analyses were performed using a Kratos XPS spectrometer (Shimadzu) equipped with a monochromated Al Kα X-ray source (hν = 1486.6 eV). During the measurements, the base pressure inside the XPS chamber was kept constant at around 6.7 × 10−7 Pa. High-resolution XPS spectra were recorded with a 0.1 eV step. The deconvolution of the spectra was conducted by use of the CasaXPS software (www.casaxps.com) using the Tougaard background. Charge correction was done based on the center position of the C sp2 spectral component at 284.5 eV91. This contribution was detected on the films’ surface after the softest possible cleaning by argon ions. Fitting was performed using Shirley background, Gaussian-Lorentzian function for adventitious carbon peak at about 284.5 eV, and Doniach-Sunjic (DS) asymmetric tail function for the peak at nearly 283.9 eV92.
The UV spectrum from the carbon film was recorded using a vacuum ultraviolet spectrometer McPherson VUVAS 1000. The VUV reflectivity measurements were performed in the spectral band of 120–240 nm with 1 nm step; the PMT detector voltage was equal to 480 V. The spectrum was reconstructed according to the Tauc equation, and the curve obtained in such a way was extrapolated from the linear region by use of the Tauc method93,94,95,96 to calculate the value of optical bandgap. Herewith the intersection with linear background was taken as described elsewhere97.
The FTIRS measurements were conducted on Vertex 70 (Bruker Optics, Germany) device using rock solid interferometer, mercury (Hg) IR-source, DLaTGS IR-Detector, and the KBr Beam-splitter. Beam spot of 3 mm size was placed in the middle of the sample. There was no integration sphere installed. Sample was fixed on undoped (100)-silicon wafer by a carbon double-side tape. The attenuated total reflection measurement did not deliver enough intensity due to the extreme surface roughness. The attributed regions were taken from literature and database98,99,100.
KP and PYS measurements were performed. Prior to the measurements the samples were annealed at 100 °C for 1 h in a glovebox with water and oxygen contamination levels below 1 ppm. The transport of samples was in a sealed plastic bag whereas the installation onto the measurement table took place in a glovebox as well.
Electrical conductivity of carbon film and its temperature dependence were obtained by means of 4-point measurements in Van der Pauw geometry eliminating contact resistance. The film was bound by tiny indium wires from the corners of the sample which was in turn glued with the help of thermal paste onto sapphire holder to isolate sample electrically and provide good thermal conductivity. Electrical resistivity of the film was measured at different temperatures sweeping from 302 °K to 2 °K and back in 31 steps in each direction. The conductivity of the material was calculated implying film’s average thickness of 1 μm.
The Hall effect was measured in the same configuration. Due to low conductivity, the AC-mode with current of 0.01 mA and frequency of 57.1 Hz was used. The magnetic field was ranged from −1 to +1 T. The VH was found to be inconsistent and negligibly low. We suggest the nonplanar geometry of the film (see Fig. S1, Fig. S2, Fig. S3) to be the main reason of a fault.
Data availability
The experimental data are available and can be shared upon reasonable request. To facilitate the reader in the reproduction of the simulation results, we add the input file “Supplementary Data 1”.
References
Ma, X., Liu, X., Xi, X., Yao, Q. & Fan, J. Influence of crystallization temperature of fluorescence of n-diamond quantum dots. Nanotechnology 31, 505712 (2012).
Lonsdale, K., Milledge, H. J. & Nave, E. X-ray studies of synthetic diamond. Miner. Mag. 32, 158–201 (1959).
Palatnik, L. S., Guseva, M. B., Babaev, V. G., Savtchenko, N. F. & Falko, I. I. On γ–carbon. Sov. Phys. JETP 60, 520–524 (1984).
Konyashin, I. et al. Nanocrystals of face-centred cubic carbon, i-carbon and diamond obtained by direct conversion of graphite at high temperatures and static ultra-high pressures. Diam. Relat. Mater. 109, 108017 (2020).
Hirai, H. & Kondo, K. Modified phases of diamond formed under shock compression and rapid quenching. Science 253, 772–774 (1991).
Burkhard, G., Dan, K., Tanabe, Y., Sawaoka, A. & Yamada, K. Formation of cubic carbon by dynamic shock com pression of a diamond/amorphous carbon powder mixture. J. Appl. Phys. 33, 5875–5885 (1994).
Endo, S., Idani, N., Oshima, R., Takano, K. & Wakatsuki, M. X-ray diffraction and transmission-electron microscopy of natural polycrystalline graphite recovered from high pressure. Phys. Rev. B 49, 22–27 (1994).
Rossi, M., Vitali, G., Terranova, M. L., Sessa, V. Experimental evidence of different crystalline forms in chemical vapor deposited diamond films. Appl. Phys. Lett. 63, 2765–2767 (1993).
Jarkov, S., Titarenko, Y., Churilov, G. Electron microscopy studies of fcc carbon particles. Carbon 36, 595–597 (1998).
Konyashin, I. et al. A new carbon modification: ‘n-diamond’ or face-centred cubic carbon? Diam. Relat. Mater. 10, 99–102 (2001).
Konyashin, I. et al. Carbon modification with fcc crystal structure. Vacuum 66, 175–178 (2002).
Konyashin, I. et al. A new hard allotropic form of carbon: dream or reality? Int. J. Refract. Met. Hard Mater. 24, 17–23 (2006).
Gogotsi, Y., Wetz, S., Ersoy, D. A. & McNallan, M. J. Conversion of silicon carbide to crystalline diamond-structured carbon at ambient pressure. Nature 411, 283–285 (2001).
Welz, S., Gogotsi, Y. & McNallan, M. J. Nucleation, growth and graphitization of diamond nanocrystals during chlorination of carbides. J. Appl. Phys. 93, 4207–4214 (2003).
Santiago, P. et al. Diamond polytypes in Mexican crude oil. Energy Fuels 18, 390–395 (2004).
Israde-Alcantara, I. et al. Evidence from central Mexico supporting the Younger Dryas extraterrestrial impact hypothesis. Proc. Natl Acad. Sci. USA 109, E738–E747 (2012).
Kennett, D. J., Kennett, J. P. & West, A. Nanodiamonds in the Younger Dryas sediment layer. Science 323, 94–98 (2009).
Yang, Z. Q. et al. TEM and Raman characterisation of diamond micro- and nanostructures in carbon spherules from upper soils. Diam. Relat. Mater. 17, 937–943 (2008).
Kennett, D. J. et al. Shock-synthesized hexagonal diamonds in Younger Dryas boundary sediments. Proc. Natl Acad. Sci. USA 106, 12623–12628 (2009).
Kinzie, C. R. et al. Nanodiamond-rich layer across three continents consistent with major cosmic impact at 12, 800 Cal BP. J. Geol. 122, 475–505 (2014).
Kurbatov, A. et al. Discovery of a nanodiamond-rich layer in the Greenland ice sheet. J. Glaciol. 56, 747757 (2010).
Wen, B., Zhao, J. J. & Li, T. J. Synthesis and crystal structure of n-diamond. Int. Mater. Rev. 52, 131–151 (2007).
Shikata, S. et al. Precise measurements of diamond lattice constant using Bond method. Jpn. J. Appl. Phys. 57, 111301 (2018).
Pickard, C. J., Milman, V. & Winkler, B. Is there theoretical evidence for a metallic carbon polymorph with space group symmetry Fm3m at ambient conditions? Diam. Relat. Mater. 10, 2225–2227 (2001).
Murrieta, G., Tapia, A. & deCoss, R. Structural stability of carbon in the face-centred-cubic (Fm3m) phase. Carbon 42, 771–774 (2004).
Ki, D. et al. Ab initio structure determination of n-diamond. Sci. Rep. 5, 13447 (2015).
Wen, B., Melnik, R., Yao, S. & Li, T. Hydrogen-doped cubic diamond and the crystal structure of n-diamond. Chem. Phys. Let. 516, 230–232 (2011).
Qiang, L. et al. First principles study on structure and electronic properties of FCC crystals for group IV. J. At. Mol. Phys. 32, 153–157 (2015).
Authier, A. International Tables for Crystallography Volume D: Physical Properties of Crystals (Kluwer Academic Publisher, 2013).
Wallace, D. M. Thermodynamics of Crystals (John Wiley & Sons Inc, United States of America, 1972).
Greczynski, G. & Hultman, L. Reliable determination of chemical state in x-ray photoelectron spectroscopy based on sample-work-function referencing to adventitious carbon: Resolving the myth of apparent constant binding energy of the C 1s peak. Appl. Surf. Sci. 451, 99–103 (2018).
Morgan, D. J. XPS insights: asymmetric peak shapes in XPS. Surf. Interface Anal. 55, 567–571 (2023).
Chen, X., Wang, X. & Fang, D. A review on C1s XPS-spectra for some kinds of carbon materials. Fuller. Nanotubes Carbon Nanostruct. 28, 1048–1058 (2020).
Elliot, S. R. AC conduction in amorphous chalcogenide and pnictide semiconductors. J. Adv. Phys. 36, 135–217 (1987).
Mamunya, Y. (Ed.: Yellampalli, S.) Carbon Nanotubes as Conductive Filler in Segregated Polymer Composites—Electrical Properties (IntechOpen, 2011).
El Hasnaoui, M., Kreit, L., Costa, L. C. & Achour, M. E. Investigations of temperature effect on the conduction mechanism of electrical conductivity of copolymer/carbon black composite. Appl. Microsc. 47, 121–125 (2017).
Schwarze, M. et al. Molecular parameters responsible for thermally activated transport in doped organic semiconductors. Nat. Mater. 18, 242–248 (2019).
Rezek, B., Stehlik, S. Surface potential of nanodiamonds investigated by KPFM. Micro and Nano Technologies (in: Nanodiamonds) 273–300 (2017).
Diederich, L., Küttel, O. M., Aebi, P. & Schlapbach, L. Electron affinity and work function of differently oriented and doped diamond surfaces determined by photoelectron spectroscopy. Surf. Sci. 418, 219–239 (1998).
Chemin, A. et al. Surface-mediated charge transfer of photogenerated carriers in diamond. Small Methods 18, e2300423 (2023).
Edmonds, M. T. et al. Work function, band bending, and electron affinity in surface conducting (100) diamond. Phys. Status Solidi A 208, 2062–2066 (2011).
Rusu, M. et al. Electronic structure of the CdS/Cu (In, Ga)Se2 interface of KF- And RbF-treated samples by kelvin probe and photoelectron yield spectroscopy. ACS Appl. Mater. Interfaces 13, 7745–7755 (2021).
Wort, C. J. H. & Balmer, R. S. Diamond as an electronic material. Mater. Today 11, 22–28 (2008).
Ballantyne, J. M. Effect of phonon energy loss on photoemissive yield near threshold. Phys. Rev. B 6, 1436–1455 (1972).
Maier, F., Ristein, J. & Ley, L. Electron affinity of plasma-hydrogenated and chemically oxidized diamond (100) surfaces. Phys. Rev. B Condens. Matter Mater. Phys. 64, 165411 (2001).
Zhang, Y. & Yang, W. Comment on “generalized gradient approximation made simple”. Phys. Rev. Lett. 80, 890–890 (1998).
Belviso, F. et al. Viewpoint: atomic-scale design protocols toward energy, electronic, catalysis, and sensing applications. Inorg. Chem. 58, 14939–14980 (2019).
Nemoshkalenko V. V. & Aleshi V. G. Structure of the photoelectron spectra of core electrons. Electron Spectroscopy of Crystals, Physics of Solids and Liquids 323–337 (Springer, Boston, MA, 1979). ISBN 978-1-4613-2901-5.
Kawai, H., Takeda, T., Fujiwara, K., Inabe, T. & Suzuki, T. Exceptionally large difference in bond length among conformational isomorphs of a hexaphenylethane derivative with a dispiropyracene skeleton. Cryst. Growth Des. 5, 2256–2260 (2005).
Oliva, J. M., Allan, N. L., Schleyer, P. V. R., Viñas, C. & Teixidor, F. Strikingly long C···C distances in 1,2-disubstituted ortho-carboranes and their dianions. J. Am. Chem. Soc. 127, 13538–13547 (2005).
Yang, J.-X. & Wang, Y. A. How long can a C–C σ-single bond be? J. Phys. Chem. A. 125, 933–942 (2021).
Silvi, B. & Savin, A. Classification of chemical bonds based on topological analysis of electron localization functions. Nature 371, 683–686 (1994).
Alvares, S. A cartography of the van der Waals territories. Dalton Trans. 42, 8617–8636 (2013).
Alonso, M. et al. Understanding the fundamental role of π-π, σ-σ, and σ-π interactions in shaping carbon-based materials. Chem. Eur. J. 20, 4931–4941 (2014).
Tiekink, E. R. T. and Zukerman, J. (Eds.). The Importance of π-Interactions in Crystal Engineering: Frontiers in Crystal Engineering (Wiley, 2012).
Kertesz, M. Pancake bonding: an unusual pi-stacking interaction. Chem. Eur. J. 25, 400–416 (2019).
Tian, Y.-H. & Kertesz, M. Is there a lower limit to the CC bonding distances in neutral radical π-dimers? The case of phenalenyl derivatives. J. Am. Chem. Soc. 132, 10649 (2010).
Tretiak, S. Triplet state absorption in carbon nanotubes: a TD-DFT study. Nano Lett. 7, 2201–2206 (2007).
Zimmerman, P. M., Bell, F., Casanova, D. & Head-Gordon, M. Mechanism for singlet fission in pentacene and tetracene: from single exciton to two triplets. J. Am. Chem. Soc. 133, 19944–19952 (2011).
Kryshtal, A. P., Minenkov, A. A. & Ferreira, P. J. Interfacial kinetics in nanosized Au/Ge films: an in situ TEM study. Appl. Surf. Sci. 409, 343–349 (2017).
Balcı, M. H. et al. Solution-based synthesis of simple fcc-Si nano-crystals under ambient conditions. Dalton Trans. 42, 2700–2703 (2013).
Balmer, R. S. et al. Unlocking diamond’s potential as an electronic material. Philos. Trans. R. Soc. A 366, 251–265 (2008).
Millán, J., Godignon, P., Perpiňà, X., Pérez-Tomás, A. and Rebollo, J. A survey of wide bandgap power semiconductor devices. IEEE Trans. Power Electron. 29, 2155–2163 (2014).
Rafiqul Islam, M., Galib, R. H., Sarkar, M. and Chowdhury, S. Wide-bandgap semiconductor device technologies for high-temperature and harsh environment applications In: (Edited by Sharif, A.) Harsh Environment Electronics: Interconnect Materials and Performance Assessment, 1st edn (Wiley-VCH Verlag GmbH & Co. KGaA, 2019).
Tsao, J. Y. et al. Ultrawide-bandgap semiconductors: research opportunities and challenges. Adv. Electron. Mat. 4, 1600501 (2018).
Porter L. M., Das K., Dong Y., Melby J. H. & Virshup A. R. Contacts to wide-band-gap semiconductors. In: Bhattacharya, P., Fornari, R., Kamimura, H. (Eds.) Comprehensive Semiconductor Science and Technology. 44–85 (Elsevier, 2011).
Roychoudhury, R. et al. Growth and characterization of phosphorus-doped diamond films using trimethyl phosphite as the doping source. J. Appl. Phys. 81, 3644–3646 (1997).
Nebel C. E., Stutzmann, M. Transport properties of diamond: carrier mobility and resistivity, Vol. 26 (eds Nazare M. H. & Neves A. J.) Properties, Growth and Applications of Diamond. IEE Emis. Data Reviews Series, 2001, UK: Institute of Engineering and Technology, p. 45.
Konyashin I., Palmer N., Colard P.-O., Twitchen D. Carbon material. Patent application WO2023/020723A1 (2022).
Fioretti, A. N. & Morales-Masis, M. Bridging the p-type transparent conductive materials gap: synthesis approaches for disperse valence band materials. J. Photonics Energy 10, 042002 (2020).
Battaglia, C., Cuevas, A. & De Wolf, S. High-efficiency crystalline silicon solar cells: status and perspectives. Energy Environ. Sci. 9, 1552–1576 (2016).
Correa-Baena, J.-P. et al. Promises and challenges of perovskite solar cells. Science 358, 739–744 (2017).
Daus, A. A., Han, S., Knobelspies, S., Cantarella, G., Vogt C. Flexible CMOS electronics based on p-type Ge2Sb2Te5 and n-type InGaZnO4 semiconductors. In Proc. IEEE International Electron Devices Meeting (IEDM) 8.1.1–8.1.4. (2017).
Nomura, K., Kamiya, T., & Hosono, H. Ambipolar oxide thin-film transistor.Adv. Mater. 23, 3431–3434 (2011).
Liu, F., Cui, Y., Qu, M. & Di, J. Effects of hydrogen atoms on surface conductivity of diamond film. AIP Adv. 5, 041307 (2015).
Schneider, C. A., Rasband, W. S. & Eliceiri, K. W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 (2012).
Abramoff, M. D., Magalhaes, P. J. & Ram, S. J. Image processing with ImageJ. Biophotonics Int. 11, 36–42 (2004).
Goedecker, S., Teter, M. & Hutter, J. Separable dual-space Gaussian pseudopotentials. Phys. Rev. B 54, 1703–1710 (1996).
Perdew, J. P. & Zunger, A. Self-interaction correction to density-functional approximations for many-electron systems. Phys. Rev. B 23, 5048–5079 (1981).
Hedin, L. & Lundqvist, B. I. Explicit local exchange-correlation potentials. J. Phys. C: Solid State Phys. 4, 2064–2083 (1971).
Perdew, J. P. & Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B 45, 13244–13249 (1992).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Hammer, B., Hansen, L. B. & Nørskov, J. K. Improved adsorption energetics within density-functional theory using revised Perdew-Burke-Ernzerhof functionals. Phys. Rev. B 59, 7413–7421 (1999).
Hamprecht, F. A., Cohen, A. J., Tozer, D. J. & Handy, N. C. Development and assessment of new exchange-correlation functionals. J. Chem. Phys. 109, 6264–6271 (1998).
Boese, A. D., Doltsinis, N. L., Handy, N. C. & Sprik, M. New generalized gradient approximation functionals. J. Chem. Phys. 112, 1670–1678 (2000).
Wu, Z. & Cohen, R. E. More accurate generalized gradient approximation for solids. Phys. Rev. B 73, 235116 (2006).
Cooper, V. R. Van der Waals density functional: an appropriate exchange functional. Phys. Rev. B 81, 161104 (2010).
Van Voorhis, T. & Scuseria, G. E. A novel form for the exchange-correlation energy functional. J. Chem. Phys. 109, 400–410 (1998).
Tran, F. & Blaha, P. Accurate band gaps of semiconductors and insulators with a semilocal exchange-correlation potential. Phys. Rev. Lett. 102, 226401 (2009).
Boese, A. D. & Handy, N. C. New exchange-correlation density functionals: the role of the kinetic-energy density. J. Chem. Phys. 116, 9559–9569 (2002).
Räsänen, E., Pittalis, S. & Proetto, C. R. Universal correction for the Becke–Johnson exchange potential. J. Chem. Phys. 132, 044112 (2010).
Lesiak, B. et al. C sp2/sp3 hybridisations in carbon nanomaterials – XPS and (X)AES study. Appl. Surf. Sci. 452, 223–231 (2018).
Doniach, S. & Sunjic, M. Many-electron singularity in X-ray photoemission and X-ray line spectra from metals. J. Phys. C Solid State Phys. 3, 285–291 (1970).
Kubelka, P. & Munk, F. An article on optics of paint layers. Z. Tech. Phys. 12, 593–601 (1931).
Tauc, J., Grigorovici, R. & Vancu, A. Optical properties and electronic structure of amorphous germanium. Phys. Status Solidi 15, 627–637 (1966).
Tauc, J. Optical properties and electronic structure of amorphous Ge and Si. Mater. Res. Bull. 3, 37–46 (1968).
Tauc, J. High absorption region. In: J. Tauc (Ed.), Amorphous and Liquid Semiconductors. 173–178 (Springer US, London and New York, 1974).
Suram, K., Newhouse, P. F. & Gregoire, J. M. High throughput light absorber discovery, part 1: an algorithm for automated Tauc analysis Santosh. ACS Comb. Sci. 18, 673–681 (2016).
Fang, C. et al. Preparation of “natural” diamonds by HPHT annealing of synthetic diamonds. Cryst. Eng. Comm. 20, 505–511 (2018).
Kim, K. P., Song, W. S., Park, M. K. & Hong, S. J. Surface analysis of amorphous carbon thin film for etch hard mask. J. Nanosci. Nanotechnol. 21, 2032–2038 (2021).
Acknowledgements
R.M. acknowledges the financial support in purchasing the X-ray diffractometer by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation), grant number AOBJ 649257. Prof. E. List-Kratochvil is appreciated for providing access to the various high-end analyses (SEM, AFM, Raman and fluorescence spectroscopy, laser microscopy). A.C. acknowledges the support of the Ministry of Education, Youth and Sports of the Czech Republic through the e-INFRA CZ (ID:90254) and access to the computational infrastructure of the OP VVV-funded project CZ.02.1.01/0.0/0.0/16_019/0000765 “Research Center for Informatics”; A.C. also acknowledges that this work was co-funded by the European Union under the project “Robotics and advanced industrial production” (reg. no. CZ.02.01.01/00/22_008/0004590). A.B. acknowledges CzechNanoLab Research Infrastructure supported by MEYS CR (LM2023051). Dr. D. Sidorenko and Dr. T. Sviridova are thanked for the contribution at the early stages of this work. Mr. M. Weise and Dr. S. Seeger are acknowledged for the FTIRS measurements.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
I.K. supervised the PACVD experiments and measurements, and wrote several parts of the manuscript; R.M. performed XRD, SEM, AFM, and Raman measurements and conducted treatments of their and some other results including work on texts; A.C. organized, supervised and performed the majority of the ab initio calculations, and wrote the corresponding part of the manuscript; A.B. conducted Raman, XPS, electron diffraction and HRTEM studies; M.R. performed the KP and PYS measurements and performed their analysis. A.K. performed some of the preliminary screening of the functionals; T.P. provided critical discussions on the theoretical results; D.T. organized and supervised the deposition trials and measurements of electric properties; N.P. performed the PACVD experiments and obtained samples of fcc-carbon needed for different examinations; B.S. and P.-O.C. were responsible for the acquisition of projects.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Materials thanks Xiaotian Zhang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Zakaria Al Balushi and Aldo Isidori. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Konyashin, I., Muydinov, R., Cammarata, A. et al. Face-centered cubic carbon as a fourth basic carbon allotrope with properties of intrinsic semiconductors and ultra-wide bandgap. Commun Mater 5, 115 (2024). https://doi.org/10.1038/s43246-024-00547-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s43246-024-00547-8
- Springer Nature Limited