
Abstract
The optimization of the quantum efficiency of single-molecule light-driven rotary motors typically relies on chemical modifications. While, in isolated conditions, computational methods have been frequently used to design more efficient motors, the role played by the solvent environment has not been satisfactorily investigated. In this study, we used multiscale nonadiabatic molecular dynamics simulations of the working cycle of a 2-stroke photon-only molecular rotary motor. The results, which display dynamics consistent with the available transient spectroscopy measurements, predict a considerable decrease in the isomerisation quantum efficiency in methanol solution with respect to the gas phase. The origin of such a decrease is traced back to the ability of the motor to establish hydrogen bonds with solvent molecules. The analysis suggests that a modified motor with a reduced ability to form hydrogen bonds will display increased quantum efficiency, therefore extending the set of engineering rules available for designing light-driven rotary motors.
Similar content being viewed by others

Introduction
Among other molecular motors (i.e., molecular-level devices capable of transducing the energy of external stimuli to directed atomic motion), light-driven rotary motors1,2,3 (LDRMs; for a full list of acronyms see the Supplementary Note 1) have the ability to operate by consuming only light (and, optionally, heat) without emanating waste products that may ultimately impair their function4,5,6,7,8. This makes LDRMs useful in the fields such as nanotechnology, optogenetics, synthetic biology, nanomedicine, functional materials, where they have been applied8,9,10,11,12,13,14,15,16,17.
The most common design of LDRMs exploits the overcrowded alkene (OA) motif1,2,3 although alternative designs can be found in the literature18,19,20,21,22. Typically, OA LDRMs (i.e., molecules based on carbon-rich scaffold, where strong steric repulsion causes a nonplanar arrangement of substituents at a double C=C bond) complete a full 360° revolution in the four steps shown in Fig. 1, where two EP → ZM and ZP → EM, photoisomerisation steps (in other words, transitioning between conformations characterised by the trans (E) or cis (Z) arrangement of substituents at the central C=C bond accompanied by helical distortion in the positive (P) or negative (M) sense of rotation) are interlaced by two thermally activated helix inversion (THI) steps. The latter steps, where the upper moiety of the LDRM (the rotor blade) moves over the lower moiety (the stator blade) changing helicity from M to P, are needed to reset the molecular conformation in a geometry suitable for maintaining the rotation of the rotor in the same direction (e.g., the counterclockwise direction, CCW, in Fig. 1) with respect to the stator3.

a The main structures in the mechanism of the motor detected experimentally in ref. 3. b Profiles of the S1 (the red curve) and S0 (the blue curve) potential energy surfaces along the rotation path. The main structures are shown by the red and blue dots, accompanied by 3D geometries (without hydrogens) and conformational labels. The hourglass symbols show the positions of conical intersections (CI). The curved greyscale arrows show the sequence of geometric transformations. The straight upward magenta arrows show the photoexcitation events. The 3D structures shown in the figure were plotted using the data from ref. 27.
The initial rotation EP → ZM occurs on the first singlet excited (S1) state potential energy surface (PES), where photoexcitation of the EP structure breaks the π-component of the exocyclic double bond that connects the two blades and allows rotation around the central axle. The steric repulsion between the rotor and the stator in the fjord region (i.e., the region where the two blades approach each other the closest) drives the rotor towards a geometry, where the two blades become (nearly) orthogonal. In this geometry, the S1 and ground (S0) state PESs approach one another and eventually cross, thus entering a conical intersection23,24,25 (CoIn), where the population is transferred nonadiabatically to S026,27,28. After that, the rotary motion continues on the S0 PES ending in a Z structure.
However, the molecule may settle in a conformation (ZM) not allowing for a prompt rotation in the same direction (CCW, in Fig. 1) and has to be reset through THI to a more stable ZP conformation; which completes half of the motor working cycle. After absorption of the second photon by the ZP structure, the same two-step sequence is activated and ends up in the EP structure.
The outlined mechanism implies that the mirror symmetry between the EP and EM, and ZP and ZM conformations should be broken, which is achieved by introducing a chiral centre;29 with the R chirality in Fig. 1. When EP and ZP structures are more stable than EM and ZM, respectively, the rotation proceeds in the CCW direction; this is typical of LDRMs with R-chiral centre3,29. The direction is dictated by the slope of the S1 PES, at the geometry of the vertical excitation S1 ← S0; which occurs due to the specific folding of the blades, see Fig. 1a, imposed by the steric overcrowding in the fjord region and around the chiral carbon atom. Thus, the point and/or axial chirality are essential constituents of OA LDRMs3,29,30.
Although OA LDRMs have already found their way into a number of applications, their rotational efficiency remains relatively low. This is measured by the low quantum yield of the photoisomerisation steps (Φiso, the ratio of the number of isomerisation events to the number of absorbed photons) dominated by branching of the S1 population into productive (i.e., propagating towards products) and unproductive (i.e., going back to reactants) fractions immediately after decay through the CoIn funnel. In addition to that, the presence of the THI steps allows LDRMs to operate efficiently only in specific temperature ranges31.
So far, the optimisation of LDRMs has been mostly driven by chemical intuition8,32 focusing primarily on optimising the THI step and their photochemistry remained less amenable to chemical modification33,34. One prospective way to improve the characteristics of LDRMs is to eliminate the two THI steps from the classic working cycle thus achieving “photon-only" LDRMs. This idea has resulted in several theoretical suggestions30,35,36,37, the synthesis of a hemithioindigo-based motor22 and phosphororganic LDRM38, which require three and four photochemical steps to complete their 360° cycles, respectively. However, because of the very low Φiso value of each step, their rotational efficiency remains unsatisfactory. For example, hemithioindigo LDRM absorbs on average~1900 photons to complete a full rotation22.
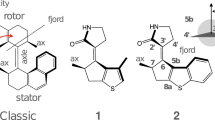
Recently, some of us reported the design and synthesis of a photon-only LDRM featuring, perhaps, the simplest working cycle39. The motor, E-3’-(2-methyl-2,3-dihydro-1H-benzo[b]cyclopenta[d]thiophen-1-ylidene)pyrrolidin-2’-one (1), abbreviated as MTDP, see Fig. 2 for chemical formula, was characterised via time-resolved spectroscopy that corroborated the theoretical conclusion that the two THI steps are seamlessly overcome at room temperature yielding a cycle with only two photochemical steps.

The red arrow shows the direction in which rotation of the upper part occurs with respect to the lower part of the motor. The inset in the upper left part shows the definition of the central torsion angle θ.
The absence of stable ZM and EM intermediates, when starting from EP and ZP reactants, respectively, in MTDP (see Fig. 3 for a schematic representation of the working cycle) was attributed to (i) the strain of the 3-methylene-1-cyclopentene (MCP) stator moiety incorporating the stereogenic centre and (ii) a decrease in steric repulsion in the fjord region39. Therefore, as seen in Fig. 3, MTDP should be able to perform a 360° rotation of its rotor blade by absorbing only two photons.

a The working cycle of the motor. b The profiles of the S1 (red) and S0 (blue) potential energy surfaces along the rotation path. The red and blue dots show the positions of the main structures, labelled by conformational labels and accompanied by 3D geometries. The positions of conical intersections (CI) are shown by hourglass symbols. The curved greyscale arrows show the sequence of transformations during the working cycle. The straight upward magenta arrows show the photoexcitation events. The 3D structures shown in the figure were plotted using the data from ref. 39.
The simulation of MTDP photodynamics in the gas phase predicted that the motor should have a very high Φiso for both isomerisation steps, on the order of 0.939. However, the value of Φiso of the first EP → ZP photoisomerisation step measured in methanol solution was dramatically lower (Φiso = 0.25 ± 0.05). Therefore, it was concluded, supported by simulations in solution, that the solvent considerably reduces the rotational efficiency in both photochemical steps39.
Although MTDP in methanol has shown a Φiso value higher than several classical LDRM (usually much less than 0.233), the atomistic understanding of the solvent effect may indicate how to increase its value further. In this paper, we focus on the understanding of how the surrounding solvent molecules impair the rotational function. In doing so, we hope to learn what modifications of the MTDP structure would mitigate these influences and improve the functionality of LDRMs via suitable chemical modifications. To achieve such a goal, we use quantum mechanics/molecular mechanics (QM/MM) modelling and nonadiabatic molecular dynamics (NAMD) simulations of the two photoisomerisation events responsible for the decreased Φiso. The QM method employed in these simulations, the state-interaction state-averaged spin-restricted ensemble-referenced Kohn-Sham (SI-SA-REKS or SSR) method, has been previously employed in ref. 39; more details of the methodology can be found in the Supporting Information. Based on the analysis of the QM/MM NAMD results, we propose the compound 2 (see Fig. 2) as an MTDP derivative with improved rotational efficiency.
Results and discussion
The ground- and excited-state PESs of MTDP have been documented in ref. 39, both in the gas phase and in solution. The most stable gas-phase structure of MTDP is the EP conformer, which is ca. 4 kcal mol−1 below ZP (for R diastereomer; S is a mirror image). On S0 PES, the two structures are separated by barriers of ca. 60 kcal mol−1, which is consistent with the high thermal stability of the EP conformer and the photostationary state (PSS) in the solution phase39. No other local minima were obtained in the ground electronic state under isolated conditions.
Under ambient conditions, MTDP exists as a pure E structure (as a racemic mix of the R-EP and S-EM diastereomers), and its geometry was obtained by X-ray crystallography. The optimised geometry of the ground state of EP is in agreement with the experiment within 0.014 Å39. Due to the equivalence between the R and S forms, in the following, only the R form will be discussed.
On the S1 PES of MTDP (the vertical excitation wavelength is ca. 280 nm for both isomers), there is a noticeable slope inclined in the direction of the CCW torsion around the central C6=\({{{{{{{{\rm{C}}}}}}}}}_{3{\prime} }\) bond. At ca. 270° and 90° of torsion, CoIns occur with the ground electronic state, which lie at ca. 72 kcal mol−1 for both isomerisation paths, EP → ZP and ZP → EP39. Qualitatively, not quantitatively, similar S1 and S0 PESs were obtained in methanol solution modelled by the QM/MM technique and two different levels of QM theory39.
The gas phase NAMD simulations of photoisomerisation of MTDP produced quite rapid decay dynamics with the S1 state lifetime \({\tau }_{{{{{{{{{\rm{S}}}}}}}}}_{{{{{{{{\rm{1}}}}}}}}}}=413\pm 13\) fs for EP → ZP, and \({\tau }_{{{{{{{{{\rm{S}}}}}}}}}_{{{{{{{{\rm{1}}}}}}}}}}=298\pm 9\) fs for ZP → EP. Both photoisomerisation steps showed perfect unidirectionality of rotation (all trajectories rotated in the CCW direction) and rather high Φiso values of 0.87 ± 0.05 and 0.91 ± 0.04 for the two isomerisation steps.
However, as anticipated above, the gas-phase theoretical values did not agree with the experimentally measured Φiso of the EP → ZP step in methanol solution that also showed a longer excited state lifetime on the order of 600 fs39. A limited set of QM/MM NAMD trajectories (only 21 and 40 trajectories were propagated using surface hopping based on exact factorisation (SHXF) and fewest switches surface hop (FSSH) methods, respectively) of the EP → ZP step in ref. 39 confirmed that the solvent has a detrimental effect on the Φiso (by lowering it to 0.35 ± 0.10) and on the S1 lifetime (by lengthening it to 562 ± 87 fs)39.
In the present work, using exclusively the SHXF method, a greater number of QM/MM NAMD trajectories are propagated, and both photoisomerisation steps are equivalently simulated. More specifically, for each step of MTDP photoisomerisation, 30 initial conditions were prepared as described in the Supplementary Note 3. The trajectories were propagated on the S1 PES for a maximum simulation time of 1.5 ps, or until a nonadiabatic transition S1 → S0 occurred, and then continued on the S0 PES until a final conformation was reached (up to 4–5 ps). Although the number of propagated trajectories is limited due to the duration of the simulations, the statistical averages obtained were refined by bootstrapping with 104 bootstrap replicas of the original data set; this enables the determination of reliable margins of error in the statistical averages40.
As mentioned above, the QM/MM NAMD simulations are carried out using the SHXF method41,42,43, which has been shown to be an accurate first principle formulation of the semi-classical trajectory surface hopping formalism41,44,45. As in ref. 39 the QM part of the simulations employed the SSR quantum-chemical method46,47,48; see Supplementary Note 2 for the acronym and theoretical details. This method is based on ensemble density functional theory (eDFT)49,50,51,52,53,54,55,56,57,58 to obtain ground and excited potential energies, where the relevant multi-configurational characteristics of the electronic states are incorporated in a fashion reminiscent of the more traditional wavefunction-based methods46,47,48,59,60,61. The method has previously been used to study the dynamics of photosteps in various LDRMs26,27,36,62,63,64, where its predictions helped modulate the photochemistry of target molecules34,36,62,63,64. In addition to LDRM, SSR was used in studies of the dynamics of photoinduced isomerisation in different retinal proteins, which provided additional support for its predictive ability65,66,67. Most importantly, SSR enabled the design of 1 and its predictions were corroborated by the experiments39. However, some limitations were recently reported for protonated Schiff bases that are related to the specific choice of functional68.
As documented in Supplementary Fig. 2, the initial values of the dihedral angle θ (see Fig. 2 for definition) are distributed around the values of 180.5° (standard deviation, std, 7.0°) for the EP structure and 5.2° (std 8.3°) for the ZP structure. The respective gas phase ground state equilibrium values are 181.1° and 4.9°, respectively.
The time evolution of the dihedral angle θ is shown in Fig. 4 along with the S1 population dynamics of the two photoisomerisation steps. The S1 population is defined as a fraction of the trajectories residing in the S1 state at a given instance of time.

Panel a shows the time evolution of the excited-state population n for the half-loop EP → ZP. The red curve is fitted to the computed populations (the blue dots) by a monoexponential function \({n}_{{{{{{{{\rm{fit}}}}}}}}}(t)={e}^{-({t}_{0}-t)/\tau }\), where t0 is the latency time and τ is the exponential decay constant. The excited-state lifetime \({t}_{{{{\mbox{S}}}}_{1}}\) is shown in the inset. The margin of error of the fitted quantities was obtained by bootstrapping with 104 replicas40. Panel b shows the time evolution of the central dihedral angle θ. The blue curves show the productive trajectories (i.e., undergoing EP → ZP isomerisation), the black curves show the unproductive trajectories (i.e., turning back, EP → EP), the grey curves show the trajectories rotating in the wrong direction (i.e., CW, instead of CCW). The red crosses show the S1 → S0 nonadiabatic transitions. The red dots show the terminal points of the trajectories, where the final value of the angle θ was reached. Horizontal blue lines show the division between the EP/EM and ZP/ZM rotamers. Panels c) and d) show the same characteristics of the ZP → EP half-loop.
Although we only found a minor prevalence of θ > 180° in the initial conditions of the EP → ZP step, all but two trajectories propagated in the CCW direction of torsion indicating that θ is not a good index for describing the molecular helicity (i.e. P or M). Indeed, the torsion angle alone does not take into account the tilt of the \({{{{{{{{\rm{C}}}}}}}}}_{3{\prime} }\)=C6 bond with respect to the stator blade, which also characterises the helicity of the motor. As an additional measure of helicity, the dihedral angle η = ∠\({{{{{{{{\rm{C}}}}}}}}}_{3{\prime} }\)–C6–C5b–C5a can be taken (see Fig. 2), which describes the tilt of the central double bond: The positive value of η corresponds the bond tilting away from the observer; characteristic of the P helicity. The negative value of η corresponds to tilting towards the observer; i.e., the M helicity. With this additional dihedral angle, nearly all geometries at the start of the trajectories belong in the P helicity; see Supplementary Fig. 3. In accord with helicity, the preferred direction of torsion (CCW) is imposed by the slope on the S1 PES in the vicinity of the FC geometry (see Fig. 4A and B of ref. 39 for gas-phase and Supplementary Fig. 22 for QM/MM PESs of the motor 1). However, in some minor cases we hypothesise that the effect of the solvation shell causes the initial momenta of the nuclei, which are directed in the opposite (CW) sense of rotation and are large enough to override the motion in the direction imposed by the slope on the PES; thus causing motor’s rotation in the undesired CW sense (see the Supplementary multimedia files). The presence of the two renegade trajectories reduces unidirectionality (a fraction that propagates in the same direction) to 0.92 ± 0.05. The cause of this reduction and possible ways of ameliorating it will be the subject of future work.
In the EP → ZP step, the majority of the S1 → S0 surface hops occur within 522 ± 70 fs, which results in the S1 lifetime \({\tau }_{{{{{{{{{\rm{S}}}}}}}}}_{{{{{{{{\rm{1}}}}}}}}}}=485\pm 71\) fs obtained by a monoexponential fit; see Fig. 4. As also observed in ref. 39, in the productive trajectories, the time required to reach the final ZP conformation (θ > 360°, which is the same as θ > 0°) ranges between 1 and 2.5 ps, although one trajectory required nearly 5 ps. Longer simulation times are avoided in this study. This is because the size of the solvent droplet (906 MeOH molecules) is limited and one has to avoid a situation where the dissipation of the thermal energy gained by the solvent molecules (i.e., the displacement of solvent molecules due to the isomerisation motion) reaches the droplet boundary. This is consistent with the notion that at short simulation times, the dynamics is not affected as the intermolecular vibrational energy distribution occurs, typically, on a timescale of several picoseconds69.
The calculated Φiso of the EP → ZP step is 0.33 ± 0.09, which is qualitatively in line with the experimentally measured value of 0.25 ± 0.0539. This value is considerably reduced from the theoretical gas phase Φiso = 0.87 ± 0.05. Together with the theoretical S1 lifetime (485 ± 71 fs) which is also qualitatively in line with the experimental estimate of 600 fs (no error bars were given in ref. 39) this implies that the photoisomerisation mechanism of this step is described by the theory (this result is similar to that of the FSSH method also used in ref. 39 for the description of the EP → ZP step only).
The ZP → EP step displays somewhat faster and previously unexplored relaxation dynamics, \({\tau }_{{{{{{{{{\rm{S}}}}}}}}}_{{{{{{{{\rm{1}}}}}}}}}}=341\pm 28\) fs, and somewhat greater Φiso = 0.43 ± 0.09; see Fig. 4. A somewhat higher theoretical value of Φiso of this step is consistent with the experimentally observed photostationary state (PSS) of 55:45 for the ratio of the EP:ZP concentrations (at PSS, the ratio of concentrations is inversely proportional to the ratio of quantum yields)39. The Φiso value in MeOH is more than twofold reduction of the gas phase value (0.91 ± 0.04), and it is obviously caused by the solvent. Interestingly, the unidirectionality of this step in MeOH (0.97 ± 0.03) also deteriorates slightly from the perfect (1.0) unidirectionality in gas phase simulations39. This notwithstanding, the productive trajectories reach their final (EP) conformations within 1.2–3.4 ps, which is slightly longer than in the previous photoisomerisation step (EP → ZP).
The QM/MM NAMD simulations of both photochemical steps of MTDP in methanol solution indicate that there is indeed a considerable decrease in Φiso. Visual analysis of the geometries along the QM/MM NAMD trajectories suggests that this may be the ability of MeOH to establish hydrogen bonds (H-bonds) with proton donor and acceptor sites in the rotor blade that is responsible for such a dramatic deterioration of motor characteristics. As seen in Fig. 2, 1 has one H-acceptor site, carbonyl oxygen \({{{{{{{{\rm{O}}}}}}}}}_{6{\prime} }\) and one H-donor site, \({{{{{{{{\rm{H}}}}}}}}}_{1{\prime} }\), which can participate in H bonding with neighbouring MeOH molecules.
Because photochemically induced torsion occurs on an ultrafast timescale (~0.5 ps), the positions of the solvent molecules remain largely the same during torsion and only minor reorientation of the surrounding MeOH molecules may occur within this time frame. Therefore, if there are persistent H-bonds between MTDP and MeOH, they can slow down the movement of the rotor blade at the initial stage of isomerisation and deter progression of the rotary motion in the CCW direction near the CoIn regions. In such a case, the most likely outcome will be to turn back to restoring the reactant conformation, which results in an unproductive trajectory.
To test this hypothesis, the shortest distances between the H-bonding sites of 1 and MeOH were calculated for all productive and unproductive trajectories in the geometries of the S1 → S0 hopping. In particular, the shortest distances between amino hydrogen \({{{{{{{{\rm{H}}}}}}}}}_{1{\prime} }\) of 1 and oxygen from MeOH, and carbonyl oxygen \({{{{{{{{\rm{O}}}}}}}}}_{6{\prime} }\) and hydrogen from MeOH were inspected. Supplementary Fig. 4 shows distributions of these distances for the two isomerisation steps of 1.
As seen in Supplementary Fig. 4, at the moments of surface hops, many trajectories have distances (d) between the H-bonding sites of 1 and MeOH, which fall in the range of typical H-bonds, d < 2.0Å. Many other trajectories have somewhat stretched H-bonds, 2.0 < d < 2.5Å. On average, the H bonds with the carbonyl oxygen \({{{{{{{{\rm{O}}}}}}}}}_{6{\prime} }\) of 1 are shorter than the H-bonds with the amino hydrogen \({{{{{{{{\rm{H}}}}}}}}}_{1{\prime} }\); see Supplementary Fig. 4.
The greatest differences in the H-bond distances between the productive and unproductive trajectories are observed for the \({{{{{{{{\rm{O}}}}}}}}}_{6{\prime} }\) sites of 1. For the EP → ZP step, the productive trajectories have on average the MeOH⋯\({{{{{{{{\rm{O}}}}}}}}}_{6{\prime} }\) distances of 2.620 ± 1.094Å (the mean ± the standard deviation), and the unproductive trajectories have 2.066 ± 0.519Å. In unproductive trajectories, the shortest H-bonding distances are more narrowly distributed around the average value, which suggests that the occurrence of a short MeOH⋯\({{{{{{{{\rm{O}}}}}}}}}_{6{\prime} }\) distance does indeed indicate that the trajectory may become unproductive. For productive trajectories, the distribution is much broader and many trajectories have MeOH⋯\({{{{{{{{\rm{O}}}}}}}}}_{6{\prime} }\) distances longer than 2.5Å.
The \({{{{{{{{\rm{N}}}}}}}}}_{1{\prime} }\)–\({{{{{{{{\rm{H}}}}}}}}}_{1{\prime} }\cdots \,\)O distances in the photostep EP → ZP are more narrowly distributed, 2.415 ± 0.511Å for unproductive and 2.577 ± 0.475Å for productive trajectories, and only a relatively small number of EP → ZP trajectories have distances < 2Å; see Supplementary Fig. 4. This suggests that these H-bonds exert much less influence on the outcome of this photostep.
For the ZP → EP photostep, the relative influence of the two types of H-bonds, MeOH⋯\({{{{{{{{\rm{O}}}}}}}}}_{6{\prime} }\) and \({{{{{{{{\rm{H}}}}}}}}}_{1{\prime} }\cdots \,\)O, is reversed. There, the \({{{{{{{{\rm{H}}}}}}}}}_{1{\prime} }\cdots \,\)O interactions seem to have a more significant effect on isomerisation productivity than the MeOH⋯\({{{{{{{{\rm{O}}}}}}}}}_{6{\prime} }\) interactions. Indeed, the difference of the \({{{{{{{{\rm{H}}}}}}}}}_{1{\prime} }\cdots \,\)O distances between the productive and unproductive trajectories, 2.634 ± 0.804Å and 2.323 ± 0.472Å, respectively, is noticeably greater, than the difference of the MeOH⋯\({{{{{{{{\rm{O}}}}}}}}}_{6{\prime} }\) distances, 2.184 ± 0.513Å and 1.924 ± 0.273Å, respectively. In addition to that, the \({{{{{{{{\rm{H}}}}}}}}}_{1{\prime} }\cdots \,\)O distances display a much broader distribution of lengths for productive trajectories, and for many productive trajectories these distances are considerably longer than 2.5Å. Therefore, it is plausible to assume that the \({{{{{{{{\rm{H}}}}}}}}}_{1{\prime} }\cdots \,\)O interactions affect the outcome of the ZP → EP photostep, likely, due to interfering with the rotary movement of the rotor blade at the moment of S1 → S0 surface hops.
The effect of the MeOH⋯\({{{{{{{{\rm{O}}}}}}}}}_{6{\prime} }\) interactions on the quantum yield of the ZP → EP photostep is less obvious, as the average distances for productive and unproductive trajectories are fairly close. However, the unproductive trajectories have somewhat shorter lengths of these distances and are more narrowly distributed across the trajectories. Therefore, these interactions still have a certain, albeit a bit milder, influence on the balance between productive and unproductive ZP → EP trajectories.
The presented analysis suggests that blocking the possibility of establishing H-bonds with solvent molecules may help to improve the quantum efficiency (i.e., the balance between productive and unproductive trajectories) of the MTDP motor. Of the two H-bonding sites in the rotor blade of MTDP, the \({{{{{{{{\rm{O}}}}}}}}}_{6{\prime} }\) and \({{{{{{{{\rm{H}}}}}}}}}_{1{\prime} }\), the former may be difficult to modify chemically, however the latter can be eliminated by methylation at the \({{{{{{{{\rm{N}}}}}}}}}_{1{\prime} }\) position. Because the \({{{{{{{{\rm{H}}}}}}}}}_{1{\prime} }\cdots \,\)O interactions exerted greater influence on QY of the ZP → EP photostep of MTDP, it is expected that, in this photostep, the proposed methylation may result in obtaining Φiso closer to the gas phase value. As for the other photostep, the effect of such a modification seems less obvious, because the \({{{{{{{{\rm{H}}}}}}}}}_{1{\prime} }\cdots \,\)O H-bonds did not act as the major deterrent to its Φiso.
The suggested modification is achieved in the MMTDP motor 2 (E-1-methyl-3’-(2-methyl-2,3-dihydro-1H-benzo[b]cyclopenta[d]thiophen-1-ylidene)pyrrolidin-2’-one) shown in Fig. 2. Indeed, because the amino hydrogen \({{{{{{{{\rm{H}}}}}}}}}_{1{\prime} }\) is now replaced by a methyl group \({{{{{{{{\rm{C}}}}}}}}}_{1{\prime} }\)H3, the H-bonding with the MeOH oxygen is no longer possible. In addition to that, the presence of a bulkier group (Me- instead of H-) displaces MeOH molecules somewhat farther away from the \({{{{{{{{\rm{N}}}}}}}}}_{1{\prime} }\) atom and further reduces the remaining H-bonding ability of the amino group of 2.
The QM/MM NAMD simulations of 2 were set up in the same way as in the case of 1; see Supplementary Note 3 for details. Similarly to 1, 30 trajectories were propagated on the S1 PES for a maximum time of 1.5 ps or until the surface hop S1 → S0 occurs. After the hop, the trajectories were propagated on the S0 PES until they reached a stable final conformation.
The results of the simulations are summarised in Fig. 5. The time evolution of the S1 population became noticeably longer for 2 than it was for 1; especially, for the EP → ZP photostep. The time to reach the final isomerisation product has also been lengthened, and it takes up to 5 ps for the trajectories to settle in the final structure; see Fig. 5. This is consistent with the increase in rotation friction caused by the presence of a larger group in 2, which protrudes beyond the frame of the rotor blade. In this way, the undertaken simulations hint at possible characteristics of the motor under an increased load.

a The time evolution of the excited-state population n for the EP → ZP step. The red curve is fitted to the computed populations (the blue dots) by a monoexponential function nfit(t) = \({e}^{-({t}_{0}-{t})/ \tau }\), where t0 is the latency time and τ is the exponential decay constant. The excited-state lifetime \({t}_{{{{\mbox{S}}}}_{1}}\) is shown in the inset. The margin of error of the fitted quantities was obtained by bootstrapping with 104 replicas40. b The time evolution of the central dihedral angle θ. The blue curves show the productive trajectories (i.e., undergoing EP → ZP isomerisation), the black curves show the unproductive trajectories (i.e., turning back, EP → EP), the grey curves show the trajectories rotating in the wrong direction (i.e., CW, instead of CCW). The red crosses show the S1 → S0 nonadiabatic transitions. The red dots show the terminal points of the trajectories, where the final value of the angle θ was reached. Horizontal blue lines show the division between the EP/EM and ZP/ZM rotamers. Panels c) and d) show the characteristics of the ZP → EP step.
The most noticeable outcome of the simulations is that Φiso did not decrease for both photosteps. For the EP → ZP step Φiso remained unchanged, 0.33 ± 0.09. However, it increased noticeably for the ZP → EP photostep, from 0.43 ± 0.09 to 0.60 ± 0.09. This increase is beyond the statistical margin of error (evaluated by bootstrapping) and is consistent with the hypothesis that blocking the H-bonding capacity of the amino group of 1 should increase the quantum efficiency of the ZP → EP photostep.
As regards the other characteristics of the photoreactions, the unidirectionality of rotation in the EP → ZP step deteriorated slightly; now there are three renegade trajectories (instead of two) propagating in the CW direction, which is a drop from unidirectionality 0.92 ± 0.05 for 1 to 0.90 ± 0.05 for 2. However, such a decrease is well within the statistical margin of error, and, probably, a much greater number of simulations would be needed to narrow it down.
Interestingly, the effect of the MeOH⋯\({{{{{{{{\rm{O}}}}}}}}}_{6{\prime} }\) interactions on the outcome of both photosteps of 2 has also decreased compared to 1. The shortest lengths of MeOH⋯\({{{{{{{{\rm{O}}}}}}}}}_{6{\prime} }\) distances at the moment of the S1 → S0 hop in the productive and unproductive trajectories become 2.543 ± 0.999Å and 2.616 ± 0.745Å for the EP → ZP step, and 2.345 ± 0.837Å and 2.315 ± 0.592Å for the ZP → EP step; see Supplementary Fig. 4. This suggests that in 2, some other factors than the MeOH⋯\({{{{{{{{\rm{O}}}}}}}}}_{6{\prime} }\) interaction may be responsible for the unchanged Φiso of the EP → ZP step. Quite likely, this could be increased friction in torsion caused by the methyl group protruding from the rotor blade of 2. However, more simulations and analyses would be needed to uncover the origin of the lower Φiso of the EP → ZP photostep. Investigation of this is deferred to future work.
As a hypothesis to be investigated in future work, the decreased influence of the MeOH⋯\({{{{{{{{\rm{O}}}}}}}}}_{6{\prime} }\) bond on the dynamics of 2 can be assigned to an increase in attraction between \({{{{{{{{\rm{O}}}}}}}}}_{6{\prime} }\) and hydrogens of the methyl group at C7, which competes with the former influence. This is equivalent to an intramolecular C–H⋯O H-bond70,71,72. In S1, the charge on \({{{{{{{{\rm{O}}}}}}}}}_{6{\prime} }\) becomes slightly more negative (by approximately -0.016e) in MMTDP. This results in strengthening of the intramolecular H-bond and in exerting a slightly stronger force opposing motion in the direction of isomerisation. Besides influencing the quantum yield, this interaction is likely to be responsible for the increased latency time of the EP → ZP photostep of 2, which is needed by the rotor blade to rotate towards the CoIn region. This mechanistic hypothesis requires further investigation, which is deferred to future work.
Conclusions
In conclusion, the set of engineering rules to design and prepare prospective 2-stroke LDRMs, such as MTDP and MMTDP, was extended to include some factors that influence the quantum efficiency of motors in a medium. In particular, this involves the ability of a motor molecule to operate efficiently in a solvent capable of forming hydrogen bonds with the atoms in the motor backbone. Through modifying one of the centres to which solvent molecules can form H-bonds, a noticeably higher quantum efficiency of one of the photosteps (by ca. 40%) was predicted from multiscale modelling.
Besides blocking the H-bonding ability, methylation of the amino nitrogen of MTDP (see Fig. 2) can potentially provide a handle to which a harness can be attached to utilize the rotary movement of LDRM in a molecular device. According to the presented simulations, attaching a harness to the 2-stroke LDRM does not deteriorate its operational efficiency, despite the increased friction due to the solvent.
Therefore, the rules for designing efficient LDRMs that remain operational at a wide range of temperatures (excluding very low ones) can be formulated as follows:
-
1.
Ensure the desired axial mode of rotation (as opposed to precessional motion typical for classic overcrowded alkene LDRMs)26,27,73 by balancing the energetic preference for homolytic and heterolytic breaking of the central π-bond (the axle)34,36,62,64,73. This prescription can be achieved by introducing more electronegative substituents into one of the motor blades, which shifts the balance towards heterolytic π-bond breaking34,73. Besides providing for a cleaner axial mode of rotation, this prescription can potentially increase the quantum efficiency and shorten the excited state lifetime by making the respective conical intersection seam more accessible; as, in this case, it typically has more peaked topography and occurs at the bottom of local minimum on the excited state PES74,75,76. Although this prescription was not directly addressed in this work, it has been amply discussed in the previous publications on the design of LDRMs36,62,64,73, and the MTDP and MMTDP LDRMs presented here satisfy these requirements36,39.
-
2.
Eliminate the THI steps in the working cycle of LDRM by: i) Introducing a chiral strained unit; e.g., MCP unit of the MTDP and MMTDP motors that incorporates the chiral centre and directs photoinduced rotation in a single direction. ii) Reducing the steric strain in the fjord region; e.g., due to the use of pentalene-like units in the stator blade of MTDP and MMTDP39.
-
3.
Block, as much as possible, the ability of LDRM to establish hydrogen bonds with the solvent molecules. Because H-bonds are typically stronger than other noncovalent interactions, they more efficiently transmit the influence of the Brownian motion of the solvent on the LDRM. The results of the present work furnish evidence that Φiso can increase due to blocking some unwanted noncovalent interactions, such as H-bonds. This may also involve changing the solvent from polar protic to polar aprotic (e.g. as in acetonitrile). However, the experimental measurements in ref. 39 were taken in methanol solution and, therefore, the present work focuses on this solvent to enable connection to the existing experimental measurements. Simulations in an aprotic solvent (acetonitrile) will be reported in a future publication.
Although more computational modelling combined with experimental characterisation is needed to address all aspects of the functioning of LDRMs, the presented results should foster the invention of new molecular designs with application-defined functionality.
Methods
We used the state-interaction state-averaged spin-restricted ensemble-referenced Kohn-Sham (SI-SA-REKS, or SSR) method46,47,48,62,77 to obtain the total energies and forces on the nuclei. In the SSR method, ensemble density functional theory (eDFT)49,50,51,52,53,54,55,56,57,58 is used to introduce the strong nondynamic correlation into the description of the ground and excited electronic states of molecules and to obtain the excitation energies in a time-independent fashion, reminiscent of the multiconfigurational methods of wavefunction theory. With the use of eDFT, a seamless incorporation of the multireference effects into the computations is achieved and the results of the standard KS computations are recovered for the weakly correlated (single-reference) systems. At the same time, it provides a much-improved description of the systems with dissociating chemical bonds, biradical and polyradical electronic states, and electronic states with pronounced charge transfer47,48. In ensemble representation, fractional occupation numbers (FONs) of several frontier KS orbitals occur, which can be used to characterise the strength of the multiconfigurational correlation effects.
The surface hopping from exact factorisation (SHXF) method41,44,45 combines the electronic equations derived from the exact factorisation of the electronic-nuclear wavefunction78,79,80,81,82 with the conventional trajectory surface hopping (TSH) formalism83. A seamless incorporation of the effect of nuclear quantum momentum is achieved from the exact factorisation, which introduces dependence on the shape of nuclear distribution into the classical equations of motion for the nuclei. In this way, a seamless decoherence of nuclear trajectories is achieved41.
All quantum chemical calculations are performed using the local version of the GAMESS-US program (2018.v3),84,85 which implements the SSR method and the analytic derivative formalism86. All calculations employ the 6-31G* basis set87 and the BH&HLYP exchange-correlation density functional88,89,90. The QM/MM calculations were performed by GAMESS-US interfaced with the TINKER (v6.3) program91. The force field calculations of the solvent molecules employed the OPLS-AA force field92. The NAMD simulations were performed by the pyUNIxMD program44,45, a standalone code that implements the SHXF method41. More information on the computational methods and the details of computations can be found in the Supplementary Note 2. Additional results of the theoretical simulations are reported in the Supplementary Note 3.
Data availability
Supplementary Files contain the raw data for the figures in the main article and multimedia files animating different types of trajectories occurring during the dynamics. Supplementary Data 1: A zipped folder with the raw data for Figs. 4 and 5 of the main article. Supplementary Movie 1: A film animating a productive trajectory for the EP MTDP motor isomerising in the CCW direction. Supplementary Movie 2: A film animating an unproductive trajectory for the EP MTDP motor isomerising in the CCW direction. Supplementary Movie 3: A film animating an unproductive trajectory for the EP MTDP motor isomerising in the CW direction.
Code availability
The computer code used in this research is available from the corresponding author (MF) upon request.
References
Koumura, N., Zijlstra, R. W. J., Delden, R. A., Harada, N. & Feringa, B. L. Light-driven monodirectional molecular rotor. Nature 401, 152–155 (1999).
Koumura, N., Geertsema, E. M., Gelder, M. B., Meetsma, A. & Feringa, B. L. Second generation light-driven molecular motors. unidirectional rotation controlled by a single stereogenic center with near-perfect photoequilibria and acceleration of the speed of rotation by structural modification. J. Am. Chem. Soc. 124, 5037–5051 (2002).
Pollard, M. M., Meetsma, A. & Feringa, B. L. A redesign of light-driven rotary molecular motor. Org. Biomol. Chem. 6, 507–512 (2008).
Credi, A. Artificial molecular motors powered by light. Aust. J. Chem. 59, 157–169 (2006).
Balzani, V., Credi, A. & Venturi, M. Light powered molecular machines. Chem. Soc. Rev. 38, 1542–1550 (2009).
Roke, D., Wezenberg, S. J. & Feringa, B. L. Molecular rotary motors: Unidirectional motion around double bonds. Proc. Natl. Acad. Sci. 115, 9423–9431 (2018).
García-López, V., Liu, D. & Tour, J. M. Light-activated organic molecular motors and their applications. Chem. Rev. 120, 79–124 (2020).
Corra, S., Curcio, M. & Credi, A. Photoactivated artificial molecular motors. JACS Au 3, 1301–1313 (2023).
Liu, D. et al. Near-infrared light activates molecular nanomachines to drill into and kill cells. ACS Nano 13, 6813–6823 (2019).
Ayala Orozco, C. et al. Visible-light-activated molecular nanomachines kill pancreatic cancer cells. ACS Appl. Mater. Interfaces 12, 410–417 (2020).
Gunasekera, R. S. et al. Molecular nanomachines can destroy tissue or kill multicellular eukaryotes. ACS Appl. Mater. Interfaces 12, 13657–13670 (2020).
Zheng, Y. et al. Optoregulated force application to cellular receptors using molecular motors. Nat. Commun. 12, 3580 (2021).
Zhu, Q. et al. Multistate switching of spin selectivity in electron transport through light-driven molecular motors. Adv. Sci. 8, 2101773 (2021).
Santos, A. L. et al. Light-activated molecular machines are fast-acting broad-spectrum antibacterials that target the membrane. Sci. Adv. 8, 2055 (2022).
Lan, R. et al. Amplifying molecular scale rotary motion: The marriage of overcrowded alkene molecular motor with liquid crystals. Adv. Mater. 34, 2109800 (2022).
Stähler, C. et al. Light-driven molecular motors embedded in covalent organic frameworks. Chem. Sci. 13, 8253–8264 (2022).
Gao, C., Vargas Jentzsch, A., Moulin, E. & Giuseppone, N. Light-driven molecular whirligig. J. Am. Chem. Soc. 144, 9845–9852 (2022).
Greb, L. & Lehn, J.-M. Light-driven molecular motors: Imines as four-step or two-step unidirectional rotors. J. Am. Chem. Soc. 136, 13114–13117 (2014).
Greb, L., Eichhöfer, A. & Lehn, J.-M. Synthetic molecular motors: Thermal N inversion and directional photoinduced C=N bond rotation of camphorquinone imines. Angew. Chem. Int. Ed. 54, 14345–14348 (2015).
Guentner, M. et al. Sunlight-powered kHz rotation of a hemithioindigo-based molecular motor. Nat. Comm. 6, 8406–8 (2015).
Wilcken, R. et al. Complete mechanism of hemithioindigo motor rotation. J. Am. Chem. Soc. 140, 5311–5318 (2018).
Gerwien, A., Mayer, P. & Dube, H. Photon-only molecular motor with reverse temperature-dependent efficiency. J. Am. Chem. Soc. 140, 16442–16445 (2018).
Atchity, G. J., Xantheas, S. S. & Ruedenberg, K. Potential energy surfaces near intersections. J. Chem. Phys. 95, 1862–1876 (1991).
Yarkony, D. R. Diabolical conical intersections. Rev. Mod. Phys. 68, 985–1013 (1996).
Bernardi, F., Olivucci, M. & Robb, M. A. Potential energy surface crossings in organic photochemistry. Chem. Soc. Rev. 25, 321–328 (1996).
Kazaryan, A. et al. Understanding the dynamics behind the photoisomerization of a light-driven fluorene molecular rotary motor. J. Phys. Chem. A 114, 5058–5067 (2010).
Kazaryan, A., Lan, Z., Schäfer, L. V., Thiel, W. & Filatov, M. Surface hopping excited-state dynamics study of the photoisomerization of a light-driven fluorene molecular rotary motor. J. Chem. Theory Comput. 7, 2189–2199 (2011).
Conyard, J. et al. Ultrafast dynamics in the power stroke of a molecular rotary motor. Nature Chem. 4, 547–551 (2012).
Marchand, G. et al. Directionality of double-bond photoisomerization dynamics induced by a single stereogenic center. J. Phys. Chem. Lett. 6, 599–604 (2015).
Wang, J. & Durbeej, B. Toward fast and efficient visible-light-driven molecular motors: A minimal design. ChemistryOpen 7, 583–589 (2018).
Geertsema, E. M., Molen, S. J., Martens, M. & Feringa, B. L. Optimizing rotary processes in synthetic molecular motors. Proc. Nat. Acad. Sci. 106, 16919–16924 (2009).
Pooler, D. R. S., Lubbe, A. S., Crespi, S. & Feringa, B. L. Designing light-driven rotary molecular motors. Chem. Sci. 12, 14964–14986 (2021).
Conyard, J., Cnossen, A., Browne, W. R., Feringa, B. L. & Meech, S. R. Chemically optimizing operational efficiency of molecular rotary motors. J. Am. Chem. Soc. 136, 9692–9700 (2014).
Pooler, D. R. S. et al. Effect of charge-transfer enhancement on the efficiency and rotary mechanism of an oxindole-based molecular motor. Chem. Sci. 12, 7486–7497 (2021).
García-Iriepa, C. et al. Chiral hydrogen bond environment providing unidirectional rotation in photoactive molecular motors. J. Chem. Phys. Lett. 4, 1389–1396 (2013).
Filatov, M., Paolino, M., Min, S. K. & Choi, C. H. Design and photoisomerization dynamics of a new family of synthetic 2-stroke light driven molecular rotary motors. Chem. Commun. 55, 5247–5250 (2019).
Ma, J., Zhao, D., Jiang, C., Lan, Z. & Li, F. Effect of temperature on photoisomerization dynamics of a newly designed two-stroke light-driven molecular rotary motor. Int. J. Mol. Sci. 23, 9694 (2022).
Boursalian, G. B. et al. All-photochemical rotation of molecular motors with a phosphorus stereoelement. J. Am. Chem. Soc. 142, 16868–16876 (2020).
& Filatov, M. et al. Towards the engineering of a photon-only two-stroke rotary molecular motor. Nat. Commun. 13, 6433 (2022).
Nangia, S., Jasper, A. W., Miller, T. F. & Truhlar, D. G. Army ants algorithm for rare event sampling of delocalized nonadiabatic transitions by trajectory surface hopping and the estimation of sampling errors by the bootstrap method. J. Chem. Phys. 120, 3586–3597 (2004).
Ha, J.-K., Lee, I. S. & Min, S. K. Surface hopping dynamics beyond nonadiabatic couplings for quantum coherence. J. Phys. Chem. Lett. 9, 1097–1104 (2018).
Min, S. K., Agostini, F., Tavernelli, I. & Gross, E. K. U. Ab initio nonadiabatic dynamics with coupled trajectories: A rigorous approach to quantum (de)coherence. J. Phys. Chem. Lett. 8, 3048–3055 (2017).
Agostini, F., Min, S. K., Abedi, A. & Gross, E. K. U. Quantum-classical nonadiabatic dynamics: Coupled- vs independent-trajectory methods. J. Chem. Theory Comput. 12, 2127–2143 (2016).
Lee, I. S. et al. Pyunixmd: A python-based excited state molecular dynamics package. J. Comp. Chem. 42, 1755–1766 (2021).
Kim, T. I., Ha, J.-K. & Min, S. K. Coupled- and independent-trajectory approaches based on the exact factorization using the pyUNIXMD package. Top. Curr. Chem. 380, 8 (2022).
Filatov, M. Assessment of density functional methods for obtaining geometries at conical intersections in organic molecules. J. Chem. Theory Comput. 9, 4526–4541 (2013).
Filatov, M. Spin-restricted ensemble-referenced kohn-sham method: basic principles and application to strongly correlated ground and excited states of molecules. WIREs Comput. Mol. Sci. 5, 146–167 (2015).
Filatov, M.: Ensemble DFT approach to excited states of strongly correlated molecular systems. In: Ferré, N., Filatov, M., Huix-Rotllant, M. (eds.) Density-functional Methods for Excited States. Top. Curr. Chem., Springer, Heidelberg vol. 368, pp. 97–124. (2016)
Valone, S. M. A one-to-one mapping between one-particle densities and some n-particle ensembles. J. Chem. Phys. 73, 4653–4655 (1980).
Lieb, E. H. Density functionals for coulomb systems. Int. J. Quantum Chem. 24, 243–277 (1983).
Perdew, J. P., Parr, R. G., Levy, M. & Balduz Jr, J. L. Density-functional theory for fractional particle number: Derivative discontinuities of the energy. Phys. Rev. Lett. 49, 1691–1694 (1982).
Englisch, H. & Englisch, R. Hohenberg-Kohn theorem and non-v-representable densities. Physica A121, 253–268 (1983).
Englisch, H. & Englisch, R. Exact density functionals for ground-state energies. I. General results. Phys. Stat. Sol. (b) 123, 711–721 (1984).
Englisch, H. & Englisch, R. Exact density functionals for ground-state energies II. Details and remarks. Phys. Stat. Sol. (b) 124, 373–379 (1984).
Gross, E. K. U., Oliveira, L. N. & Kohn, W. Rayleigh-Ritz variational principle for ensembles of fractionally occupied states. Phys. Rev. A 37, 2805–2808 (1988).
Gross, E. K. U., Oliveira, L. N. & Kohn, W. Density-functional theory for ensembles of fractionally occupied states. I. Basic formalism. Phys. Rev. A 37, 2809–2820 (1988).
Oliveira, L. N., Gross, E. K. U. & Kohn, W. Density-functional theory for ensembles of fractionally occupied states. II. Application to the He atom. Phys. Rev. A 37, 2821–2833 (1988).
Oliveira, L. N., Gross, E. K. U. & Kohn, W. Ensemble-density functional theory. Int. J. Quantum Chem.: Quantum Chem. Symp. 24, 707–716 (1990).
Filatov, M. & Shaik, S. A spin-restricted ensemble-referenced Kohn-Sham method and its application to diradicaloid situations. Chem. Phys. Lett. 304, 429–437 (1999).
Moreira, Id. P. R., Costa, R., Filatov, M. & Illas, F. Restricted ensemble-referenced Kohn-Sham versus broken symmetry approaches in density functional theory: Magnetic coupling in Cu binuclear complexes. J. Chem. Theory Comput. 3, 764–774 (2007).
Kazaryan, A., Heuver, J. & Filatov, M. Excitation energies from spin-restricted ensemble-referenced Kohn-Sham method: A state-average approach. J. Phys. Chem. A 112, 12980–12988 (2008).
Nikiforov, A., Gamez, J. A., Thiel, W. & Filatov, M. Computational design of a family of light-driven rotary molecular motors with improved quantum efficiency. J. Phys. Chem. Lett. 7, 105–110 (2016).
Paolino, M. et al. Design, synthesis, and dynamics of a green fluorescent protein fluorophore mimic with an ultrafast switching function. J. Am. Chem. Soc. 138, 9807–9825 (2016).
Filatov, M., Paolino, M., Min, S. K. & Kim, K. S. Fulgides as light-driven molecular rotary motors: Computational design of a prototype compound. J. Phys. Chem. Lett. 9, 4995–5001 (2018).
Liang, R., Liu, F. & Martínez, T. J. Nonadiabatic photodynamics of retinal protonated Schiff base in channelrhodopsin 2. J. Phys. Chem. Lett. 10, 2862–2868 (2019).
Yu, J. K., Liang, R., Liu, F. & Martínez, T. J. First-principles characterization of the elusive I fluorescent state and the structural evolution of retinal protonated Schiff base in bacteriorhodopsin. J. Am. Chem. Soc. 141, 18193–18203 (2019).
Liang, R., Yu, J. K., Meisner, J., Liu, F. & Martínez, T. J. Electrostatic control of photoisomerization in channelrhodopsin 2. J. Am. Chem. Soc. 143, 5425–5437 (2021).
Barneschi, L. et al. Assessment of the electron correlation treatment on the quantum-classical dynamics of retinal protonated Schiff base models: XMS-CASPT2, RMS-CASPT2, and REKS methods. J. Chem. Theory Comput. 19, 8189–8200 (2023).
Park, S.-M., Nguyen, P. H. & Stock, G. Molecular dynamics simulation of cooling: Heat transfer from a photoexcited peptide to the solvent. J. Chem. Phys. 131, 184503 (2009).
Desiraju, G. R. The C-H⋯O hydrogen bond: Structural implications and supramolecular design. Acc. Chem. Res. 29, 441–449 (1996).
Gu, Y., Kar, T. & Scheiner, S. Fundamental properties of the CH⋯O interaction: Is it a true hydrogen bond? J. Am. Chem. Soc. 121, 9411–9422 (1999).
Scheiner, S. Weak H-bonds. comparisons of CH⋯O to NH⋯O in proteins and PH⋯N to direct P⋯N interactions. Phys. Chem. Chem. Phys. 13, 13860–13872 (2011).
Filatov, M. & Olivucci, M. Designing conical intersections for light-driven single molecule rotary motors: From precessional to axial motion. J. Org. Chem. 79, 3587–3600 (2014).
Ben-Nun, M., Molnar, F., Schulten, K. & Martínez, T. J. The role of intersection topography in bond selectivity of cis-trans photoisomerization. Proc. Nat. Acad. Sci. 99, 1769–1773 (2002).
Virshup, A. M., Chen, J. & Martínez, T. J. Nonlinear dimensionality reduction for nonadiabatic dynamics: The influence of conical intersection topography on population transfer rates. J. Chem. Phys. 137, 22–519 (2012).
Liu, L., Liu, J. & Martínez, T. J. Dynamical correlation effects on photoisomerization: Ab initio multiple spawning dynamics with MS-CASPT2 for a model trans-protonated Schiff base. J. Phys. Chem. B 120, 1940–1949 (2016).
Filatov, M., Martínez, T. J. & Kim, K. S. Using the GVB Ansatz to develop ensemble DFT method for describing multiple strongly correlated electron pairs. Phys. Chem. Chem. Phys. 18, 21040–21050 (2016).
Hunter, G. Conditional probability amplitudes in wave mechanics. Int. J. Quantum Chem. 9, 237–242 (1975).
Abedi, A., Maitra, N. T. & Gross, E. K. U. Exact factorization of the time-dependent electron-nuclear wave function. Phys. Rev. Lett. 105, 123002 (2010).
Abedi, A., Maitra, N. T. & Gross, E. K. U. Correlated electron-nuclear dynamics: Exact factorization of the molecular wave-function. J. Chem. Phys. 137, 22–530 (2012).
Abedi, A., Agostini, F., Suzuki, Y. & Gross, E. K. U. Dynamical steps that bridge piecewise adiabatic shapes in the exact time-dependent potential energy surface. Phys. Rev. Lett. 110, 263001 (2013).
Agostini, F. et al. The exact electronic back-reaction on classical nuclei in non-adiabatic charge transfer. J. Chem. Phys. 142, 084303 (2015).
Tully, J. C. Molecular dynamics with electronic transitions. J. Chem. Phys. 93, 1061 (1990).
Schmidt, M. W. et al. General atomic and molecular electronic structure system. J. Comput. Chem. 14, 1347–1363 (1993).
Gordon, M.S., Schmidt, M.W.: Theory and Applications of Computational Chemistry, the First Forty Years, 1167-1189. Elsevier, Amsterdam (2005)
Filatov, M., Liu, F. & Martínez, T. J. Analytical derivatives of the individual state energies in ensemble density functional theory method. I. General formalism. J. Chem. Phys. 147, 034113 (2017).
Krishnan, R., Binkley, J. S., Seeger, R. & Pople, J. A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 72, 650–654 (1980).
Becke, A. D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 38, 3098–3100 (1988).
Lee, C., Yang, W. & Parr, R. G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 37, 785–789 (1988).
Becke, A. D. A new mixing of Hartree-Fock and local density-functional theories. J. Chem. Phys. 98, 1372–1377 (1993).
Ponder, F. M. & Richards, J. W. Tinker molecular modeling package. J. Comput. Chem. 8, 1016–1024 (1987).
Caleman, C. et al. Force field benchmark of organic liquids: Density, enthalpy of vaporization, heat capacities, surface tension, isothermal compressibility, volumetric expansion coefficient, and dielectric constant. J. Chem. Theory Comput. 8, 61–74 (2012).
Acknowledgements
This research was supported by the Korean National Research Foundation (NRF) funded by the Korean government (Ministry of Science and ICT (MSIT)) (NRF-2023M3K5A1094813, RS-2023-00257666). M.F. and S.K.M. were supported by the Institute for Basic Science (IBS-R019-D1) The computational work for this research was performed on the Olaf supercomputer supported by the IBS Research Solution Centre. M.O. is grateful to the NSF CHE-SDM A for Grant No. 2102619. M.O. and M.P. are also grateful for EU funding within the MUR PNRR National Centre for Gene Therapy and Drugs based on RNA Technology (Project no. CN00000041 CN3 RNA) Spoke 6. M.O. is also grateful to BGSU and the Centre for Photochemical Sciences for additional support.
Author information
Authors and Affiliations
Contributions
M.F. and M.P. have designed the molecule. M.F. and D.K. prepared the initial data for the simulations. M.F., E.K., and S.K.M. performed the simulations and analysed the results. M.F., M.O., and S.K.M. have written the manuscript.
Corresponding authors
Ethics declarations
Competing interests
Seung-Kyu Min is Guest Editor for the Photoactivated Molecular Switches and Motors Collection hosted by Communications Physics but was not involved in the editorial review of, or the decision to publish this article. All other authors declare no competing interests.
Inclusion & Ethics
The research reported here complies with the ethics & inclusion guidelines of the Nature Portfolio Journals.
Peer review
Peer review information
Communications Physics thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Filatov(Gulak), M., Paolino, M., Kaliakin, D. et al. Impact of solvation on the photoisomerisation dynamics of a photon-only rotary molecular motor. Commun Phys 7, 219 (2024). https://doi.org/10.1038/s42005-024-01716-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42005-024-01716-4
- Springer Nature Limited